

Abstract
Deciphering the complexity and heterogeneity of cancer benefits from integration of proteomic level data into systems biology efforts. The opportunities available as a result of advances in proteomic technologies, the successes to date and the challenges involved in integrating diverse datasets are addressed in this review.
1. Introduction
While cancer is considered a disease of the genome, a mechanistic understanding of tumor initiation and progression is unlikely to be fully elucidated without integration of proteomics into the systems biology of cancer. Furthermore, as the most functional compartment encoded in the genome, the proteome remains a major source of cancer biomarkers and therapeutic targets. Substantial progress has been made in the field of proteomics to allow comprehensive profiling of complex proteomes, in-depth analysis of proteins in specific cellular compartments (Figure 1), in the microenvironment and in biological fluids. Mining the proteome extends beyond assessment of protein concentrations. It encompasses numerous types of modifications (Figure 2) that impact activity and functional states.
Figure 1. Proteomic profiling of cancer cells.
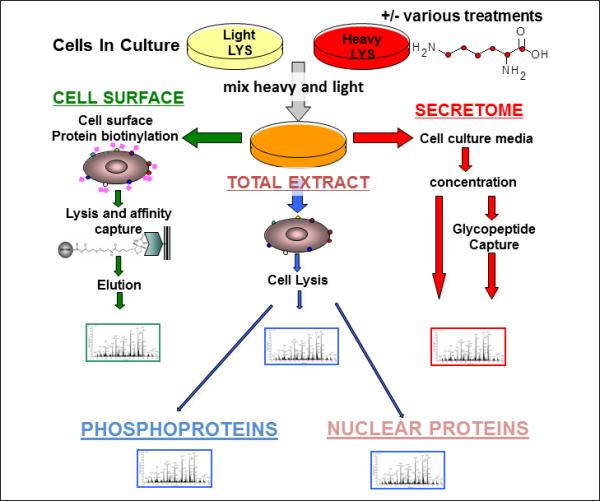
Freshly isolated tumor cells or cancer cell lines to be compared are cultured in the presence of isotopically labeled amino acids. Media, biotinylated cell surface proteins and whole lysates are isolated separately and analyzed. Cell lysates may be further fractionated for separate analysis of sub-compartments (e.g. nuclear proteins), or protein subgroups (e.g. phosphoproteins).
Figure 2.
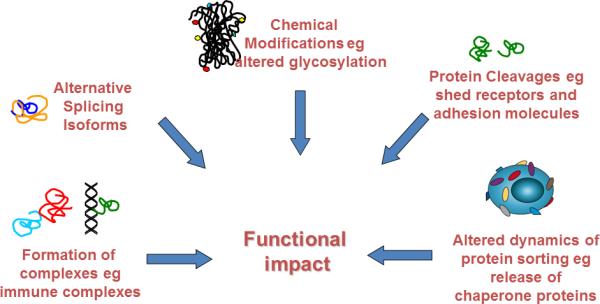
The wide world of protein modifications that affect function and yield biomarkers and therapeutic targets
2. The diversity of approaches to interrogate the proteome
2.1 Mass spectrometry
Remarkable advances in mass spectrometry for protein analysis have occurred in the past two decades. The use of mass spectrometry simply for mass peak profiling without protein identification has become rather obsolete. Current instruments based on electrospray ionization provide very high sensitivity and scan speed that allow identification of a major protein form for virtually all proteins translated from expressed genes in a cell population1 and allow comprehensive analysis of the proteome of serum and plasma and biological fluids across seven or more logs of protein abundance.2 Likewise improvements in Matrix Assisted Laser Desorption Ionization mass spectrometry have been introduced in part through the development of novel probes and optimized liquid matrices that have substantially improved sensitivity, which is particularly relevant to modified peptides.3 The characterization of common post-translational modifications in proteins notably phosphorylation and glycosylation is currently feasible.4, 5 While comprehensive proteomic profiling by mass spectrometry has limited throughput, quantitative profiling of a pre-defined set of peptide products of proteins in complex mixtures through the use of multiple reaction monitoring (MRM) is currently feasible for exploratory and confirmation studies for proteins of interest that occur in moderate abundance.6
2.2 Parallel approaches to mass spectrometry
Proteins in complex mixtures may be captured and identified using affinity capture agents spotted onto microarrays which provides a high throughput approach to interrogation of the proteome.7 Coverage is dependent on the diversity of the capture agents, primarily antibodies that are available. A reverse process to interrogate the proteome involves arraying lysates, proteins and peptides. Recombinant proteins and synthetic peptides may be spotted onto microarrays and alternatively peptides and proteins may be synthesized directly on the microarrays.8-10 Aside from assessment of protein levels and interactions, the development of activity based probes allows a determination of protein functionality and alterations in protein activity associated with tumor initiation and progression that would be otherwise difficult to assess through genomics or through quantification of protein levels and modifications.11
3. Contribution of proteomics to systems biology of cancer
Here we review the added contribution of proteome level studies to the molecular profiling of cancer to elucidate pathways, networks and processes that inform about tumor development, progression and classification and that have a translational potential by yielding biomarkers for diagnostics and targets for therapeutics.12 A conceptual framework for the contribution of proteomics to the systems biology of cancer is presented in Figure 3.
Figure 3.
