

Abstract
Although Crohn’s disease (CD) etiology remains unclear, a growing body of evidence suggests that CD may include an infectious component, with Mycobacterium avium subsp. paratuberculosis (MAP) being the most likely candidate for this role. However, the molecular mechanism of the MAP involvement in CD pathogenesis remains unclear. The polymorphism of the NOD2 gene, coding for an intracellular pattern recognition receptor, is a factor of predisposition to mycobacterial infections and CD. Recent findings on NOD2 interactions and functions provide the missing pieces in the puzzle of a NOD2-mediated mechanism common for mycobacterial infections and CD. Implications of these new findings for the development of a better understanding and treatments of CD and mycobacterial infections are discussed.
Keywords: Vesicle acidity, Crohn’s disease, Mycobacteria, Sulfonated glycolipids, Autophagy, Pattern recognition receptors
Introduction
Crohn’s disease (CD) is a systemic inflammatory disease involving primarily the intestinal tract and associated with the variety of extraintestinal manifestations. Although it may affect any part of the digestive tract from the mouth to the anus, it most commonly affects the last part of the small intestine (ileum) and/or the large intestine (colon and rectum). The etiology of CD remains unclear. However, there is phenotypical, epidemiological and clinical evidence of Mycobacterium avium subsp. paratuberculosis (MAP) involvement in CD development [1-3]. This enteric pathogen is significantly associated with CD [4-7]. MAP causes paratuberculosis (Johne’s disease), a chronic, contagious bacterial disease that primarily affects the small intestines of ruminants. Johne’s disease affects approximately 68% and 32% of cows in the USA and the UK, respectively [7,8]. Live MAP is found even in pasteurized cow’s milk, suggesting that dairy products and beef, widely present in the “western” diet, can play a role in MAP transmission to human population [1]. The lack of evidence for horizontal or vertical transmission of CD suggests that MAP is a zoonotic agent or an opportunistic pathogen in humans [9]. The signs of Johne’s disease in ruminants are similar to the symptoms of CD. Moreover, CD demonstrates a striking similarity of symptoms to intestinal tuberculosis caused by M. tuberculosis, Mtb [10-12].
The systemic characters of CD and mycobacterial infections suggest that the underlying pathological processes are defects in basic cellular signaling mechanisms common to different cell types. However, these molecular mechanisms remain unclear. As a result, CD remains incurable and its incidence increases around the globe, which makes CD a global health problem with high societal costs and a substantial health-related quality-of-life burden [13,14]. The recent rapid growth of CD incidence in Asia may be related to the westernization of the diet and an improved hygiene [15].
The polymorphism of Nucleotide binding and Oligomerization Domain 2 (NOD2) is a genetic predisposition factor for both CD and mycobacterial infections [16-20]. However, it does not seem to contribute significantly to the CD incidence in Eastern Asians, probably due to the low presence of the characteristic CD-associated NOD2 polymorphisms in this part of the world (Rs2066844; Rs2066845; Rs2066847 (Rs5743293)) [15,21,22]. These facts suggest that NOD2 polymorphism is rather secondary for disease development, which, however, does not exclude a NOD2 role in CD etiology.
It is prompting to speculate that NOD2 mediates a mechanism important for both mycobacterial infection and CD. However, until recently, little was known about what basic NOD2-dependent mechanism could link CD and mycobacterial infection and at the same time explain the characteristic features of these diseases. Several years ago it became clear that, to prove the mycobacterial hypothesis of CD, immunologists should identify the microbe-associated ligands mediating CD immune defects [23]. In the last two years, this gap in understanding of CD etiology has been filled for NOD2. This review for the first time summarizes the new findings linking NOD2, mycobacterial infection and CD development, and explains some characteristic molecular features of these diseases.
NOD2 and its ligands
The NOD2 (Blau, CARD15) gene encodes a 115-kDa cytosolic protein with multiple C-terminal leucine-rich repeats (LRR), a central NACHT (NAIP, CIITA, HET-E, TP-1) domain, and two N-terminal caspase recruitment domains (CARDs). The NACHT domain bears high homology to NTPase domains; however, the intrinsic NTPase activity of the NACHT domain is not well established. The NOD2 NACHT domain resembles the ATPase domain of the proton-pumping F1-ATPase, which in turn is highly similar to that of Vacuolar-type H+-ATPase (V-ATPase) [24,25]. The NACHT domain mediates homo- and heterotypic oligomerization, which triggers recruitment of pro-inflammatory factors (caspase-1 and RIP2) to CARDs and enhances pro-inflammatory activity at both the transcriptional and the post-transcriptional levels [26-30]. Unbound to a ligand, the LRR domain covers the NACHT domain and prevents the NACHT-mediated oligomerization [29]. The genetic polymorphism of the NOD2 LRR predisposes to CD whereas the NACHT polymorphism is associated with deregulation of NF-kB activity and development of Blau syndrome, an inflammatory disorder that primarily affects the skin, joints, and eyes [16,17,31].
