

Abstract
Down Syndrome (DS) is the most common genetic cause of mental retardation, with a reported frequency of epilepsy between 1.4-17% (1). There is a paucity of data in the literature regarding epilepsy in Down syndrome and its relation to intellectual disability.
Objectives:
The purpose of this article is to analyze the association of epilepsy in children with DS - frequency and type of seizures, treatment, outcome and to compare cognitive impairment of children with DS and epilepsy and DS without epilepsy from our cohort.
Methods:
A four years systematic retrospective analysis of the database of the Pediatric Neurology Clinic (January 2010 - December 2013) identified a cohort of 39 pediatric cases with DS and neurological symptoms, 9 of them (23%) associating epileptic seizures. Following data were analysed: clinical and neurological examination, type/s of seizures, electroencephalography (EEG), cerebral magnetic resonance imaging (MRI), psychological examination, psychiatric evaluation in selected cases, electrocardiography (ECG), cardiac ultrasonography, ophthalmologic examination.
Results:
23% (9 patients) of the children with DS of our cohort presented epilepsy. Five patients had epileptic spasms (56%), one of these further developed astatic seizures. Focal seizures were observed in three patients (33%) and absence with eyelid myoclonias in one patient (11%). Two of the nine patients with DS and epilepsy had generalized seizures, both with very good response to levetiracetam (LEV). EEG was abnormal at seizure onset, and was improved after treatment. Of the nine children with DS and epilepsy, two (22%) presented mild mental retardation and seven (78%) had moderate to severe cognitive delay. Of the 30 children with DS and without epilepsy, 21 (70%) had mild mental retardation and 9 (30%) had moderate to severe cognitive impairment.
Conclusions:
The most frequent epileptic syndrome associated with DS is West syndrome, with good response to specific antiepileptics. All children with DS from our cohort have intelectual disability, more severe in those with epilepsy. Slight improvement of intelectual and language capabilities were seen after seizures control.
INTRODUCTION
Down Syndrome (1/1000 newborns) is the most common genetic condition, characterized by a supplementary 21 chromosome (Figure 1A), or a part of it. 95% of the cases are related with mother's age (the age over 35 being a major risk factor), in 3-4% cases - a Robertsonian translocation from chromosome 14 being present and in 1-2% of the cases the mosaicism being the underlying mechanism (1).
Figure 1. Down syndrome: A. karyotype - trisomy 21; B. Facial typical features: flat facies, straight hair, upslanting palpebral fissures, epicanthal folds, small nose, low nasal bridge with upturned nares.
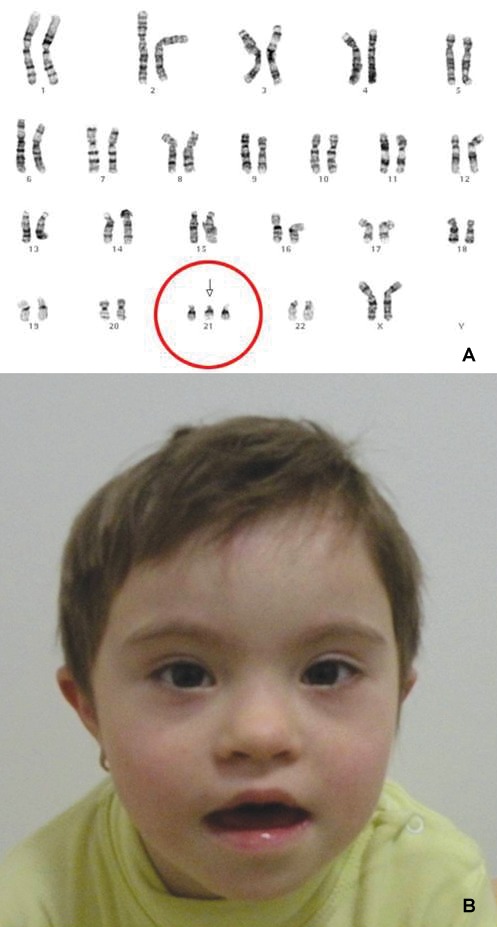
The classical phenotype of DS is defined by well-known constitutional features (Figure 1B), associated with systemic involvement that may impair the gastrointestinal, cardiac, ophthalmologic, endocrine, immune, orthopedic, or neurologic systems.