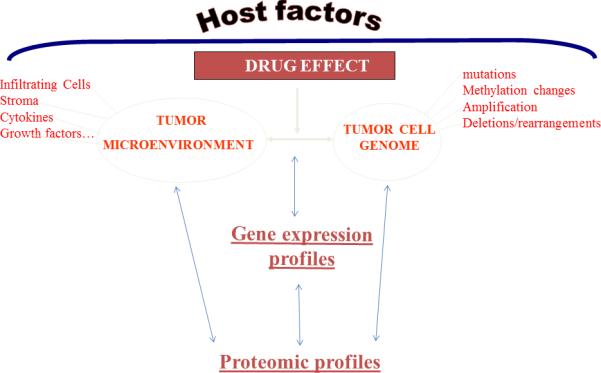
Conceptual framework for the contribution of proteomics to the systems biology of cancer
3.1 The complementary nature of proteomic and transcriptomic data
The merits of quantitative proteomics as a complement to gene expression profiling depend on whether the proteome is strictly regulated at, and predicted from the transcriptome in cancer, in which case protein levels in cell and tissues may be simply inferred from gene expression data. Relatively few studies have critically addressed this issue. An informative study was based on the NCI-60 cancer cell line panel which represents nine cancer tissue types.13 Some 65% of genes for which mRNA and protein concentration data were available exhibited a statistically significant transcript-protein correlation. In a separate study, comparative analysis of mRNA and protein expression for 98 genes in lung adenocarcinomas revealed poor concordance for most genes.14 Even if concordance between RNA and protein levels is high, the specific compartment(s) in which a protein resides or the nature of interacting proteins in a disease process cannot be inferred from transcriptomic data alone and therefore evaluation of multiple protein compartments may be necessary to elucidate a cancer biological process. A proteomic study comparing metastatic and non-metastatic cell lines revealed a multi-layered TGFβ regulatory network by integrating data from cell surface, secreted and cytosolic proteins.15 Analysis of proteins from the separate proteomic compartments did not identify a dominant regulatory pathway, but analyzing the combined proteomic compartments together identified TGFβ as the most highly significant network. This combined analysis further revealed a complex regulation of TGFβ, as a few TGFβ-interacting proteins from each compartment coalesced into a regulatory network by integration of the data.
Discordance between RNA and protein levels may reflect in part the role of translational control. The extent to which translational control in cancer may favor certain mRNAs over others has been explored in some detail. In many types of human cancers, eukaryotic initiation factors (eIFs) are either overexpressed or ectopically activated by Ras-MAPK and PI3K-mTOR signaling cascades.16 An isogenic cellular model of colorectal cancer transition from invasive carcinoma to metastasis was utilized to examine the role of translational control in metastasis.17 Changes in the level of mRNA association with polysomes occurred more than 2-fold greater than changes in the level of total cellular mRNA in the transition to metastasis. Distinct signatures of statistically over-represented gene functions in translated mRNAs were observed. An increase in the hyperphosphorylated form of the eIF4E-BP1 protein in the metastatic cell line was found that may have contributed to increased activation of cap-dependent translation of certain mRNAs. A multitude of kinases have been investigated specifically for their role in 4EBP1 phosphorylation and stability in cancer, leading to the identification of several kinases that may be involved in cancer development.18 EIF4B, another translation initiation factor, has been found to regulate translation of proliferative and pro-survival mRNAs. Depletion of eIF4B in cancer cells attenuated proliferation and sensitized cells to genotoxic stress-driven apoptosis.19
It follows from these findings that integration of quantitative proteomics with transcriptomic profiling enhances the potential for delineating molecular signatures associated with cancer. A combined analysis of the transcriptome and proteome of lung adenocarcinomas yielded greater insights than either alone. Eleven components of the glycolytic pathway were shown to be associated with poor survival in the combined mRNA-protein datasets20. Proteomic analysis of prostate tumor tissue extracts, and integration with genomic data allowed construction of a multiplex gene signature representing progression of indolent cancer to aggressive disease.21
Integration of transcriptomic and proteomic profiles from mouse models of cancer with human cancer profiles can overcome limitations associated with human biospecimen based investigations as demonstrated in a recent study.22 Global comparisons of plasma proteomic profiles from a large set of various cancer mouse models revealed plasma protein signatures of lung cancer (Figure 4). Further integration of gene expression profiles from human lung tumor and profiles following gene knock-out manipulations in human lung cancer cell lines, NKX2-1/TITF1-regulated proteins were significantly elevated in lung adenocarcinoma mouse models, indicating that NKX2-1/TITF1, known as a lineage specific oncogene and a master regulator of lung development, regulates production of proteins that are released into circulation with tumor development. In addition, pathway analysis of plasma protein profiles revealed EGFR signature in an EGFR mutant lung adenocarcinoma mouse model, supported by comprehensive proteomic profile of subcellular compartments of EGFR mutant vs non-mutant lung adenocarcinoma cell lines (Figure 4). Integration of tumor gene expression profiles with plasma protein profiles identified neuroendocrine signature in plasma from a small cell lung cancer mouse model (Figure 4). These findings suggest that plasma protein profile may reflect functionally relevant pathways or characteristics in tumor. Importantly, concordant findings were observed in human lung cancer plasmas. This study demonstrated that integrative analysis of multiple omics data from various materials is a powerful tool to identify relevant signatures in human cancer.
Figure 4. plasma protein signatures of lung cancer derived from analysis of mouse models of cancer.
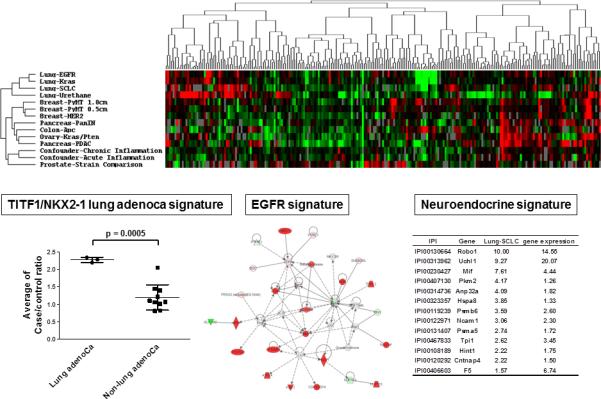
The heat map displays clustering together of lung cancer mouse models’ plasma proteome data. Below is a comparison between human lung adenocarcinoma plasmas and human lung non-adenocarcinoma plasmas of levels of proteins associated with an NKX2.1 signature identified in lung adenocarcinoma. An EGFR related protein network identified in plasma from an EGFR mutant lung cancer mouse model is shown. A list of proteins associated with a neuroendocrine signature in small cell lung cancer plasma is presented numbers refer to plasma protein case/control ratios and corresponding gene expression ratios in tumor tissue from the SCLC mouse model compared to controls.