Increased NOD2 expression alone can activate pro-inflammatory NF-kB activity, suggesting a default character of this NOD2 activity [28,32]. The baseline gene expression of NOD2 is very low in different cell types, reflecting the specific and powerful characters of the NOD2 regulated processes [33]. Indeed, NOD2 gene expression is up-regulated under stress conditions such as hypoxia or the presence of bacterial lipopolysaccharides, both known to regulate the transcriptional activity of hypoxia-inducible factor type 1 (HIF-1) [32,34-36].
NOD2 also mediates autophagy, a catabolic intracellular process of partial cytoplasm sequestration into double-membrane autophagosomes that fuse with lysosomes to digest the sequestered material [37,38]. Muramyl dipeptide (N-acetylmuramyl-L-alanyl-D-isoglutamine), a fragment of the bacterial cell wall, seems to be an unspecific NOD2 activator inducing both pro-inflammatory and autophagy activities [26-29,37,38]. The processes of inflammation and autophagy are antagonistic to each other [39]. For NOD2, it may mean that NOD2 mediates inflammation by default if it is not involved in autophagy.
NOD2 belongs to the family of pattern recognition receptors (PRRs) serving as innate immunity sensors. PRRs recognize a limited number of conservative immunogenic epitopes (patterns) including endogenous damage-associated molecular patterns, DAMPs [40-42]. Autophagy-inducing cytoplasmic PRRs can specifically recognize host glycans from the outer leaflets of membranes when membrane damage (i.e. caused by pathogens) exposes the outer glycans to the cytoplasm [43]. These findings may shed additional light on the sentinel role of NOD2 at the host membranes [44].
3-O-sulfogalactocerebroside (sulfatide), a sphingolipid normally present on the outer membrane leaflet, has recently been identified as the first NOD2 DAMP mediating NOD2 involvement in autophagy [32]. Of interest, hypoxia also stimulates the gene expression of GAL3ST1 (Galactose-3-O-sulfotransferase 1), whose protein product catalyzes the conversion of 3’-phosphoadenosine-5’-phosphosulfate (PAPS) + galactosylceramide to adenosine 3’,5’-bisphosphate + sulfatide [32]. These findings are in line with others showing that renal carcinoma cells, known for their deregulated activity of HIF-1, have elevated sulfatide and sulfotransferase activities [45,46]. Thus, the co-expression of NOD2 and GAL3ST1 prepares vulnerable membranes to effective recognition by NOD2 and subsequent autophagy if the membranes become damaged.
NOD2 vesicle-associated function
Intracellular vesicle-associated acidity grows under hypoxia [47]. This vesicle acidity is mediated by the V-ATPase proton-pumping catalytic activity. These newly formed vesicles need to protect their acidity because the hypoxia-associated ATP deficiency can induce vesicle leakage [48]. However, the V-ATPase function is not limited to proton pumping. Assembled but inactive V-ATPase mediates vesicle content storage, whereas its disassembly mediates vesicle fusion and content release (including leakage) [49-52]. NOD2 deficit decreases the intracellular vesicle acidity but not vesicle acidification, suggesting a NOD2 role in vesicle content storage. NOD2 interacts with assembled, catalytically inactive V-ATPase until the NOD2 – V-ATPase complex reaches sulfatide-rich membranes, where the V-ATPase disassembles (Figure 1) [32]. These and more recent findings directly link NOD2 function to intracellular vesicles [53].
Figure 1.
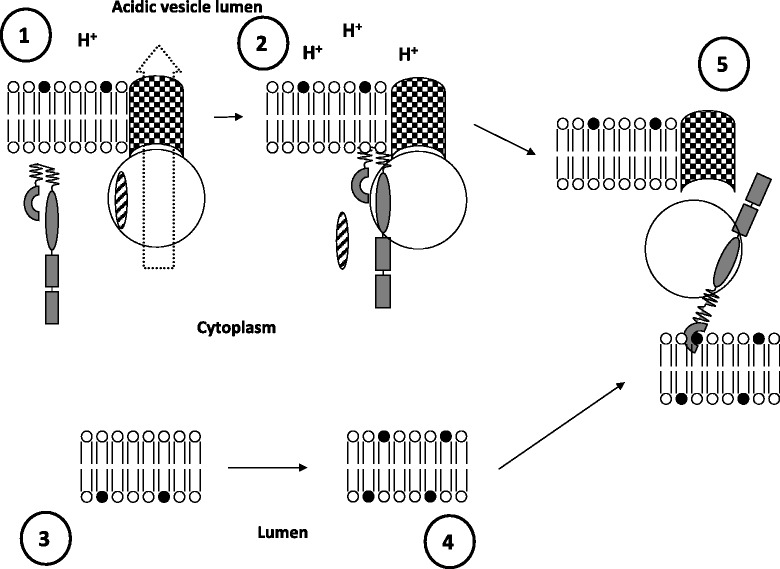
The model of NOD2 vesicle-associated function. 1) Catalytically active V-ATPase consisting of transmembrane V0 and cytoplasmic V1 (big circle) sectors pumps (big dotted arrow) protons from the cytoplasm into the vesicle. Cytoplasmic NOD2 (grey figure) is in the self-inhibiting state; 2) NOD2 may substitute for the catalytic V1A subunit (stripped oval) in the V-ATPase complex, when pumping is stopped and the rest of V-ATPase complex remains assembled. 3) A normal membrane keeps sulfatide (black-head “lipid”) on the outer (opposite to the cytoplasm) leaflet. 4) When the membrane is damaged, it exposes sulfatide to the cytoplasm. 5) The sulfatide exposure to the cytoplasm is recognized by NOD2, which induces V-ATPase complex disassembly and opens the fusion-mediating V0 sector, making the vesicle fusion-competent.
