

Abstract
Purpose
To determine underlying correlations in multiplex ligation-dependent probe amplification (MLPA) data and their significance regarding survival following treatment of choroidal melanoma (CM).
Methods
MLPA data were available for 31 loci across four chromosomes (1p, 3, 6, and 8) in tumor material obtained from 602 patients with CM treated at the Liverpool Ocular Oncology Center (LOOC) between 1993 and 2012. Data representing chromosomes 3 and 8q were analyzed in depth since their association with CM patient survival is well-known. Unsupervised k-means cluster analysis was performed to detect latent structure in the data set. Principal component analysis (PCA) was also performed to determine the intrinsic dimensionality of the data. Survival analyses of the identified clusters were performed using Kaplan–Meier (KM) and log-rank statistical tests. Correlation with largest basal tumor diameter (LTD) was investigated.
Results
Chromosome 3: A two-cluster (bimodal) solution was found in chromosome 3, characterized by centroids at unilaterally normal probe values and unilateral deletion. There was a large, significant difference in the survival characteristics of the two clusters (log-rank, p<0.001; 5-year survival: 80% versus 40%). Both clusters had a broad distribution in LTD, although larger tumors were characteristically in the poorer outcome group (Mann–Whitney, p<0.001). Threshold values of 0.85 for deletion and 1.15 for gain optimized the classification of the clusters. PCA showed that the first principal component (PC1) contained more than 80% of the data set variance and all of the bimodality, with uniform coefficients (0.28±0.03). Chromosome 8q: No clusters were found in chromosome 8q. Using a conventional threshold-based definition of 8q gain, and in conjunction with the chromosome 3 clusters, three prognostic groups were identified: chromosomes 3 and 8q both normal, either chromosome 3 or 8q abnormal, and both chromosomes 3 and 8q abnormal. KM analysis showed 5-year survival figures of approximately 97%, 80%, and 30% for these prognostic groups, respectively (log-rank, p<0.001). All MLPA probes within both chromosomes were significantly correlated with each other (Spearman, p<0.001).
Conclusions
Within chromosome 3, the strong correlation between the MLPA variables and the uniform coefficients from the PCA indicates a lack of evidence for a signature gene that might account for the bimodality we observed. We hypothesize that the two clusters we found correspond to binary underlying states of complete monosomy or disomy 3 and that these states are sampled by the complete ensemble of probes. Consequently, we would expect a similar pattern to emerge in higher-resolution MLPA data sets. LTD may be a significant confounding factor. Considering chromosome 8q, we found that chromosome 3 cluster membership and 8q gain as traditionally defined have an indistinguishable impact on patient outcome.
Introduction
Uveal melanoma (UM) is the most common primary intraocular malignancy, which is fatal in approximately 50% of patients because of untreatable diffuse metastases, which usually involve the liver. The risk of UM metastasis can be determined with a high degree of accuracy, when considering the clinical, histomorphological, and genetic features of these tumors during treatment [1]. The Liverpool Ocular Oncology Center (LOOC) is one of three referral centers for treating adult ocular tumors in England. Here, genetic prognostication testing for patients with UM is routinely performed, as part of the patient management protocol.
Choroidal melanoma (CM) represents the majority of UM involving the choroid and potentially the adjacent ciliary body but not the iris. Data were collected on patients with CM treated at the LOOC, including basic demographics, follow-up, and outcome and, since 2006, relative chromosome expression measurements across 43 chromosome sites (loci) representing four chromosomes (1p, 3, 6, and 8), using the technique known as multiplex ligation-dependent probe amplification (MLPA SALSA P027.B1, MRC, Holland [1]. Loci are identified on each chromosome corresponding to certain genes. The loci data themselves constitute relative measurements of the amount of target DNA present in the tumor biopsy tissue compared with a reference sample. These measurements are therefore unit-less: A normal result is close to unity, whereas low (e.g., <0.65) or high (e.g., >1.35) probe readings indicate loss or gain, respectively, at the corresponding loci. These decision thresholds have been recommended by the equipment manufacturer but are not necessarily optimal. In particular, a reading of between 0.65 and 0.85 is usually interpreted as borderline loss and 1.15–1.35 as borderline gain.
Non-random genetic changes may arise as partial or complete deletions (monosomy) or amplifications of chromosomes (polysomy). There is convincing evidence that monosomy 3 and polysomy 8q are strongly associated with a poor survival prognosis in CM [2,3]. It would be useful to determine whether any of the MLPA loci predict clinical outcome in an individual patient and how the ensemble of MLPA data, itself a collection of continuous variables, is related to the categorical states of monosomy, disomy. or polysomy.