3.2 Interrogating the proteome adds functionality to cancer molecular profiling
There is increasing reliance on profiling protein post-translational modifications notably phosphorylation as part of an overall strategy to identify molecular features associated with aggressive tumors and metastasis, and targets for therapeutics and mechanisms of therapeutic response and resistance. Quantitative phosphoproteome and transcriptome analysis of MCF-7 human breast cancer cells stimulated with ligand yielded an understanding of tamoxifen resistance at a systems level.23 Following stimulation of ligands an enrichment in phospho-proteins was observed in sensitive compared with tamoxifen-resistant cells. Parallel analysis of transcriptomic data suggested that deregulated activation of GSK3β (glycogen-synthase kinase 3β) and MAPK1/3 signaling might be associated with altered activation of cAMP-responsive element-binding protein and AP-1 transcription factors in tamoxifen-resistant cells. This hypothesis derived from the combined phosphoproteome and transcriptome data was validated by reporter assays and by testing in human clinical samples which revealed that inhibitory phosphorylation of GSK3β at serine 9 was significantly lower in cancer patients that relapsed following treatment with tamoxifen. In another study breast tumor lysate arrays were interrogated using 146 antibodies to proteins relevant to breast cancer to determine whether such a functional proteomics approach improves breast cancer classification and can predict pathological complete response in patients receiving neoadjuvant taxane and anthracyclinetaxane-based systemic therapy in independent training and testing sets.24 Six breast cancer subgroups associated with different recurrence-free survival were identified based on a 10-protein biomarker panel in the training set and confirmed in the test set.
An approach that integrates phosphoprotein profiling was used to identify and quantify clinically relevant, drug-specific biomarkers for phosphatidylinositol 3-kinase (PI3K) pathway inhibitors that target AKT, phosphoinositide-dependent kinase 1 (PDK1), and PI3K-mammalian target of rapamycin (mTOR).25 A total of 375 PI3K pathway-relevant phosphopeptides containing AKT, PDK1, or mitogen-activated protein kinase substrate recognition motifs were interrogated, of which 71 were drug-regulated, some by all three inhibitors. Phosphospecific antibodies were produced against specific, drug-regulated phosphorylation sites as biomarker tools for PI3K pathway inhibitors.
Using the Met receptor as the major model system, a study combined multiplex phosphoproteomics, genome-wide expression profiling, and functional assays in various cancer cells addicted to oncogenic receptor tyrosine kinases.26 Met blockade was found to affect a limited subset of Met downstream signals. Only a restricted signature of transducers and transcriptional effectors downstream of Ras or phosphoinositide 3-kinase (PI3K) was inactivated. Met inhibition led to cell-cycle arrest as did Inhibition of Ras-dependent signals and PI3K-dependent signals also resulted in cell-cycle arrest. Inhibition of Met without inactivation of Ras or PI3K signaling did not affect proliferation. Interestingly a similar signature was observed by inhibition of epidermal growth factor receptor in a different cellular context. These findings pointed to the critical role of Ras and PI3K as determinants of therapeutic response.
Integrated proteomic and genomic profiling have yielded insights into the process of metastasis.In mice. Lkb1 deletion and activation of Kras (G12D) results in lung tumors with metastases. Integrated genomic and proteomic profiles of tumors from this model identified gene and phosphoprotein signatures associated with Lkb1 loss and progression to invasive and metastatic lung tumors.27 The combined inhibition of SRC, PI3K, and MEK1/2 in this model resulted in synergistic tumor regression.
Epithelial-mesenchymal transition (EMT) is a process associated with invasion and metastasis of epithelial tumors. Protein, phosphoprotein, phosphopeptide and RNA transcript abundance was assessed to develop a systems view of EMT.28 Findings included a coordinate metabolic reduction in a cluster of 17 free-radical stress pathway components which correlated with reduced glycolytic and increased oxidative phosphorylation enzyme capacity, consistent with reduced cell cycling and reduced need for macromolecular biosynthesis in the mesenchymal state. An attenuation of EGFR autophosphorylation and of IGF1R, MET and RON signaling with EMT was observed. In parallel, increased prosurvival autocrine IL11/IL6-JAK2-STAT signaling, autocrine fibronectin-integrin α5β1 activation, autocrine Axl/Tyro3/PDGFR/FGFR RTK signaling and autocrine TGFβR signaling was observed. Seemingly, paradoxical findings were observed in proteomic profiling of leiomyosarcoma, a common mesenchymal tumor type. Expression of the epithelial marker E-cadherin was significantly elevated in a subset of leiomyosarcomas and was correlated with better survival.29 The epithelial gene expression signature at the mRNA level was also associated with better survival. Transcriptome data revealed an inverse correlation between E-cadherin and E-cadherin repressor Slug (SNAI2) in leiomyosarcoma which was validated at the protein level. Knockdown of Slug expression in leiomyosarcoma cells significantly increased E-cadherin; decreased the mesenchymal markers vimentin and N-cadherin and significantly decreased cell proliferation, invasion, and migration. These studies based on proteome and phosphoproteome profiling integrated with genomic and transcriptomic findings have provided novel insights into tumor invasion and metastasis and determinants of therapeutic response and resistance. However they have covered a rather narrow spectrum of the proteome with a focus on a single post-translational modification namely phosphorylation. Undoubtedly additional insights may derive from assessment 30 of a wider spectrum of post-translational modification. A case in point is the cross talk between phosphorylation and O-linked beta-N-acetylglucosamine (O-GlcNAcylation) at the same amino acid residues which can be interrogated with current proteomic technologies. O-GlcNAcylation is a ubiquitous, reversible process that modifies serine and threonine residues. A recent study identified 141 previously unknown O-GlcNAc sites on proteins that function in spindle assembly and cytokinesis, many of which are either identical to known phosphorylation sites.31 Induced overexpression of O-GlcNAc transferase increased the inhibitory phosphorylation of cyclin-dependent kinase 1 (CDK1) and reduced the phosphorylation of CDK1 target proteins. Advances in glycomics and glycoproteomics are likely to impact our understanding of the role of glycans and aberrant glycosylation in cancer.32,30 Other chemical modifications of proteins and the role of proteases and hydrolases also remain under-explored.).