At high ATP concentrations, the catalytic activity of V-ATPase may compensate for the lack of NOD2 functionality by pumping the leaked protons back into the vesicles. This makes NOD2 protein dispensable for normal conditions, which is supported by the very low NOD2 gene expression in normal conditions. However, stress conditions associated with a deficit of ATP production (e.g. hypoxia) will increase the need for the NOD2-mediated energy-saving mechanism of proton storage in vesicles.
The induction of “fusion-competent” vesicles after NOD2-sulfatide interaction suggests their accumulation in close proximity to damaged membranes. These vesicles may provide membrane material and eventually direct autophagosome growth specifically around the damaged membranes, without sequestration of undamaged areas (Figure 2). On the other hand, the presence of sulfatide-mimicking agents at a distance from the sulfatide-exposing membranes will inhibit the specificity of NOD2 function and induce unspecific fusion and vesicle content release (leakage).
Figure 2.
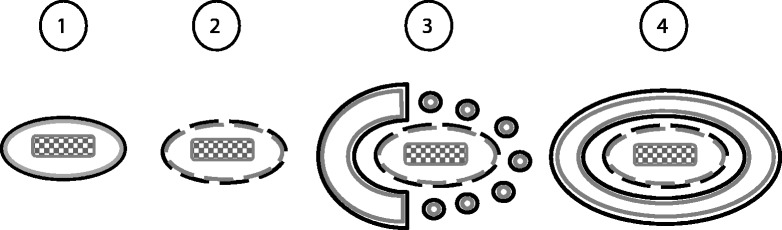
The activated by NOD2 vesicles in autophagy. 1) A phagosome containing a microbe (checkered figure) becomes damaged and 2) exposes sulfatide to the cytoplasm. 3) Autophagy is initiated and fusion-competent vesicles are accumulated around the damaged phagosome due to V-ATPase disassembly triggered by sulfatide-NOD2 interaction. 4) The fusion competent vesicles fuse with the autophagosome providing material and directing autophagosome growth specifically around the damaged phagosome. The outer (sulfatide-containing) membrane leaflet is grey; the inner membrane leaflet is black.
NOD2 and mycobacterial immune escape
Macrophages, professional antigen-presenting immune cells expressing one of the highest amounts of NOD2 in the body, are the preferred hosts for MAP and Mtb. Whether active mycobacteria remain inside phagosomes or translocate from phagosomes into the cytosole has been a matter of debate in recent years [54]. In both cases, mycobacteria should perforate the phagosomal membrane to egress into the cytoplasm or to get access to the cytoplasm nutrients [55,56]. Obviously, to survive, mycobacteria induce mechanisms that prevent or subvert the membrane-damage-associated activation of NOD2.
Mtb lipid virulence factors may have evolved to mimic host lipids and thereby directly influence innate immune responses of macrophages via interactions with specific signaling pathways [56]. Mycobacteria synthesize a specific sulfolipid (SL-1) that mimics sulfatide in binding to NOD2 [32]. This suggests that SL-1 interaction with NOD2 can activate unspecific NOD2-mediated processes of V-ATPse disassembly, making intracellular vesicles leaky and/or activating unspecific fusion of these vesicles. This clarifies the SL-1-mediated inhibition of: 1) lysosome fusion with Mtb-containing phagosomes and 2) lysosome maturation [57-59]. Subverting NOD2 activity in the vesicle–associated function of NOD2 (i.e. autophagy), SL-1 inevitably activates NOD2-mediated inflammation, which can explain (at least partly) the characteristic inflammation present in mycobacterial infection [60,61].
Mtb lipids are abundantly produced during macrophage infection and are actively trafficked out of mycobacterial phagosomes [62]. Moreover, mycobacterial lipids can be found in extracellular vesicles and could be observed in uninfected ‘bystander’ cells, which expand the bacteria’s sphere of influence beyond the membranes of the infected host cell [62]. For CD pathogenesis, it means that MAP-infected intestinal cells may contaminate with SL-1 surrounding intestinal cells, such as enterochromaffin (EC) cells, Paneth cells, and their progenitor stem cells, all of which are known to be affected in CD [63,64].