Comprehensive clinical, histomorphological, and genetic data exist for 602 patients with CM, but the high dimensionality of the data set means the data are sparse, and therefore relatively difficult to analyze with conventional statistical techniques. In this paper, we investigate the underlying structure of the data and correlation between MLPA variables, to reduce dimensionality and identify novel predictors of survival.
The main questions we addressed were as follows: What are the best thresholds in MLPA analysis for loss or gain? How are monosomy and polysomy best defined? Are there any special loci acting as “signature genes”? How well does MLPA analysis predict patient survival?
Methods
Patients entered into this study included all those with CM who were treated at the Royal Liverpool University Hospital between 1993 and 2013. MLPA analysis came into routine use in 2006. However, this analysis included a few patients who were treated before 2006 and for whom tumor samples were available. Exclusion criteria were bilateral CM. Of the four chromosomes analyzed, the focus of this work was chromosomes 3 and 8q, given their importance to CM patient survival. There were 13 loci within chromosome 3 and 4 loci in 8q (Table 1).
Table 1. The measurement loci for Chromosome 3p and 8q.
| Chromosomal Region | Gene |
|---|---|
| 3p25.3 |
FANCD2(i) |
| 3p24.3 |
FANCD2(ii) |
| 3p25.3 |
VHL |
| 3p22.1 |
MLH1 |
| 3p22 |
CTNNB1 |
| 3p21.3 |
SEMA3B |
| 3p14.2 |
FHIT(i) |
| 3p14.2 |
FHIT(ii) |
| 3p12.2 |
ROBO1 |
| 3q12 |
CPO |
| 3q21.3 |
RHO |
| 3q25.1 |
MME |
| 3q29 |
OPA1 |
| 8q11.23 |
RP1 |
| 8q24.12 |
MYC(i) |
| 8q24.12 |
MYC(ii) |
| 8q24.2 | DDEF1 |
Data were analyzed using MATLAB® (The Mathworks Inc., Natick, MA) to determine the distribution of patients with CM within each locus. Within chromosome 3, the 13 loci measured for each patient were interpreted as 13 orthogonal spatial dimensions, and the 602 patients represented points situated within this 13-dimensional space. Three-dimensional (3D) projections of the full data set were depicted given a choice of three specific loci.
Cluster analysis was used to identify latent structure in these data. Principal component analysis (PCA) was also performed to reduce the dimensionality of the data. The technique transformed the highly correlated combined input variables into a (potentially smaller) set of uncorrelated (and orthogonal) variables, which, when ordered, accounted for progressively smaller amounts of variance within the data. Geometrically, this amounted to rotating and translating the coordinate axes to align first with the direction of maximum spread (the first principal component), and then similarly for subsequent components, subject to the constraint of orthogonality. Higher-order principal components exhibiting negligible variance were discarded, and a projection of the data set was established that maximally exposed the structure/variance already detected. A scree plot was used to determine the optimum number of principal components to describe the data.
Finally, chromosome data were correlated with the largest basal tumor diameter. Survival of any groups of patients identified by the clustering and PCA was determined with Kaplan–Meier curves and log-rank statistics using SPSS version 20 (IBM, NY).
This study adhered to the tenets of the Declaration of Helsinki. (Please note that there is no particular statement for ARVO on human subjects: it also refers to the Declaration of Helsinki. There is only a separate one for the "Use of Animals in Ophthalmic and Vision Research). The Institutional Research Governance Board approved this study for service evaluation.
Results
Cluster analysis
A summary of the demographics of the patients with CM is shown in Table 2. Initial projections suggested PCA showed that the data points for chromosome 3 were distributed over a small range, typically between 0 and 2–2.5. In each projection, the presence of two distinct clusters within the data was (subjectively) discerned: One shifted toward higher probe measurements, and the other approximated zero. These clusters were better separated in some projections than in others, but the presence of two regions of high density was clear. This was confirmed with simple histogram plots of selected loci, where bimodal distributions were always observed.
Table 2. Clinicopathologic features of patients included in the study.