Aside from quantitative assessment of protein levels and modification, activity-based profiling has substantial functional relevance to our understanding of cancer. Selective pharmacological probes are currently available that allow interrogation of enzyme activity in tumor cells that complement quantitative proteomics and other molecular profiling approaches to delineate key alteration and druggable targets in cancer.33
3.3 Building and mining protein networks and pathways
A fundamental contribution of systems biology is understanding networks and pathways that regulate cell processes and that may be dysregulated in cancer. Proteomics is integral to this understanding as evidenced from studies that explored a multitude of cancer types. To better understand prostate tumor progression, concurrent quantification of gene expression and protein levels was obtained following treatment of LNCaP prostate cancer cells. 34 The resulting data was integrated with a global network of protein interactions which identified the network of growth factor regulation of cell cycle as the main response module for androgen treatment in LNCap cells. The findings from this study suggested that growth factor signaling represented a secondary effect of the initial androgen stimulus likely transmitted from multiple growth factor receptors through pathways that constitute an interconnected network module. The implication is that a combination of targeted therapeutics are necessary to affect tumor growth. Resistance to cisplatin is a major issue in ovarian cancer treatment. A systems biology approach was used to examine global protein level and network level changes by comparing proteomics profiles between cisplatin-resistant cell lines and cisplatin-sensitive cell lines.35 A list of 119 differentially expressed proteins was assembled which was expanded into a cisplatin-resistant activated subnetwork. Significant enrichment of proton-transporting ATPase and ATP synthase complexes was observed. Sub-network protein interaction function categories were examined using two-dimensional visualization matrixes. Significant cellular physiological responses were found to result from endogenous, abiotic, and stress-related signals that correlated with known mechanisms of action of cisplatin. A colorectal cancer (CRC) study tested the hypothesis that small changes in the mRNA expression of multiple genes in the neighborhood of a protein-hub can be synergistically associated with significant changes in the activity of that protein and its network neighbors.36. It was further hypothesized that proteomic targets with significant fold change between phenotype and control may be used to “seed” a search for small sub-networks that are functionally associated with these targets. Proteomic targets having significant expression changes in CRC from two independent proteomic screens were selected. Random walk based models of network crosstalk were used to develop novel reference models to identify sub-networks that are statistically significant in terms of their functional association with these proteomic targets. Synergistic changes in the activity of identified sub-networks were assessed based on genome-wide screens of mRNA expression in CRC. Cross-classification experiments to predict disease class yielded excellent performance using only a few subnetworks pointing to the utility of this approach to discover pertinent sub-networks.
Phosphoproteomics was applied to the identification of Ras-regulated phosphorylation events through the analysis of immortalized human bronchial epithelial cells with and without the expression of oncogenic Ras.37 A majority of the Ras-targeted events identified consisted of a [pSer/Thr]-Pro motif, that indicated involvement of proline-directed kinases. Integrating the phosphorylated signatures into the Pathway Interaction Database, yielded Ras-regulated pathways, including MAPK and other novel signaling cascades. Another independent study also used phosphoproteomics to identify tyrosine phosphorylated proteins in isogenic human bronchial epithelial cells and human lung adenocarcinoma cell lines, expressing either of the two mutant alleles of EGFR (L858R and Del E746-A750), or a mutant KRAS allele, which occur commonly in human lung adenocarcinomas.38 Tyrosine phosphorylation of signaling molecules was greater in cells expressing the mutant EGFRs than in cells expressing wild type EGFR or mutant KRAS. Bayesian network analysis revealed that polymerase transcript release factor might be a potentially important component of the ERBB signaling network.
Proteomics has been featured in numerous studies of the systems biology of breast cancer. Resistance to endocrine treatment, such as anti-estrogens, often occurs in breast cancer and estrogen receptor (ER)-positive breast cancer research has been viewed as an ideal example of how systems biology can be applied to better understand this disease.39 A search for mechanisms leading to the development of antiestrogen-resistance was based on analysis of the gene and protein expression patterns of the human breast carcinoma cell line T47D and its resistant derivative T47D-r.40 Thirty-eight proteins were found to be reproducibly up- or down-regulated in T47D-r versus T47D with concordant differential expression at the RNA level including cathepsin D, Rab11a and MxA, and the secreted protein hAG-2. For 11 proteins, the corresponding mRNA was not found to be differentially expressed, and for eight proteins an inverse regulation was found at the mRNA level pointing to discordant RNA and protein levels.
To identify regulators of intracellular signaling in breast cancer, 541 kinases and kinase-related molecules were targeted with small interfering RNAs (siRNAs), and their effects on signaling was determined with lysate arrays interrogated with 42 phospho and total proteins.41 Network-based analysis identified the MAPK subnetwork of genes along with p70S6K and FRAP1 as the most prominent targets that increased phosphorylation of AKT, a key regulator of cell survival. The siRNA screen revealed bi-directionality in the AKT and glycogen synthase kinase 3 interaction which was unexpected.