In these circumstances, the NOD2 polymorphism associated with decreased sulfatide recognition makes the host predisposed to mycobacterial infections. When mycobacterial infection is established, the double pressure on the NOD2 vesicle function from the polymorphism and SL-1 substantially increases the chance of defects in acidic vesicle homeostasis. Notably, the 1007 fs NOD2 polymorphism most commonly associated with predisposition to CD, only slightly decreases the NOD2 binding to sulfatide, suggesting that SL-1 presence plays a more important role in CD development than does genetic predisposition [32]. Indeed, only about 5% of NOD2 mutation homozygotes develop CD, suggesting crucial roles for additional factors (like mycobacterial infection) in CD development. Of interest, sulfonated compounds like dextan sulfate and 2,4,6-trinitrobenzenesulfonic acid are used the most frequently for experimental colitis induction.
Vesicle-associated abnormalities and CD specific features
We found NOD2 in cell-division-specific structures associated with the massive fusion of intracellular vesicles providing the membrane material for cell division [32], [65]. Cell division and a high level of autophagy, where the latter maintains the stemness, are typical stem cell features supported by the expression and functional activities of proteins mediating these processes [66]. NOD2 has an important biological role in bone marrow CD34+ hematopoietic cells [67]. Intestinal crypt Lgr5+ stem cells also express Nod2 mediating gut epithelial regeneration [68]. The latter suggests that NOD2 regulates the Notch signaling pathway, a key cell communication pathway that suppresses production of secretory intestinal cells (i.e. EC cells) in favor of higher gut epithelial cell production [69]. Notch activity is promoted by the fusion of Notch receptor-containing endosomes with V-ATPase-containing lysosomes [70-72]. All this suggests that the SL-1-associated unspecific activation of NOD2 in intestinal stem cells may increase production of EC cells, which are responsible for 90% of the body serotonin (5-hydroxytryptamine).
CD-affected intestines have higher numbers of EC cells and levels of serotonin [63,73]. Enteric serotonin is a major gastrointestinal paracrine hormone and neurotransmitter mediating the peristaltic activity, blood clotting and bone metabolism, all impaired in CD [74-78]. The systemic character of serotonin action in the body suggests that imbalances of serotonin in CD can be among the factors mediating the systemic character of the disease. Serotonin imbalances are also found in leprosy and tuberculosis [79,80].
V-ATPase generates the proton membrane potential that is used by vesicular monoamine transporters to sequester newly synthesized or externally up-taken serotonin into intracellular vesicles [81]. SL-1-induced vesicle content leakage will lead to a prolonged exposure of non-sequestered monoamines to cytoplasmic (mitochondrial) monoamine oxidases. This results in an increased conversion of monoamines into toxic aldehydes, causing cell damage and inflammation. These effects in turn enhance EC cell production from intestinal stem cells, making the pathological process self-sustaining [82-84].
NOD2 and other genetic and non-genetic factors of predisposition to CD
Mechanisms mediating serotonin release from cells become very important when serotonin sequestration is defective. Indeed, genetic polymorphisms of polyspecific organic cation transporters OCTN1/2, translocating cytoplasmic serotonin through the cytoplasmic membrane, are among the CD predisposition factors [85,86]. Moreover, the CD-associated OCTN1 and NOD2 gene polymorphisms are additive for CD development [87].
Only about 10 to 20 percent of patients have a family history of CD, suggesting the main role of environmental factors in CD development. Similar to SL-1 competing with sulfatide for NOD2 binding, other factors affecting sulfatide synthesis or accessibility can trigger NOD2 functional deficiency.
CD is more common in urban areas. In general, these areas are better supplied with potable water, which, even after chlorination, may serve as a transmission route of MAP [88]. Chlorate ion (ClO3−), often used for or formed as a byproduct in water chlorination, is a well-known inhibitor of PAPS synthesis and consequently sulfatide synthesis. Exposure of cells to sodium chlorate has a similar effect on autophagy as NOD2 deficiency [32]. Thus, the hypothesis of MAP transmission via potable water should include water chlorination as a risk factor.
Conclusion
The absence of a clear mechanistic explanation of the role of MAP in CD has been one of the main obstacles in transformation of their well-known association into causation. NOD2, an intracellular pattern recognition receptor playing a role in mycobacterial infections and CD, has been suspected as a possible link between them. This review summarizes very recent findings on the NOD2 ligand and functional specificities that establish the causative link between mycobacteria and CD via mycobacteria-specific inhibition of NOD2 function. Moreover, these findings clarify the role of other genetic and environmental factors of predisposition to systemic CD. Further development of these NOD2 findings may provide novel therapeutic targets for CD and other mycobacteria-related pathologies.
Acknowledgments
I am thankful to Prof. Buurman and Prof. Vooijs from Maastricht University, The Netherlands, for their helpful comments and to Prof. Wieczorek and Dr. Huss from Osnabruck University, Germany, for their theoretical support in V-ATPase processes understanding.
Abbreviations
- CD
Crohn’s disease
- MAP
Mycobacterium avium subsp. paratuberculosis
- Mtb
M. tuberculosis
- NOD2
Nucleotide binding and Oligomerization Domain 2
- V-ATPase
Vacuolar-type H+-ATPase
- EC cells
Enterochromaffin cells
- PRR
Pattern recognition receptor
- GAL3ST1
Galactose-3-O-sulfotransferase 1
- DAMP
Damage-associated molecular patterns
- LRR
Leucine rich repeats
- NACHT
NAIP, CIITA, HET-E, TP-1
- CARD
Caspase recruitment domain
- OCTN1(2)
Organic cation transporter, novel, type 1(2)
- PAPS
3’-phosphoadenosine-5’-phosphosulfate
- HIF-1
Hypoxia-inducible factor type 1
Footnotes
Competing interests
The author declares that he has no competing interests.