| Feature | n (%) | Median (Range) |
|---|---|---|
| Sex
Male
Female |
352 (58.5%)
250 (41.5%) |
|
| Age, year |
63.11 (15.75 – 94.4) |
|
| Largest basal tumour diameter, mm |
14.3 (4.1 – 23.8) |
|
| Epithelioid cellularity
Absent
Present
Unknown |
231 (38.4%)
341 (56.6%)
30 (5%) |
|
| Anterior margin
post-ora
pre-ora |
401 (66.6%)
201 (33.4%) |
|
| Extra-ocular spread
No
Yes |
525 (87.2%)
77 (12.8%) |
|
| Ciliary body involvement
No
Yes
Unknown |
401 (66.7%)
200 (33.2%)
1 (0.1%) |
|
| TNM stage
T1
T2
T3
T4
Unknown |
96 (15.9%)
159 (26.4%)
224 (37.2%)
119 (19.8%)
4 (0.7%) |
|
| Treatment
Brachytherapy
Scleral resection
Enucleation
Proton beam therapy
Endoresection
Treated elsewhere
Other |
87 (14.4%)
48 (8%)
312 (51.8%)
89 (14.8%)
38 (6.3%)
18 (3%)
10 (1.7%) |
|
| Followup time, year | 3.08 (0.07 – 19.7) |
The results of a two-means cluster analysis of the chromosome 3 data are shown in Appendix 1 and Appendix 2. These appendices show the 3D projections and the histograms, with the data classified into one or other of the clusters, indicated by the markers in different shades of gray. Since the output of this iterative algorithm depends (in principle) upon the initial, random allocation of data to the classes, the routine was executed five times, and on each occasion, the total distance to the centroids in the final iteration was calculated and found to be stable at 165.27. Stability of this parameter across trials suggests that a global minimum of convergence was found. These appendices show that the first cluster (C1) generally corresponds to lower MLPA measurements, with its centroid indicating deletions on all loci, whereas the second cluster (C2) has a centroid which indicates normal readings (close to unity) on all probes.
A plot of the silhouette scores for the two-cluster solution is given in Appendix 3, and the global mean silhouette score is plotted against the number of clusters. The silhouette score reached a maximum of 0.8 at two clusters, indicating this choice was appropriate. There was a large, significant difference in the survival characteristics of the two clusters (log-rank, p<0.001; 5-year survival: 80% versus 40%).
The 3D scatter plots in Appendix 1 show that in each projection the data are arrayed diagonally through the variable space, suggesting a degree of pair-wise correlation between variables. Robustly, a test for monotonic correlation between each pair of variables (13 ×12 / 2 = 78 possible combinations) showed strong evidence against the null hypothesis of zero correlation in every case with a Spearman coefficient that was always at least 0.62, and, excluding OPA1 (3q29), always at least 0.70 (p<0.001). Correlation seemed to be slightly weaker for many of the pairs involving the OPA1 probe (Table 3).
Table 3. Spearman correlation coefficients for the different loci in chromosome 3. All correlations were significant with p<0.001.
| Gene | Spearman r |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FANCD2(ii) | VHL | MLH1 | CTNNB1 | SEMA3B | FHIT(i) | FHIT(ii) | ROBO1 | CPO | RHO | MME | OPA1 | |
| FANCD2(i) |
0.878 |
0.848 |
0.824 |
0.791 |
0.823 |
0.780 |
0.771 |
0.748 |
0.804 |
0.786 |
0.780 |
0.687 |
| FANCD2(ii) |
- |
0.871 |
0.834 |
0.781 |
0.783 |
0.852 |
0.820 |
0.743 |
0.800 |
0.778 |
0.808 |
0.676 |
| VHL |
- |
- |
0.822 |
0.701 |
0.763 |
0.749 |
0.726 |
0.705 |
0.781 |
0.743 |
0.723 |
0.620 |
| MLH1 |
- |
- |
- |
0.704 |
0.754 |
0.810 |
0.794 |
0.717 |
0.774 |
0.768 |
0.806 |
0.629 |
| CTNNB1 |
- |
- |
- |
- |
0.801 |
0.794 |
0.759 |
0.847 |
0.795 |
0.722 |
0.779 |
0.698 |
| SEMA3B |
- |
- |
- |
- |
- |
0.737 |
0.708 |
0.746 |
0.753 |
0.806 |
0.737 |
0.675 |
| FHIT(i) |
- |
- |
- |
- |
- |
- |
0.836 |
0.809 |
0.816 |
0.757 |
0.862 |
0.686 |
| FHIT(ii) |
- |
- |
- |
- |
- |
- |
- |
0.772 |
0.770 |
0.746 |
0.843 |
0.667 |
| ROBO1 |
- |
- |
- |
- |
- |
- |
- |
- |
0.798 |
0.747 |
0.803 |
0.642 |
| CPO |
- |
- |
- |
- |
- |
- |
- |
- |
- |
0.769 |
0.802 |
0.673 |
| RHO |
- |
- |
- |
- |
- |
- |
- |
- |
- |
- |
0.813 |
0.632 |
| MME | - | - | - | - | - | - | - | - | - | - | - | 0.708 |
An initial review of the four loci in chromosome 8q did not reveal any obvious multimodal features, unlike chromosome 3, although in each case there was a clear single peak at unity and a non-symmetric distribution skewed toward higher probe values. Similarly, a 3D scatter plot did not suggest any obviously distinct groups within the data, though some degree of correlation between the variables was seen. Calculating Spearman correlation coefficients between each pair of probes showed significant correlations between all combinations (p<0.001), and in particular between MYC(i) and MYC(ii); (8q24.12); where r=0.9255, (p<0.001; Table 4). Spearman coefficients were characteristically smaller (0.23–0.25) for all pairs involving DDEF1 (8q24.2), compared with at least 0.58 for all other combinations.