The insulin-like growth factor (IGF-1) signaling network was analyzed in the ER- MDA-MB231 breast cancer cell line.42 Lysate arrays were utilized to measure changes in protein phosphorylation after IGF-1 stimulation. A computational procedure integrated mass action modeling with particle swarm optimization to train the model against the experimental data and infer model parameters. The trained model was used to identify drug combinations that minimally increased phosphorylation of other proteins elsewhere in the network. Experimental testing of predictions based on the model revealed that optimal drug combinations inhibited cell signaling and proliferation, compared with non-optimal combination of inhibitors which increased phosphorylation of nontargeted proteins and rescued cells from cell death. These studies, though limited in their interrogation of the proteome clearly demonstrate the added value of proteome level data.
4. Methods and tools to assist in integrating proteomics into systems biology of cancer
An overview of publicly available databases relevant to proteomics studies in cancer research has been presented recently.43 The review covers general use protein databases, gene/protein expression databases, gene mutation and single nucleotide polymorphisms databases, tumor antigen databases, protein-protein interaction, and biological pathway databases. Another recent comprehensive review has focused on major protein bioinformatics databases and resources that are relevant to comparative proteomics research.44 It encompasses sequence databases, family databases, structure databases, function databases, and both gel electrophoresis and mass spectrometry based proteomics databases. Here we highlight some of the recently developed databases that have a relatively broad scope or that focus on a particular cancer type.
The Human Protein Atlas project aims to create a map of protein expression patterns in normal cells, tissues and cancer based on antibody profiling.45 Currently, more than 11,000 unique proteins corresponding to over 50% of all human protein-encoding genes have been analyzed. All protein expression data, including underlying high-resolution images, are published on the free and publically available Human Protein Atlas portal (http://www.proteinatlas.org). The database is particularly useful when candidates are identified in a particular study and there is a need to determine levels of expression in different normal and tumor tissues and cell populations. The Quantitative Assay Database (QuAD)46 provides methods and reagents for monitoring cancer related signaling pathways and biological processes with mass spectrometry through multiple reaction monitoring. The purpose of the QuAD is to also share methods and disseminate reagents for studying cancer biology spanning from cell lines to limited amounts of clinical samples. It is envisioned that additional information using other analytical platforms will be incorporated as the database grows.
dbDEPC47 is a curated database that contains over 4,000 protein entries, from 331 experiments across 20 types of human cancers.48 The database may be used to search for particular proteins to determine their range of expression and changes as may be related to genomic aberrations. Information is provided pertaining to experimental design, and tools are available for filtering and for network analysis.
There are also numerous databases with a restricted focus. Apoptosis is implicated in a large number of human diseases. Many quantitative proteome studies of apoptosis have been performed to gain insight into proteins involved in this process. A research group in Norway has established an ApoptoProteomics database for storage, browsing, and analysis of the outcome of large scale proteome analyses of apoptosis derived from human, mouse and rat.49 This database has also been used to identify hundreds of caspase substrates from apoptosis. The Pancreatic Expression database50 typifies an effort focused on a single disease. It is a component of the European Union project,51 whose mission is to develop novel molecular diagnostic tools for the prevention and diagnosis of pancreatic cancer. The database currently includes over 60,000 measurements derived from transcriptomics, proteomics, genomics and miRNA profiles from various sources.52 The data model is generic allowing its application to other cancers.
EBI has established a biological sample oriented database, BioSD.53 The database is intended to: 1) record and manage sample information consistently and to link sample information to assay data across multiple resources; 2 ) minimize data entry efforts for the user, enabling submission of sample descriptions only once; 3) support cross database sample queries by sample description; and 4) build a continuously growing set of consistently annotated samples that are used in multiple experiments.
A protocol has been developed for the identification of alternatively spliced peptide sequences from tandem mass spectrometry datasets.54 This approach is suitable for human and mouse datasets. Application of the method was illustrated with a study of the Kras activation-Ink4/Arf deletion mouse model of human pancreatic ductal adenocarcinoma. A novel integrated approach, named CAERUS, has been described for the identification of gene signatures to predict cancer outcomes based on the domain interaction network in the human proteome.43 A model was developed to score each protein by quantifying the domain connections to its interacting partners and the somatic mutations present in the domain. Proteins were then defined as gene signatures if their scores were above a preset threshold. The correlation of expression levels between a gene signature and its neighboring proteins was determined. The results of the quantification for each subject were then used to predict cancer outcome by a modified naïve Bayes classifier. A list of cancer-associated gene signatures and domains was also compiled to provide testable hypotheses. The utility of this approach was demonstrated for breast and ovarian cancer datasets.
The integrated analysis of diverse datasets including proteomics represents a statistical and computational challenge. Integration of diverse data may be hampered by lack of standardization of identifier nomenclature among proteins, genes, and microarray datasets. A study compared three freely available internet-based identifier mapping resources for mapping UniProt accessions (ACCs) to Affymetrix probesets identifications (IDs): DAVID, EnVision, and NetAffx55 which uncovered a high level of discrepancy among the mapping resources. Methods continue to be developed that address various needs related to data integration and mining, a sampling of which is presented here. A Partial Least Squares Regression (PLSR)-based data integration strategy, has been proposed for simultaneous analysis of proteomic data, gene expression data and classical clinical parameters.56 PLSR allows visualization of complex datasets and collapses multidimensional data into fewer relevant dimensions for data interpretation. PLSR facilitates identification of functionally important modules by performing comparison to databases on known biological interactions. A novel Bayesian model was developed that extends the applicability of multivariate, multi-way ANOVA-type methods to multi-source data.57 The method has the capability of finding covariate-related dependencies between the sources. It estimates the multivariate covariate effects and their interaction effects for the discovered groups of variables and partitions the effects to those shared between the sources and to source-specific ones. Other methods have been proposed for cross-platform biological data integration, such as eScience-Bayes strategy,58 an adjusted RV coefficient approach,59 and a sparse simultaneous component method.60
5. Looking ahead
Advances in proteomics technologies and development of related resources are likely to contribute substantially to the systems biology of cancer and to translate knowledge into applications from diagnostics to therapeutics. Important issues for the field include feasibility of interrogating the entire complement of proteins expressed in cells, tissues and biological fluids that span 6-8 logs of protein abundance; ability to determine not only quantitative levels of proteins sets but also their post-translational modifications and functional states. Given the remarkable advances in mass spectrometry and array technologies we have witnessed in the past decade, it is likely that such rate of growth will be maintained for the foreseeable future. Such advances in broad proteomic profiling technologies need to be paralleled with similar advances in our ability to interrogate defined panels of proteins with the necessary sensitivity, specificity and quantitative reproducibility for translational applications.