References
- 1.Greenstein RJ. Is Crohn’s disease caused by a mycobacterium? Comparisons with leprosy, tuberculosis, and Johne’s disease. Lancet Infect Dis. 2003;3:507–514. doi: 10.1016/S1473-3099(03)00724-2. [DOI] [PubMed] [Google Scholar]
- 2.Uzoigwe JC, Khaitsa ML, Gibbs PS. Epidemiological evidence for Mycobacterium avium subspecies paratuberculosis as a cause of Crohn’s disease. Epidemiol Infect. 2007;135:1057–1068. doi: 10.1017/S0950268807008448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gitlin L, Borody TJ, Chamberlin W, Campbell J. Mycobacterium avium ss paratuberculosis-associated diseases: piecing the Crohn’s puzzle together. J Clin Gastroenterol. 2012;46:649–655. doi: 10.1097/MCG.0b013e31825f2bce. [DOI] [PubMed] [Google Scholar]
- 4.Feller M, Huwiler K, Stephan R, Altpeter E, Shang A, Furrer H, et al. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: a systematic review and meta-analysis. Lancet Infect Dis. 2007;7:607–613. doi: 10.1016/S1473-3099(07)70211-6. [DOI] [PubMed] [Google Scholar]
- 5.Chiodini RJ, Chamberlin WM, Sarosiek J, McCallum RW. Crohn’s disease and the mycobacterioses: a quarter century later. Causation or simple association? Crit Rev Microbiol. 2012;38:52–93. doi: 10.3109/1040841X.2011.638273. [DOI] [PubMed] [Google Scholar]
- 6.Bull TJ, McMinn EJ, Sidi-Boumedine K, Skull A, Durkin D, Neild P, et al. Detection and verification of Mycobacterium avium subsp. paratuberculosis in fresh ileocolonic mucosal biopsy specimens from individuals with and without Crohn’s disease. J Clin Microbiol. 2003;41:2915–2923. doi: 10.1128/JCM.41.7.2915-2923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhodes G, Richardson H, Hermon-Taylor J, Weightman A, Higham A, Pickup R. Mycobacterium avium Subspecies paratuberculosis: Human Exposure through Environmental and Domestic Aerosols. Pathogens. 2014;3:577–595. doi: 10.3390/pathogens3030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nielsen SS, Toft N. A review of prevalences of paratuberculosis in farmed animals in Europe. Prev Vet Med. 2009;88:1–14. doi: 10.1016/j.prevetmed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Liverani E, Scaioli E, Cardamone C, Dal Monte P, Belluzzi A. Mycobacterium avium subspecies paratuberculosis in the etiology of Crohn’s disease, cause or epiphenomenon? World J Gastroenterol. 2014;20:13060–13070. doi: 10.3748/wjg.v20.i36.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makharia GK, Srivastava S, Das P, Goswami P, Singh U, Tripathi M, et al. Clinical, endoscopic, and histological differentiations between Crohn’s disease and intestinal tuberculosis. Am J Gastroenterol. 2010;105:642–651. doi: 10.1038/ajg.2009.585. [DOI] [PubMed] [Google Scholar]
- 11.Pulimood AB, Amarapurkar DN, Ghoshal U, Phillip M, Pai CG, Reddy DN, et al. Differentiation of Crohn’s disease from intestinal tuberculosis in India in 2010. World J Gastroenterol. 2011;17:433–443. doi: 10.3748/wjg.v17.i4.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cappell MS, Saad A, Bortman JS, Amin M. Ileocolonic tuberculosis clinically, endoscopically, and radiologically mimicking Crohn’s disease: disseminated infection after treatment with infliximab. J Crohns Colitis. 2014;8:560–562. doi: 10.1016/j.crohns.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 13.Ng SC. Epidemiology of inflammatory bowel disease: focus on Asia. Best Pract Res Clin Gastroenterol. 2014;28:363–372. doi: 10.1016/j.bpg.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Floyd DN, Langham S, Severac HC, Levesque BG. The Economic and Quality-of-Life Burden of Crohn’s Disease in Europe and the United States, 2000 to 2013: A Systematic Review. Dig Dis Sci. 2014. doi:10.1007/s10620-014-3368-z. [DOI] [PubMed]
- 15.Ng SC. Emerging Leadership Lecture: Inflammatory Bowel Disease in Asia: Emergence of a “Western Disease”. J Gastroenterol Hepatol. 2014. doi:10.1111/jgh.12859. [DOI] [PubMed]
- 16.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 17.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 18.Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, et al. Genomewide association study of leprosy. New Engl J Med. 2009;361:2609–2618. doi: 10.1056/NEJMoa0903753. [DOI] [PubMed] [Google Scholar]
- 19.Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol. 2008;181:7157–7165. doi: 10.4049/jimmunol.181.10.7157. [DOI] [PubMed] [Google Scholar]