Table 4. Spearman correlation coefficients for the different loci in chromosome 8. All correlations were significant with p<0.001.
| Gene | Spearman r |
||
|---|---|---|---|
| MYC(i) | MYC(ii) | DDEF1 | |
| RP1 |
0.583 |
0.609 |
0.240 |
| MYC(i) |
- |
0.926 |
0.241 |
| MYC(ii) | - | - | 0.253 |
Chromosome 3 deletion frequency
There is uncertainty about the prognostic significance of ambiguous MLPA results (e.g., if only three out of 13 loci on chromosome 3 were deleted). This was investigated by considering how the total number of deletions for each patient is distributed over the two clusters. In Appendix 4, the distribution of total number of deletions is indicated, stratified by cluster, for the two threshold values customarily used (i.e., 0.65 and 0.85). Using the 0.65 threshold (Appendix 4), the data are skewed toward no deletions at all, and although the majority of patients with no deletions are in cluster 2, several patients are in cluster 1 with no MLPA readings below 0.65, as indicated by the overlapping section at 0 deletions. Between one and three locus deletions, there is a fairly even split between the two clusters. This indicates that on this information alone it is very difficult to correctly classify a patient with a few or no deletions into one of these groups. Conversely, the weaker deletion threshold of 0.85 (Appendix 4) seems to offer a much smaller intersection between the two clusters: There is now a more equal split of patients between no deletions and all deletions, and furthermore, all patients with ten or more deletions belong to cluster 1, while patients with seven or fewer are in cluster 2. A small number of patients have eight or nine MLPA scores below 0.85, and these are split between the two groups.
Principal component analysis
The required rotation matrix was calculated, after the data were centered by subtracting the respective mean value from each probe reading for chromosome 3 [4]. A scree plot is given in Appendix 5 indicating the percentage of the total variance contained within the first seven principal components. Together these components account for more than 95% of the variance, with the majority (78%) in the first component. Histograms showing the distributions of data in the first three components are also shown in Appendix 5 (no significant structure was observed in the distribution of subsequent component values, which are not shown). Moreover, the bimodal distribution shown in Appendix 2 is now only seen in the first component, compared with the bimodal distributions observed along every axis of the untransformed variables, suggesting that the clusters are arrayed along some kind of diagonal in the 13-dimensional input space. The matrix coefficients for the first principal component were uniform (each fell within a small range of 0.28±0.03). Thus, along the axis of the largest variance (in which the clusters are now seen to be exclusively separated), no probe in particular distinguishes between one cluster and the other; instead, an even mix of all probes appears to be relevant. A scatter plot of the data in the first two principal components is shown in Figure 1, with the data points shaded according to the original k-means classification. The power of the PCA technique is well demonstrated; having begun with a 13-dimensional space impossible to fully visualize, it is now possible to separate these clusters by eye, placing a dividing vertical line somewhere near the origin.
Figure 1.
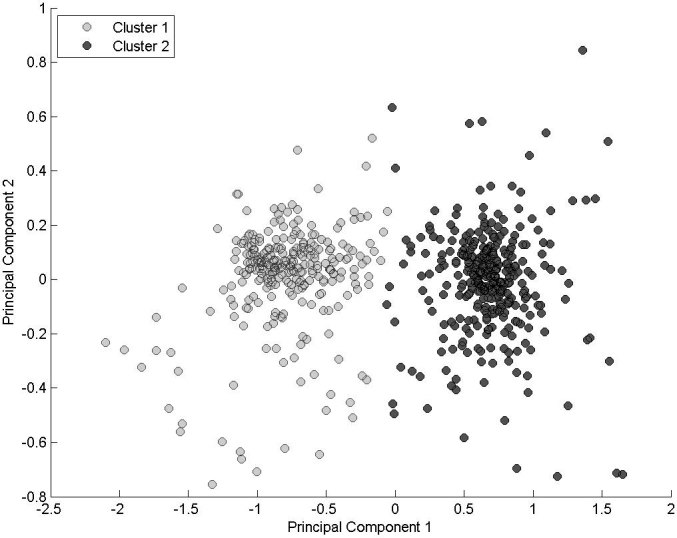
Scatter plot of the (centered) chromosome 3 data in the first two principal components, with points colored according to the cluster. This represents an optimal two-dimensional projection of the data.