Other fields that promise to contribute to the systems biology of cancer beyond genomics and proteomics include metabolomics and glycomics. The challenge is in developing efficient means for data integration and for testing derived hypotheses. However a full understanding of cancer pathogenesis will be incomplete without such a comprehensive systems based effort.
References
- 1.Olsen JV, Schwartz JC, Griep-Raming J, Nielsen ML, Damoc E, Denisov E, Lange O, Remes P, Taylor D, Splendore M, et al. A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol Cell Proteomics. 2009;8(12):2759–2769. doi: 10.1074/mcp.M900375-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Q, Faca V, Hanash S. Mining the plasma proteome for disease applications across seven logs of protein abundance. J Proteome Res. 2011;10(1):46–50. doi: 10.1021/pr101052y. [DOI] [PubMed] [Google Scholar]
- 3.Kaneshiro K, Fukuyama Y, Iwamoto S, Sekiya S, Tanaka K. Highly sensitive MALDI analyses of glycans by a new aminoquinoline-labeling method using 3-aminoquinoline/alpha-cyano-4-hydroxycinnamic acid liquid matrix. Anal Chem. 2011;83(10):3663–3667. doi: 10.1021/ac103203v. [DOI] [PubMed] [Google Scholar]
- 4.Wang H, Wong CH, Chin A, Taguchi A, Taylor A, Hanash S, Sekiya S, Takahashi H, Murase M, Kajihara S, et al. Integrated mass spectrometry-based analysis of plasma glycoproteins and their glycan modifications. Nat Protoc. 2011;6(3):253–269. doi: 10.1038/nprot.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palumbo AM, Smith SA, Kalcic CL, Dantus M, Stemmer PM, Reid GE. Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom Rev. 2011;30(4):600–625. doi: 10.1002/mas.20310. [DOI] [PubMed] [Google Scholar]
- 6.Remily-Wood ER, Liu RZ, Xiang Y, Chen Y, Thomas CE, Rajyaguru N, Kaufman LM, Ochoa JE, Hazlehurst L, Pinilla-Ibarz J, et al. A database of reaction monitoring mass spectrometry assays for elucidating therapeutic response in cancer. Proteomics Clin Appl. 2011;5(7-8):383–396. doi: 10.1002/prca.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramirez AB, Lampe P. Discovery and Validation of Ovarian Cancer Biomarkers Utilizing High Density Antibody Microarrays. Cancer Biomark. 2011 doi: 10.3233/CBM-2011-0215. [DOI] [PubMed] [Google Scholar]
- 8.Hu S, Xie Z, Onishi A, Yu X, Jiang L, Lin J, Rho HS, Woodard C, Wang H, Jeong JS, et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139(3):610–622. doi: 10.1016/j.cell.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller C, Liotta LA, Espina V. Reverse phase protein microarrays advance to use in clinical trials. Mol Oncol. 2010;4(6):461–481. doi: 10.1016/j.molonc.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu J, Choi G, Li L, Wang H, Pitteri SJ, Pereira-Faca SR, Krasnoselsky AL, Randolph TW, Omenn GS, Edelstein C, et al. Occurrence of autoantibodies to annexin I, 14-3-3 theta and LAMR1 in prediagnostic lung cancer sera. J Clin Oncol. 2008;26(31):5060–5066. doi: 10.1200/JCO.2008.16.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulick MG, Bogyo M. Application of activity-based probes to the study of enzymes involved in cancer progression. Curr Opin Genet Dev. 2008;18(1):97–106. doi: 10.1016/j.gde.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Angulo AM, Hennessy BT, Mills GB. Future of personalized medicine in oncology: a systems biology approach. J Clin Oncol. 2010;28(16):2777–2783. doi: 10.1200/JCO.2009.27.0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shankavaram UT, Reinhold WC, Nishizuka S, Major S, Morita D, Chary KK, Reimers MA, Scherf U, Kahn A, Dolginow D, et al. Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Mol Cancer Ther. 2007;6(3):820–832. doi: 10.1158/1535-7163.MCT-06-0650. [DOI] [PubMed] [Google Scholar]
- 14.Chen G, Gharib TG, Huang CC, Taylor J, Misek DE, Kardia S, Giordano TJ, Iannettoni MD, Orringer MB, Hanash SM, et al. Discordant protein and mRNA expression in lung adenocarcinomas. Molecular and Cellular Proteomics. 2002;10:1074. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- 15.Schliekelman MJ, Gibbons DL, Faca VM, Creighton CJ, Rizvi ZH, Zhang Q, Wong CH, Wang H, Ungewiss C, Ahn YH, et al. Targets of the tumor suppressor miR-200 in regulation of the epithelial-mesenchymal transition in cancer. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-11-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malina A, Cencic R, Pelletier J. Targeting translation dependence in cancer. Oncotarget. 2011;2(1-2):76–88. doi: 10.18632/oncotarget.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Provenzani A, Fronza R, Loreni F, Pascale A, Amadio M, Quattrone A. Global alterations in mRNA polysomal recruitment in a cell model of colorectal cancer progression to metastasis. Carcinogenesis. 2006;27(7):1323–1333. doi: 10.1093/carcin/bgi377. [DOI] [PubMed] [Google Scholar]
- 18.Pons B, Armengol G, Livingstone M, Lopez L, Coch L, Sonenberg N, Ramon YCS. Association between LRRK2 and 4E-BP1 protein levels in normal and malignant cells. Oncol Rep. 2012;27(1):225–231. doi: 10.3892/or.2011.1462. [DOI] [PubMed] [Google Scholar]