- 20.Pinedo PJ, Buergelt CD, Donovan GA, Melendez P, Morel L, Wu R, et al. Association between CARD15/NOD2 gene polymorphisms and paratuberculosis infection in cattle. Vet Microbiol. 2009;134:346–352. doi: 10.1016/j.vetmic.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 21.Sugimura M, Kinouchi Y, Takahashi S, Aihara H, Takagi S, Negoro K, et al. CARD15/NOD2 mutational analysis in Japanese patients with Crohn’s disease. Clin Genet. 2003;63:160–162. doi: 10.1046/j.0009-9163.2002.00174.x. [DOI] [PubMed] [Google Scholar]
- 22.Pugazhendhi S, Santhanam S, Venkataraman J, Creveaux I, Ramakrishna BS. NOD2 gene mutations associate weakly with ulcerative colitis but not with Crohn’s disease in Indian patients with inflammatory bowel disease. Gene. 2013;512:309–313. doi: 10.1016/j.gene.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 23.Lalande JD, Behr MA. Mycobacteria in Crohn’s disease: how innate immune deficiency may result in chronic inflammation. Expert Rev Clin Immunol. 2010;6:633–641. doi: 10.1586/eci.10.29. [DOI] [PubMed] [Google Scholar]
- 24.Albrecht M, Lengauer T, Schreiber S. Disease-associated variants in PYPAF1 and NOD2 result in similar alterations of conserved sequence. Bioinformatics. 2003;19:2171–2175. doi: 10.1093/bioinformatics/btg370. [DOI] [PubMed] [Google Scholar]
- 25.Futai M, Nakanishi-Matsui M, Okamoto H, Sekiya M, Nakamoto RK. Rotational catalysis in proton pumping ATPases: from E. coli F-ATPase to mammalian V-ATPase. Biochim Biophys Acta. 1817;2012:1711–1721. doi: 10.1016/j.bbabio.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Hsu LC, Ali SR, McGillivray S, Tseng PH, Mariathasan S, Humke EW, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A. 2008;105:7803–7808. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva CJ, Hoffman HM, et al. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007;82:177–183. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- 28.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 29.Tanabe T, Chamaillard M, Ogura Y, Zhu L, Qiu S, Masumoto J, et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J. 2004;23:1587–1597. doi: 10.1038/sj.emboj.7600175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferwerda G, Kramer M, de Jong D, Piccini A, Joosten LA, Devesaginer I, et al. Engagement of NOD2 has a dual effect on proIL-1beta mRNA transcription and secretion of bioactive IL-1beta. Eur J Immunol. 2008;38:184–191. doi: 10.1002/eji.200737103. [DOI] [PubMed] [Google Scholar]
- 31.Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 32.Nabatov AA, Hatzis P, Rouschop KM, van Diest P, Vooijs M. Hypoxia inducible NOD2 interacts with 3-O-sulfogalactoceramide and regulates vesicular homeostasis. Biochim Biophys Acta. 1830;2013:5277–5286. doi: 10.1016/j.bbagen.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Tsai WH, Huang DY, Yu YH, Chen CY, Lin WW. Dual roles of NOD2 in TLR4-mediated signal transduction and -induced inflammatory gene expression in macrophages. Cell Microbiol. 2011;13:717–730. doi: 10.1111/j.1462-5822.2010.01567.x. [DOI] [PubMed] [Google Scholar]
- 34.Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 35.King K, Bagnall R, Fisher SA, Sheikh F, Cuthbert A, Tan S, et al. Identification, evolution, and association study of a novel promoter and first exon of the human NOD2 (CARD15) gene. Genomics. 2007;90:493–501. doi: 10.1016/j.ygeno.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 36.Kuschel A, Simon P, Tug S. Functional regulation of HIF-1alpha under normoxia–is there more than post-translational regulation? J Cell Physiol. 2012;227:514–524. doi: 10.1002/jcp.22798. [DOI] [PubMed] [Google Scholar]
- 37.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 38.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 39.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mackey D, McFall AJ. MAMPs and MIMPs: proposed classifications for inducers of innate immunity. Mol Microbiol. 2006;61:1365–1371. doi: 10.1111/j.1365-2958.2006.05311.x. [DOI] [PubMed] [Google Scholar]
- 41.Foell D, Wittkowski H, Roth J. Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat Clin Pract Rheumatol. 2007;3:382–390. doi: 10.1038/ncprheum0531. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Q, Kang R, Zeh HJ, 3rd, Lotze MT, Tang D. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9:451–458. doi: 10.4161/auto.23691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin-8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Philpott DJ, Girardin SE. Nod-like receptors: sentinels at host membranes. Curr Opin Immunol. 2010;22:428–434. doi: 10.1016/j.coi.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 45.Sakakibara N, Gasa S, Kamio K, Makita A, Koyanagi T. Association of elevated sulfatides and sulfotransferase activities with human renal cell carcinoma. Cancer Res. 1989;49:335–339. [PubMed] [Google Scholar]