PCA analyses were repeated for chromosome 8q. The results showed that nearly all of the variance was contained within the first three components, and more than 90% in the first two components. Coefficients for these components showed a small contribution by DDEF1 (8q24.2) in the first component, but a large contribution from this same probe in the second, which is consistent with the smaller correlation between this variable and the other three. There was no obvious grouping of data in chromosome 8q as was seen with chromosome 3. Applying the k-means algorithm with a range of values of k suggested the presence of two clusters, with a mean silhouette score of 0.728, though there were several negative scores, and the visualization in principal component space was not convincing. Therefore, a threshold-based definition of 8q gain on which to base subsequent analysis was considered in conjunction with the results of the chromosome 3 analysis. Figure 2 shows the joint space formed from the first principal components of chromosome 3 and chromosome 8q. Patients with a gain in chromosome 8q (i.e. three or four probes taking a value >1.15) occurred equally in both chromosome 3 groups and tended to predominantly occupy the upper half of this plot. This indicates the possibility of the presence of three clusters when the two chromosomes are combined.
Figure 2.
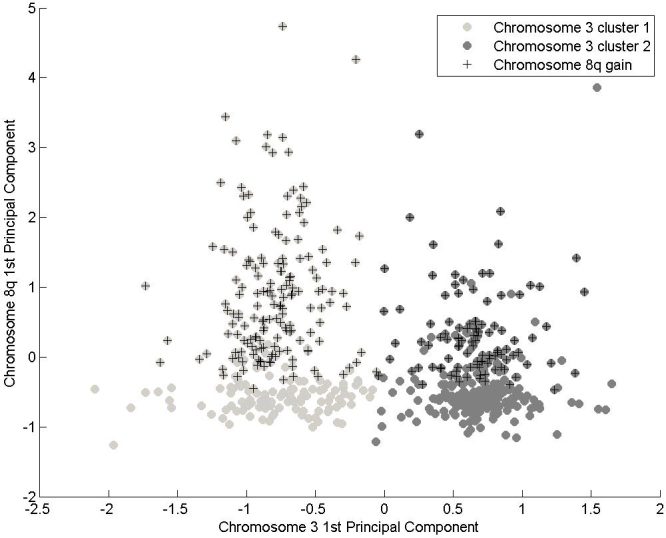
Representation of the data in the joint space formed from the first principal components of chromosome 3 and chromosome 8q.
Survival analysis
Based on these results, cases were classified as chromosome 3 deletion if they belonged to cluster C1 or normal chromosome 3 if they belonged to cluster C2. Cases were further classified as chromosome 8q gain if the three or four probes took a value >1.15; otherwise, they were classified as normal for chromosome 8q. Jointly, cases were classified into four prognostic groups: (i) normal chromosomes 3 and 8q; (ii) chromosome 3 deletion, normal chromosome 8q; (iii) normal chromosome 3, chromosome 8q gain; and (iv) chromosome 3 deletion, chromosome 8q gain.
The Kaplan–Meier plots for these four groups are shown in Figure 3. The figure shows the individuals allocated to group (i) have the best outcomes, with a cumulative survival of 97% at 5 years (95% CI=94–99.6%). This is in contrast to those in group (iv), with 30.3% 5-year cumulative survival (95% CI=20.5–40.1%). There are two middle groups, with abnormal results on only one chromosome (3 or 8q, but not both). Log-rank statistics show that the difference between each pair of survival curves is statistically significant (p<0.001) apart from the middle two (p=0.341; Table 5). This result suggests that chromosome 3 deletion or chromosome 8q gain has an essentially indistinguishable impact on outcome, when it occurs in isolation.
Figure 3.
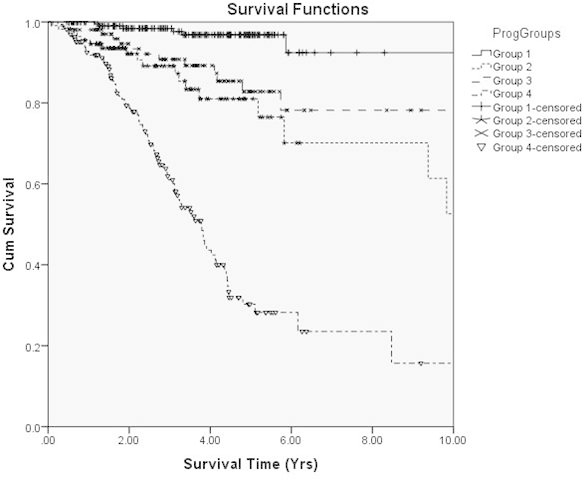
Kaplan–Meier cumulative survival functions for the four patient groups identified jointly using the k-means clustering solution (chromosome 3) and a simple thresholding condition on chromosome 8q. Group 1 represents disomy 3 and disomy 8 (n=220), group 2 represents monosomy 3 and disomy 8 (n=113), group 3 represents disomy 3 and polysomy 8 (n=104), and group 4 represents monosomy 3 and polysomy 8 (n=165).