The most common neurological findings are global developmental delay – with better outcome on motor skills, despite severe hypotonia in the first year of life, but with persistence of cognitive impairment – from mild to severe mental retardation, and also seizures, spinal ligaments laxity, acute/subacute motor deficits due to moyamoya, stroke, other neurological associated conditions.
The incidence of epilepsy in persons with DS is 1.4-17% and varies with age, globally higher comparing to the general population (~1%) (1-7), being one of the main chromosomal abnormalities that associates seizures, along with the monosomy 1p36, Wolf-Hirschhorn, Angelman, Miller-Dieker, inversion-duplication 15, ring 20 syndromes. In all these disorders affecting the autosomes, already associating intellectual disability, the epilepsy is an aggravating factor that could add more burden on cognitive impairment.
There are only a few studies that try to delineate better the epileptic phenotypes in DS. The distribution of epilepsy seems to be bimodal, with a peak in infancy and young age (epileptic spasms in West syndrome, focal epilepsies, reflex epilepsies, Lennox-Gastaut syndrome) and another peak at older age (adults), when myoclonic epilepsy with late onset and dementia is described (3-7).
Epileptic spasms have onset in the first year of life and, if early diagnosed and treated promptly, have good response to treatment and decreased negative impact on children long-term prognosis. Delayed diagnosis and treatment of infantile spasms are associated with important neurological impairment, especially on cognitive level.
Even if progression from West to Lennox-Gastaut syndrome is less common in DS, there are cases with delayed onset of the latter mentioned syndrome, characterized by polymorphic seizures – with prominent tonic seizures and severe neurological disability.
Myoclonic epilepsy with late onset is a frequent form in adults, with good response to levetiracetam therapy, altought the patients have Alzheimer-like demention. ❑
OBJECTIVE
The aim of this article is to: (1) analyze epilepsy epidemiology in patients with DS evaluated in the Pediatric Neurology Department between 2010-2013 – frequency and type of seizures, therapeutic options and response to treatment; (2) to compare the cognitive impairment in patients with DS and epilepsy and patients with DS without epilepsy. ❑
METHODS
The archive of Pediatric Neurology Clinic was retrospectively reviewed over a four years period (January 2010 – December 2013) and patients with DS were selected. The study was approved by the Local Ethics Committee. The clinical status was registered at different times: at first admission in the Clinic and at follow-up evaluations. Besides neurological examinations, other evaluations were performed during hospitalizations: awake EEG, sleep EEG (if necessary), cerebral MRI, psychological evaluation, psychiatric examination in selected cases, ECG, cardiac ultrasonography, ophthalmologic examination. In patients with DS and epilepsy we analyzed: gender, age of onset, type of seizures, aspect of EEG and cerebral MRI, electroclinical syndrome, antiepileptic treatment used and its efficacy, cognitive impairment. For statistical analysis we used SPSS17 programme. ❑
RESULTS
39 patients with DS were evaluated in our clinic between January 2010 - December 2013. Of this cohort, nine children that associated epileptic seizures were selected and analyzed (23%).
Onset and gender. Of these nine patients six were girls (67%) and three were boys (33%). All of them were admitted in our clinic for paroxysmal events that were classified as seizures. Five of them had seizures onset before the age of one year (56%) - two boys and three girls; four patients had seizures onset between three and six years (44%) – one boy and three girls.
Clinical aspect of seizures. All the five (56%) patients with seizures onset before one year (two boys and three girls) had epileptic spasms; one of them developed later astatic seizures. From the group of four with seizures onset between three and six years - one (11%) girl presented atypical absences with myoclonus after the age of six (one febrile seizure previously, at age of one), three (33%) patients (two girls and one boy) had focal seizures; one of the girls presented focal motor seizures only for a short period of time, associated with a right motor deficit, being the clinical expression of an acute left middle cerebral artery stroke.