- 46.Keefe SM, Nathanson KL, Rathmell WK. The molecular biology of renal cell carcinoma. Semin Oncol. 2013;40:421–428. doi: 10.1053/j.seminoncol.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 47.Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, et al. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. 2008;4:195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bronk SF, Gores GJ. Efflux of protons from acidic vesicles contributes to cytosolic acidification of hepatocytes during ATP depletion. Hepatology. 1991;14(4 Pt 1):626–633. doi: 10.1002/hep.1840140409. [DOI] [PubMed] [Google Scholar]
- 49.Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121:607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.El Far O, Seagar M. A role for V-ATPase subunits in synaptic vesicle fusion? J Neurochem. 2011;117:603–612. doi: 10.1111/j.1471-4159.2011.07234.x. [DOI] [PubMed] [Google Scholar]
- 51.Poea-Guyon S, Ammar MR, Erard M, Amar M, Moreau AW, Fossier P, et al. The V-ATPase membrane domain is a sensor of granular pH that controls the exocytotic machinery. J Cell Biol. 2013;203:283–298. doi: 10.1083/jcb.201303104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camacho M, Machado JD, Montesinos MS, Criado M, Borges R. Intragranular pH rapidly modulates exocytosis in adrenal chromaffin cells. J Neurochem. 2006;96:324–334. doi: 10.1111/j.1471-4159.2005.03526.x. [DOI] [PubMed] [Google Scholar]
- 53.Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Maziere A, et al. Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature. 2014;509:240–244. doi: 10.1038/nature13133. [DOI] [PubMed] [Google Scholar]
- 54.Welin A, Lerm M. Inside or outside the phagosome? The controversy of the intracellular localization of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2012;92:113–120. doi: 10.1016/j.tube.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 55.Teitelbaum R, Cammer M, Maitland ML, Freitag NE, Condeelis J, Bloom BR. Mycobacterial infection of macrophages results in membrane-permeable phagosomes. Proc Natl Acad Sci U S A. 1999;96:15190–15195. doi: 10.1073/pnas.96.26.15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stanley SA, Cox JS. Host-pathogen interactions during Mycobacterium tuberculosis infections. Curr Top Microbiol Immunol. 2013;374:211–241. doi: 10.1007/82_2013_332. [DOI] [PubMed] [Google Scholar]
- 57.Goren MB, D’Arcy Hart P, Young MR, Armstrong JA. Prevention of phagosome-lysosome fusion in cultured macrophages by sulfatides of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1976;73:2510–2514. doi: 10.1073/pnas.73.7.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mougous JD, Petzold CJ, Senaratne RH, Lee DH, Akey DL, Lin FL, et al. Identification, function and structure of the mycobacterial sulfotransferase that initiates sulfolipid-1 biosynthesis. Nat Struct Mol Biol. 2004;11:721–729. doi: 10.1038/nsmb802. [DOI] [PubMed] [Google Scholar]
- 59.Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddyx PL, Collins HL, Fok AK, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- 60.Kaufmann SH, Dorhoi A. Inflammation in tuberculosis: interactions, imbalances and interventions. Curr Opin Immunol. 2013;25:441–449. doi: 10.1016/j.coi.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 61.Scollard DM. The biology of nerve injury in leprosy. Lepr Rev. 2008;79(3):242–253. [PubMed] [Google Scholar]
- 62.Beatty WL, Rhoades ER, Ullrich HJ, Chatterjee D, Heuser JE, Russell DG. Trafficking and release of mycobacterial lipids from infected macrophages. Traffic. 2000;1:235–247. doi: 10.1034/j.1600-0854.2000.010306.x. [DOI] [PubMed] [Google Scholar]
- 63.Bishop AE, Pietroletti R, Taat CW, Brummelkamp WH, Polak JM. Increased populations of endocrine cells in Crohn’s ileitis. Virchows Arch A Pathol Anat Histopathol. 1987;410:391–396. doi: 10.1007/BF00712758. [DOI] [PubMed] [Google Scholar]
- 64.Thachil E, Hugot JP, Arbeille B, Paris R, Grodet A, Peuchmaur M, et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease. Gastroenterology. 2012;142:1097–1099. doi: 10.1053/j.gastro.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 65.McKay HF, Burgess DR. ‘Life is a highway’: membrane trafficking during cytokinesis. Traffic. 2011;12:247–251. doi: 10.1111/j.1600-0854.2010.01139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pan H, Cai N, Li M, Liu GH, Izpisua Belmonte JC. Autophagic control of cell ‘stemness’. EMBO Mol Med. 2013;5:327–331. doi: 10.1002/emmm.201201999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sioud M, Floisand Y. NOD2/CARD15 on bone marrow CD34+ hematopoietic cells mediates induction of cytokines and cell differentiation. J Leukoc Biol. 2009;85:939–946. doi: 10.1189/jlb.1008650. [DOI] [PubMed] [Google Scholar]