Table 5. Pairwise log-rank statistical test (p values in brackets) for the four prognostic groups; Group 1 represents disomy3 and disomy8 (n=220), group 2 represents monosomy3 and disomy8 (n=113), group 3 represents disomy3 and polysomy8 (n=104) and group 4 represents monosomy3 and polysomy8 (n=165).
| Prognostic Group | Log-rank statistic (p) |
||
|---|---|---|---|
| 2 | 3 | 4 | |
| 1 |
22.426 (< 0.001) |
11.353 (0.001) |
134.665 (< 0.001) |
| 2 |
- |
1.919 (0.341) |
28.869 (< 0.001) |
| 3 | - | - | 46.93 (< 0.001) |
A plot of the distribution of the largest basal tumor diameter for the two clusters is given in Figure 4. A slight bias toward larger tumors is seen for cluster 1 (poorer outcomes), and smaller tumors for cluster 2 (better outcomes); however, there is a large overlap between the two distributions, indicating a wide range of tumor diameters in both groups. A Mann–Whitney test for equality of medians showed strong evidence against the null hypothesis (p<0.001), and that therefore tumors are characteristically larger in the first cluster.
Figure 4.
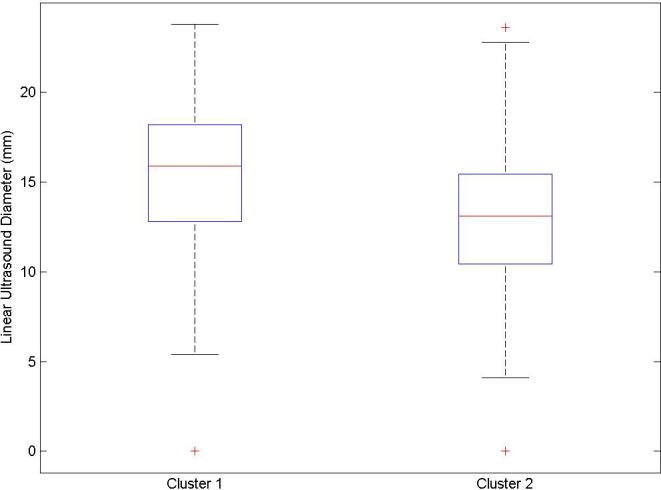
Distribution in LUD for each cluster. The Mann–Whitney test strongly indicates the medians are not equal.
Discussion
Main findings
This study shows that the MLPA chromosome 3 measurements are arrayed in two well-defined clusters. This clustering solution was validated with a commonly used metric based on silhouette values. Kaplan–Meier analysis confirmed three prognostic groups: group 1 (normal chromosomes 3 and 8q), group 2 (either 3 or 8q abnormal), and group 3 (3 and 8q abnormal). We also found that each of the 13 probes on chromosome 3 strongly correlates with all the others, although this correlation was weaker for the 13th probe; the reason is unclear. The correlation in the data indicated a lower intrinsic dimensionality in the data set than suggested by the sheer number of loci sampled. Indeed, principal component analysis showed that through suitable transformation of coordinate axes, more than 95% of the variation in the data was contained within seven orthogonal components, and all the bimodal structure within the first principal component. Moreover, this first principal component comprised a uniform mix of the 13 probes, suggesting there are no preferred loci accounting for the majority of variation in the data. The results also showed strong correlation between the clusters and tumor size. Since tumor size is a strong predictor of survival, tumor size may be a confounding factor in inferring any causal link from the Kaplan–Meier analysis. Examining the distribution of tumor stages in the three groups shows that the majority of tumors at stage T1 (57%) belong to group 1 whereas the majority of those at stage T4 (54%) are in group 4 (χ2, p<0.001). This suggests that the huge difference between the survival curves for these two groups can be at least partially attributed to lead time bias.
Chromosome 8q did not show any readily identifiable clustering pattern, although there was significant cross-correlation between the probe measurements, particularly the two probes on 8q24.12. Further, PCA showed that nearly all of the variance in the data was confined to three orthogonal components. Although there was no obvious clustering solution, the 8q data could be classified in a more traditional manner based on the suggested gain threshold of 1.15.