- 19.Shahbazian D, Parsyan A, Petroulakis E, Hershey J, Sonenberg N. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle. 2010;9(20):4106–4109. doi: 10.4161/cc.9.20.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G, Gharib TG, Wang H, Huang C-C, Kuick R, Thomas DG, Shedden KA, Misek DE, Taylor JMG, Giordano TJ, et al. Protein profiles associated with survival in lung adenocarcinoma. Proceedings of the National Academy of Sciences, USA. 2003;100(23):13537–13542. doi: 10.1073/pnas.2233850100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8(5):393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Taguchi A, Politi K, Pitteri SJ, Lockwood WW, Faca VM, Kelly-Spratt K, Wong CH, Zhang Q, Chin A, Park KS, et al. Lung cancer signatures in plasma based on proteome profiling of mouse tumor models. Cancer Cell. 2011;20(3):289–299. doi: 10.1016/j.ccr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oyama M, Nagashima T, Suzuki T, Kozuka-Hata H, Yumoto N, Shiraishi Y, Ikeda K, Kuroki Y, Gotoh N, Ishida T, et al. Integrated quantitative analysis of the phosphoproteome and transcriptome in tamoxifen-resistant breast cancer. J Biol Chem. 2011;286(1):818–829. doi: 10.1074/jbc.M110.156877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez-Angulo AM, Hennessy BT, Meric-Bernstam F, Sahin A, Liu W, Ju Z, Carey MS, Myhre S, Speers C, Deng L, et al. Functional proteomics can define prognosis and predict pathologic complete response in patients with breast cancer. Clin Proteomics. 2011;8(1):11. doi: 10.1186/1559-0275-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersen JN, Sathyanarayanan S, Di Bacco A, Chi A, Zhang T, Chen AH, Dolinski B, Kraus M, Roberts B, Arthur W, et al. Pathway-based identification of biomarkers for targeted therapeutics: personalized oncology with PI3K pathway inhibitors. Sci Transl Med. 2010;2(43):43ra55. doi: 10.1126/scitranslmed.3001065. [DOI] [PubMed] [Google Scholar]
- 26.Bertotti A, Burbridge MF, Gastaldi S, Galimi F, Torti D, Medico E, Giordano S, Corso S, Rolland- Valognes G, Lockhart BP, et al. Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci Signal. 2009;2(102):er11. doi: 10.1126/scisignal.2102er11. [DOI] [PubMed] [Google Scholar]
- 27.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, Buttarazzi MS, Sanofsky BA, McNamara KL, Brandstetter KA, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17(6):547–559. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomson S, Petti F, Sujka-Kwok I, Mercado P, Bean J, Monaghan M, Seymour SL, Argast GM, Epstein DM, Haley JD. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis. 2011;28(2):137–155. doi: 10.1007/s10585-010-9367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Eddy JA, Pan Y, Hategan A, Tabus I, Wang Y, Cogdell D, Price ND, Pollock RE, Lazar AJ, et al. Integrated proteomics and genomics analysis reveals a novel mesenchymal to epithelial reverting transition in leiomyosarcoma through regulation of slug. Mol Cell Proteomics. 2010;9(11):2405–2413. doi: 10.1074/mcp.M110.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor AD, Hancock WS, Hincapie M, Taniguchi N, Hanash SM. Towards an integrated proteomic and glycomic approach to finding cancer biomarkers. Genome Med. 2009;1(6):57. doi: 10.1186/gm57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Udeshi ND, Slawson C, Compton PD, Sakabe K, Cheung WD, Shabanowitz J, Hunt DF, Hart GW. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci Signal. 2010;3(104):ra2. doi: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hart GW, Copeland RJ. Glycomics hits the big time. Cell. 2010;143(5):672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nomura DK, Dix MM, Cravatt BF. Activity-based protein profiling for biochemical pathway discovery in cancer. Nat Rev Cancer. 2010;10(9):630–638. doi: 10.1038/nrc2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vellaichamy A, Dezso Z, JeBailey L, Chinnaiyan AM, Sreekumar A, Nesvizhskii AI, Omenn GS, Bugrim A. “Topological significance” analysis of gene expression and proteomic profiles from prostate cancer cells reveals key mechanisms of androgen response. PLOS One. 2010;5(6):e10936. doi: 10.1371/journal.pone.0010936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen JY, Shen C, Yan Z, Brown DP, Wang M. A systems biology case study of ovarian cancer drug resistance. Computational systems bioinformatics / Life Sciences Society Computational Systems Bioinformatics Conference. 2006:389–398. [PubMed] [Google Scholar]
- 36.Nibbe RK, Koyuturk M, Chance MR. An integrative -omics approach to identify functional sub networks in human colorectal cancer. PLoS Comput Biol. 2010;6(1):e1000639. doi: 10.1371/journal.pcbi.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sudhir PR, Hsu CL, Wang MJ, Wang YT, Chen YJ, Sung TY, Hsu WL, Yang UC, Chen JY. Phosphoproteomics identifies oncogenic Ras signaling targets and their involvement in lung adenocarcinomas. PLOS One. 2011;6(5):e20199. doi: 10.1371/journal.pone.0020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guha U, Chaerkady R, Marimuthu A, Patterson AS, Kashyap MK, Harsha HC, Sato M, Bader JS, Lash AE, Minna JD, et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proc Natl Acad Sci USA. 2008;105(37):14112–14117. doi: 10.1073/pnas.0806158105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zwart W, Theodorou V, Carroll JS. Estrogen receptor-positive breast cancer: a multidisciplinary challenge. Wiley interdisciplinary reviews Systems biology and medicine. 2011;3(2):216–230. doi: 10.1002/wsbm.109. [DOI] [PubMed] [Google Scholar]