- 68.Nigro G, Rossi R, Commere PH, Jay P, Sansonetti PJ. The cytosolic bacterial peptidoglycan sensor nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe. 2014;15:792–798. doi: 10.1016/j.chom.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 69.Vooijs M, Liu Z, Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology. 2011;141:448–459. doi: 10.1053/j.gastro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan Y, Denef N, Schupbach T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev Cell. 2009;17:387–402. doi: 10.1016/j.devcel.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sethi N, Yan Y, Quek D, Schupbach T, Kang Y. Rabconnectin-3 is a functional regulator of mammalian Notch signaling. J Biol Chem. 2010;285:34757–34764. doi: 10.1074/jbc.M110.158634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider M, Troost T, Grawe F, Martinez-Arias A, Klein T. Activation of Notch in lgd mutant cells requires the fusion of late endosomes with the lysosome. J Cell Sci. 2013;126(Pt 2):645–656. doi: 10.1242/jcs.116590. [DOI] [PubMed] [Google Scholar]
- 73.Spiller R. Serotonin, inflammation, and IBS: fitting the jigsaw together? J Pediatr Gastroenterol Nutr. 2007;45(Suppl 2):S115–S119. doi: 10.1097/MPG.0b013e31812e66da. [DOI] [PubMed] [Google Scholar]
- 74.Grundy D. 5-HT system in the gut: roles in the regulation of visceral sensitivity and motor functions. Eur Rev Med Pharmacol Sci. 2008;12(Suppl 1):63–67. [PubMed] [Google Scholar]
- 75.Stadnicki A. Involvement of coagulation and hemostasis in inflammatory bowel diseases. Curr Vasc Pharmacol. 2012;10:659–669. doi: 10.2174/157016112801784495. [DOI] [PubMed] [Google Scholar]
- 76.Sorrentino D. Fibrocytes, inflammation, and fibrosis in Crohn’s disease: another piece of the puzzle. Dig Dis Sci. 2014;59:699–701. doi: 10.1007/s10620-013-2888-2. [DOI] [PubMed] [Google Scholar]
- 77.DeBoer MD, Denson LA. Delays in puberty, growth, and accrual of bone mineral density in pediatric Crohn’s disease: despite temporal changes in disease severity, the need for monitoring remains. J Pediatr. 2013;163:17–22. doi: 10.1016/j.jpeds.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gershon MD. 5-Hydroxytryptamine (serotonin) in the gastrointestinal tract. Curr Opin Endocrinol Diabetes Obes. 2013;20:14–21. doi: 10.1097/MED.0b013e32835bc703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar R, Vaidya MC, Belurkar N. Blood levels of serotonin in human leprosy. Lepr India. 1980;52:532–535. [PubMed] [Google Scholar]
- 80.Stepanian ES, Blokh EL. Increased serotonin level in the blood of patients with pulmonary tuberculosis. Vrach Delo. 1966;10:56–59. [PubMed] [Google Scholar]
- 81.Henry JP, Sagne C, Bedet C, Gasnier B. The vesicular monoamine transporter: from chromaffin granule to brain. Neurochem Int. 1998;32:227–246. doi: 10.1016/S0197-0186(97)00092-2. [DOI] [PubMed] [Google Scholar]
- 82.Naoi M, Maruyama W, Inaba-Hasegawa K. Type A and B monoamine oxidase in age-related neurodegenerative disorders: their distinct roles in neuronal death and survival. Curr Top Med Chem. 2012;12:2177–2188. doi: 10.2174/156802612805219950. [DOI] [PubMed] [Google Scholar]
- 83.Marchitti SA, Deitrich RA, Vasiliou V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev. 2007;59:125–150. doi: 10.1124/pr.59.2.1. [DOI] [PubMed] [Google Scholar]
- 84.O’Hara JR, Sharkey KA. Proliferative capacity of enterochromaffin cells in guinea-pigs with experimental ileitis. Cell Tissue Res. 2007;329:433–441. doi: 10.1007/s00441-007-0430-6. [DOI] [PubMed] [Google Scholar]
- 85.Hinz M, Stein A, Uncini T. APRESS: apical regulatory super system, serotonin, and dopamine interaction. Neuropsychiatr Dis Treat. 2011;7:457–463. doi: 10.2147/NDT.S23676. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Tamai I. Pharmacological and pathophysiological roles of carnitine/organic cation transporters (OCTNs: SLC22A4, SLC22A5 and Slc22a21) Biopharm Drug Dispos. 2013;34:29–44. doi: 10.1002/bdd.1816. [DOI] [PubMed] [Google Scholar]
- 87.Tomer G, Wetzler G, Keddache M, Denson LA. Polymorphisms in the IBD5 locus are associated with Crohn disease in pediatric Ashkenazi Jewish patients. J Pediatr Gastroenterol Nutr. 2009;48:531–537. doi: 10.1097/MPG.0b013e318183138a. [DOI] [PubMed] [Google Scholar]
- 88.Pierce ES. Possible transmission of Mycobacterium avium subspecies paratuberculosis through potable water: lessons from an urban cluster of Crohn’s disease. Gut Pathog. 2009;1:17. doi: 10.1186/1757-4749-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]