Strengths and weaknesses
The main strength of this study is the large sample size of 602 patients with a median follow-up time of more than 3 years. In this study, latent structure in the data was examined to classify the patients before survival of the different classes was examined. The main weakness is that the results must be validated with an unseen data set beyond the 602 patients. One potential difficulty is that new patient data are acquired using a new assay of 19 loci in chromosome 3 instead of 13, and not all of the original 13 loci are still available. However, results presented in this study suggest that no preferred probe (or subset of probes) discriminates between the two prognostic groups identified in chromosome 3 but that all 13 probes are important as indicated by the first principal component. By increasing the number of probes, therefore, the sampling resolution of essentially binary clustering is increased, and a similar clustering pattern into two groups is expected in the new data set.
Comparison with previous studies
Previous studies that relied on fluorescent in situ hybridization (FISH) for identifying monosomy or disomy 3 showed significant differences in survival between the two classes [5-9]. Results from these studies showed that the technique had a high specificity but low sensitivity [10,11]. With MLPA analysis, chromosomal abnormalities can be detected on a larger number of loci and using a scalar measure rather than a binary measure. Therefore, there is potential for this technique to identify more prognostic groups using all the information from this analysis. In this study, we looked at the latent structure of the data to determine the possibility of the existence of more prognostic groups.
Clinical and research implications
There is scope for further research investigating whether a multinomial regression model or a Gaussian process can be fitted to this joint space, to predict probabilities of class membership in this three-way partitioning of the data of the three prognostic groups identified. An anticipated challenge is the relative sparseness of the data. As a rule, ten to 20 data points in each class are required for each predictor. For multiple classes with potentially 17 predictors and only 602 patients in total, this becomes a limiting factor. It should be possible to extract a subspace of lower dimensionality from the joint space as has been attempted here for the two chromosomes in isolation.
Finally, despite the apparent existence of up to three prognostic groups in the joint space of chromosome 3 and chromosome 8q, a few patients cannot be classified on this basis. For example, 11 patients (1.8%) survived more than 5 years, despite abnormal results for chromosomes 3 and 8q. We could not identify anything from the histopathology or available genetic information that might explain this disparity although nine of these 11 tumors had no extraocular spread. Additional variables might play a role in the survival of these patients such as psychological factors. A more focused study of these particular patients’ clinical and histopathological data is currently in progress.
Conclusion
This study shows that for MLPA data in CM, there is a strong suggestion of two groups of patients according to chromosome 3 data, and that this was confirmed with k-means clustering. A two-cluster solution was established and validated. The center points of these two clusters correspond to 13 deletions (<0.65) and 13 normal probe readings (about 1.0). Survival analysis showed statistically significant differences between the two groups. However, the difference between the median tumor diameters in each cluster was also statistically significant, which is a possible source of a confounding error.
Clusters were well characterized by considering the total number of deletions, if the deletion threshold was taken as 0.85 instead of 0.65. Patients with ten or more deletions (<0.85) out of a possible 13 are exclusively in cluster 1 (poor prognosis) and seven or fewer in cluster 2 (good prognosis). A small number of patients have eight or nine deletions, and these are split between the two groups. There was a strong correlation between the probes so the data set does not fill out the full space made up of 13 probes but is restricted to a smaller variable space. PCA supplements this by demonstrating that most of the variation in the chromosome 3 data was characterized by one new variable that is an even mix of all probes (equivalent to a mean probe value). The study was unable to identify a preferred probe or subset of probes that were exclusively abnormal in the poorer outcome cluster (i.e., no signature genes were detected). Instead, we hypothesized that the two groups of data we found correspond in some way to underlying binary states of complete monosomy or disomy 3. The new measurement kit comprises 19 probes instead of 13, with different chromosome loci. We therefore expect to see a similar two-cluster solution in the new data (i.e, we have essentially increased the sampling frequency).
For chromosome 8q, there was no obvious clustering as for chromosome 3. As with chromosome 3, each locus is correlated with every other locus. Using the current convention for chromosome 8q gain (three or four probes out of four are >1.15), and considering the joint space of chromosome 3 and chromosome 8q, three significantly different prognostic groups were confirmed: both 3 and 8q normal, both 3 and 8q abnormal, and either 3 or 8q abnormal.
Appendix 1.
To access the data, click or select the words “Appendix 1.” Some examples of 3D projections with each point classified according to the output of the kmeans algorithm, for k=2. The clusters appear better defined in some projections than others, but the intuitive overall picture seems to have been well captured by the algorithm.