- 40.Huber M, Bahr I, Kratzschmar JR, Becker A, Muller EC, Donner P, Pohlenz HD, Schneider MR, Sommer A. Comparison of proteomic and genomic analyses of the human breast cancer cell line T47D and the antiestrogen-resistant derivative T47D-r. Mol Cell Proteomics. 2004;3(1):43–55. doi: 10.1074/mcp.M300047-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Lu Y, Muller M, Smith D, Dutta B, Komurov K, Iadevaia S, Ruths D, Tseng JT, Yu S, Yu Q, et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene. 2011;30(45):4567–4577. doi: 10.1038/onc.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iadevaia S, Lu Y, Morales FC, Mills GB, Ram PT. Identification of optimal drug combinations targeting cellular networks: integrating phospho-proteomics and computational network analysis. Cancer Res. 2010;70(17):6704–6714. doi: 10.1158/0008-5472.CAN-10-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang KX, Ouellette BF. CAERUS: predicting CAncER oUtcomeS using relationship between protein structural information, protein networks, gene expression data, and mutation data. PLoS Comput Biol. 2011;7(3):e1001114. doi: 10.1371/journal.pcbi.1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C, Huang H, Wu CH. Protein bioinformatics databases and resources. Methods Mol Biol. 2011;694:3–24. doi: 10.1007/978-1-60761-977-2_1. [DOI] [PubMed] [Google Scholar]
- 45.Ponten F, Schwenk JM, Asplund A, Edqvist PH. The Human Protein Atlas as a proteomic resource for biomarker discovery. Journal of internal medicine. 2011;270(5):428–446. doi: 10.1111/j.1365-2796.2011.02427.x. [DOI] [PubMed] [Google Scholar]
- 46.The Quantitative Assay Datebase (QuAD) [ http://proteome.moffitt.org/QUAD]
- 47.a database of Differentially Expressed Proteins in Human Cancer. [ http://lifecenter.sgst.cn/dbdepc/index.do]
- 48.He Y, Zhang M, Ju Y, Yu Z, Lv D, Sun H, Yuan W, He F, Zhang J, Li H, et al. dbDEPC 2.0: updated database of differentially expressed proteins in human cancers. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arntzen MO, Thiede B. ApoptoProteomics: An integrated database for analysis of proteomics data obtained from apoptotic cells. Mol Cell Proteomics. 2011 doi: 10.1074/mcp.M111.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.PED. [ http://www.pancreasexpression.org]
- 51.MolDiag-PaCa. [ http://www.moldiagpaca.eu]
- 52.Cutts RJ, Gadaleta E, Hahn SA, Crnogorac-Jurcevic T, Lemoine NR, Chelala C. The Pancreatic Expression database: 2011 update. Nucleic Acids Res. 2011;39(Database issue):D1023–1028. doi: 10.1093/nar/gkq937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gostev M, Faulconbridge A, Brandizi M, Fernandez-Banet J, Sarkans U, Brazma A, Parkinson H. The BioSample Database (BioSD) at the European Bioinformatics Institute. Nucleic Acids Res. 2012;40(1):D64–70. doi: 10.1093/nar/gkr937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Menon R, Omenn GS. Identification of alternatively spliced transcripts using a proteomic informatics approach. Methods Mol Biol. 2011;696:319–326. doi: 10.1007/978-1-60761-987-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Day RS, McDade KK, Chandran UR, Lisovich A, Conrads TP, Hood BL, Kolli VS, Kirchner D, Litzi T, Maxwell GL. Identifier mapping performance for integrating transcriptomics and proteomics experimental results. BMC Bioinformatics. 2011;12:213. doi: 10.1186/1471-2105-12-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jorgensen KM, Hjelle SM, Oye OK, Puntervoll P, Reikvam H, Skavland J, Anderssen E, Bruserud O, Gjertsen BT. Untangling the intracellular signalling network in cancer--a strategy for data integration in acute myeloid leukaemia. J Proteomics. 2011;74(3):269–281. doi: 10.1016/j.jprot.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 57.Huopaniemi I, Suvitaival T, Nikkila J, Oresic M, Kaski S. Multivariate multi-way analysis of multi- source data. Bioinformatics. 2010;26(12):i391–398. doi: 10.1093/bioinformatics/btq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eklund M, Spjuth O, Wikberg JE. An eScience-Bayes strategy for analyzing omics data. BMC Bioinformatics. 2010;11:282. doi: 10.1186/1471-2105-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mayer CD, Lorent J, Horgan GW. Exploratory analysis of multiple omics datasets using the adjusted RV coefficient. Stat Appl Genet Mol Biol. 2011;10(1) doi: 10.2202/1544-6115.1540. Article 14. [DOI] [PubMed] [Google Scholar]
- 60.Van Deun K, Wilderjans TF, van den Berg RA, Antoniadis A, Van Mechelen I. A flexible framework for sparse simultaneous component based data integration. BMC Bioinformatics. 2011;12(1):448. doi: 10.1186/1471-2105-12-448. [DOI] [PMC free article] [PubMed] [Google Scholar]