EEG aspect. The five patients with epileptic spasms presented hypsarrhythmia at onset (Figure 2), four of them with normal EEG after treatment was initiated and one with persistent hypsarrhythmia and seizures. Of the four patients with controlled epileptic spasms, in one girl seizures reoccured at age of one year nine months - astatic seizures with slow background and bilateral spike-waves discharges, mainly located in fronto-temporal areas, later on then EEG normalizing under treatment. One girl presented at 6-year-old age atypical absences with myoclonias and the EEG showed spike-waves, polyspike-waves discharges, bilateral, hyperventilation induced, associated with episodes of unresponsiveness+/-chewing automatisms (Figure 3), then EEG normalized under antiepileptic therapy. In the group of three patients with focal seizures the EEG showed focal discharges - spike-waves type, multifocal, mainly in the contralateral hemisphere.
Figure 2. EEG interictal aspect in a child with epileptic spasms and DS (hypsarrhythmia).
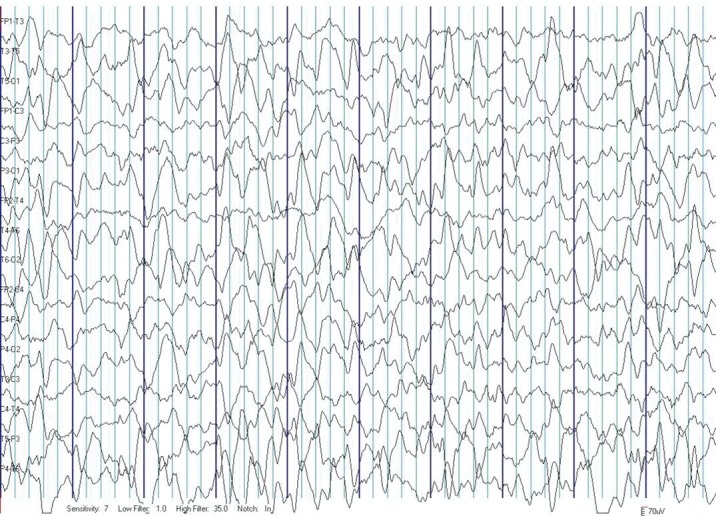
Figure 3. EEG trace - background with theta rhythm, modulated in spindles, with an average frequency of 7 Hz, an average amplitude of 100 microvolts, intricated with degraded sharp-waves discharges in bilateral frontal derivations, larger in left derivations (A); registration of ictal rhythms - bilateral epileptiform discharges with degraded sharp-waves, slow onset and ending, apparently larger in left leads, with greater frequency of discharges at onset and slower at the end, with an average frequency of 8 seconds, with impaired consciousness as clinical correspondent, without any motor signs - 3 episodes (B, C, D) - seizures - atypical absences - in the patient with DS and atypical absences with myoclonias, with onset of the age of 6 years.

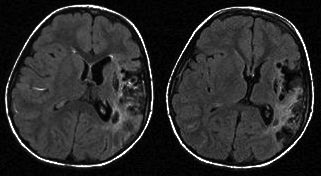
Cerebral MRI All nine patients had cerebral MRI performed. The girl with infantile spasms and astatic seizures presented an old lacuna in the right internal capsulae, probably unrelated with seizures onset or semiology (Figure 4). The girl with acute right motor deficit and focal seizures presented in fact a left medial cerebral artery (MCA) stroke (Figure 5). The other seven patients had normal cerebral MRI.
Figure 4. Cerebral MRI - old lacuna in the right internal capsulae.
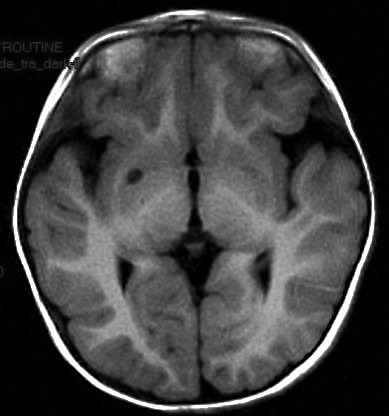
Figure 5. Cerebral MRI - aspect of left medial cerebral artery (MCA) stroke.