Appendix 2.
To access the data, click or select the words “Appendix 2.” Histograms with clusters binned separately. It can be seen that the two peaks in the bimodal distributions correspond distinctly to the K-means classification.
Appendix 3.
To access the data, click or select the words “Appendix 3.” (A) Silhouette scores plotted horizontally for the 602 data points when k=2, indicating the majority of points have a silhouette score in the range 0.6 - 0.9. (B) A plot of mean silhouette score against k, indicating the optimum clustering solution has been found at k=2.
Appendix 4.
To access the data, click or select the words “Appendix 4.” Distribution of the data in total number of deletions, stratified by cluster, where a deletion has been defined as <0.65 (A) and <0.85 (B).
Appendix 5.
To access the data, click or select the words “Appendix 5.” Principal Component Analysis of chromosome 3 data. (A): Scree plot showing the distribution of variance (and ≥95% cumulative variance) over the first seven components. Nearly 80% of the total variance is contained within the first principle component. (B, C, D): Distribution of the data in the first three principle components, the bimodality is now exposed exclusively in the first component, and the variance reduces in successive components as the distributions become progressively narrower and taller (note the change in vertical scale for constant bin widths).
References
- 1.Damato B, Eleuteri A, Taktak AF, Coupland SE. Estimating prognosis for survival after treatment of choroidal melanoma. Prog Retin Eye Res. 2011;30:285–95. doi: 10.1016/j.preteyeres.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Dopierala J, Damato BE, Lake SL, Taktak AF, Coupland SE. Genetic heterogeneity in uveal melanoma assessed by multiplex ligation-dependent probe amplification. Invest Ophthalmol Vis Sci. 2010;51:4898–905. doi: 10.1167/iovs.09-5004. [DOI] [PubMed] [Google Scholar]
- 3.Lake SL, Damato BE, Dopierala J, Baudo MM, Taktak AF, Coupland SE. Multiplex ligation-dependent probe amplification analysis of uveal melanoma with extraocular extension demonstrates heterogeneity of gross chromosomal abnormalities. Invest Ophthalmol Vis Sci. 2011;52:5559–64. doi: 10.1167/iovs.10-6845. [DOI] [PubMed] [Google Scholar]
- 4.Semlow JL. Multivariate Analysis: Principal Component Analysis and Independent Component Analysis, in Biosignal and Medical Image Processing. 2009, CRC Press: Boca Raton, FL. [Google Scholar]
- 5.Aronow M, Sun Y, Saunthararajah Y, Biscotti C, Tubbs R, Triozzi P, Singh AD. Monosomy 3 by FISH in uveal melanoma: variability in techniques and results. Surv Ophthalmol. 2012;57:463–73. doi: 10.1016/j.survophthal.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Bonaldi L, Midena E, Filippi B, Tebaldi E, Marcato R, Parrozzani R, Amadori A. FISH analysis of chromosomes 3 and 6 on fine needle aspiration biopsy samples identifies distinct subgroups of uveal melanomas. J Cancer Res Clin Oncol. 2008;134:1123–7. doi: 10.1007/s00432-008-0382-6. [DOI] [PubMed] [Google Scholar]
- 7.Damato B, Duke C, Coupland SE, Hiscott P, Smith PA, Campbell I, Douglas A, Howard P. Cytogenetics of Uveal Melanoma. A 7-Year Clinical Experience. Ophthalmology. 2007;114:1925–31. doi: 10.1016/j.ophtha.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Patel KA, Edmondson ND, Talbot F, Parsons MA, Rennie IG, Sisley K. Prediction of prognosis in patients with uveal melanoma using fluorescence in situ hybridisation. Br J Ophthalmol. 2001;85:1440–4. doi: 10.1136/bjo.85.12.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Bosch T, van Beek JG, Vaarwater J, Verdijk RM, Naus NC, Paridaens D, de Klein A, Kiliç E. Higher percentage of FISH-determined monosomy 3 and 8q amplification in uveal melanoma cells relate to poor patient prognosis. Invest Ophthalmol Vis Sci. 2012;53:2668–74. doi: 10.1167/iovs.11-8697. [DOI] [PubMed] [Google Scholar]
- 10.Damato B, Dopierala J, Klaasen A, van Dijk M, Sibbring J, Coupland SE. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Invest Ophthalmol Vis Sci. 2009;50:3048–55. doi: 10.1167/iovs.08-3165. [DOI] [PubMed] [Google Scholar]
- 11.Damato B, Dopierala JA, Coupland SE. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res. 2010;16:6083–92. doi: 10.1158/1078-0432.CCR-10-2076. [DOI] [PubMed] [Google Scholar]