Treatment & outcome. Four of the patients with WS had favorable outcome of seizures under corticotherapy with synthetic adrenocorticotropic hormone (ACTH) for six weeks, alone or with topiramate (TPM) until the age of two (two patients received TPM), or vigabatrine (VGB) for six months (one patient), with seizures control and normal EEG. One child from this group of four patients (a girl) developed astatic seizures at one years nine months, controlled with levetiracetam (LEV), with normal EEG after seizures control. One of the patients with IS had persistent seizures and hypsarrhythmia under treatment with topiramate and vigabatrine and died because of a respiratory intercurrence (we could tried only for couple of days ACTH because of the frequent respiratory problems and extreme hypotonia). The girl with atypical absences with myoclonias was treated with LEV with seizures control and normalization of EEG trace. Of the three children with focal seizures - two had partial seizures control under epileptic drugs (rare seizures, improvement of EEG aspect), while the third one (the girl with acute left MCA stroke) presented focal seizures only in the acute period, now being seizure-free and without antiepileptic medication.
Psychomotor development and cognitive impairment. Of the five patients with IS, one died because of acute respiratory insufficiency during a respiratory intercurrence and he was very hypotonic, with profound developmental delay; two of the patients presented severe mental retardation (one of them is the girl with astatic seizures) and two had moderate mental retardation. Four patients with IS presented apparently normal milestones until seizures onset, then psychomotor regression, with slight cognitive improvement after seizures control and good motor recovery. The girl with atypical absences with myoclonias had mild global developmental delay, with good recovery on motor side, but only slight improvement on cognitive level after seizures stopped. All of the three patients with focal seizures had mild global developmental delay, but after seizures onset two of them had moderate cognitive impairment and normal motor functioning; one of them - the girl with stroke - had mild cognitive impairment and right spastic hemiparesis.
Of the nine children with DS and epilepsy - one (11%) had profound mental retardation, three (33%) had severe mental retardation (quotient of inteligence (QI) <35), three (33%) had moderate cognitive impairment (QI=35-49) and two (22%) had mild cognitive delay (QI=50-70). Of the other 30 children of the cohort - DS withouth epilepsy, two (6,5%) had profound mental retardation, two (6.5%) had severe cognitive impairment, five (17%) moderate cognitive delay and 21(70%) presented mild intellectual disability.
Associated disorders. Six of the patients' mothers were over 35 years old during pregnancy, one of them was exactly 35 years old and two were younger than 35. Five of the mothers older than 35 years and one under this age had one or two miscarriages before the child with DS. The other mother, whose age was above 35, suffered from diabetes mellitus type 1.
From associated cardiac malformation point of view, only four of the children presented cardiac involvement - the one who died had atrial and ventricular septal defects, other two had small atrial septal defect (one with IS, one with focal seizures, no hemodynamic involvement) and the girl with IS and astatic seizures had small aortic insufficiency and hypothyroidism. ❑
DISCUSSIONS
Nine children (23%) from the cohort of 39 patients with DS presented epilepsy, more than mentioned in specialized literature (~13-17% maximum) (1-7), situation that could be explained by the fact that the reported cases addressed to a tertiary center and all children had DS with neurological features, especially epileptic seizures.
Six mothers were over 35 years (67%), of which five (56%) had one or two miscarriages before the birth of the child with DS - two of the major risk factors for DS.
In our cohort there were six girls (67%) and three boys (33%), although in some studies, boys and girls were equally affected or boys were predominant when DS associated IS with early onset (7,8).
Five (56%) of the nine patients had IS, four of them (44%) with remission of epileptic seizures and EEG normalization after ACTH and TPM/ VGB, comparable to the literature where the same medication was used, the epileptic spasms being reported as particularly sensitive to treatment comparing to those of different etiologies (5-7,9-12).
Two patients (22%) had generalized seizures – one girl with astatic seizures and one girl with atypical absences and myoclonias, with spectacular response to LEV – from the first doses seizures stopped and the EEG normalized. This aspect was not described in the medical literature and there isn't a specific antiepileptic drug for seizures treatment in patients with DS, but, from our clinical experience LEV is a very good therapeutical option in pediatric patients with DS and generalized seizures.
There are no pathognomonic abnormalities on the EEG in patients with DS, the electroencephalographic changes being in accordance with the electroclinical syndrome each child presented.
Seven (78%) patients had normal cerebral MRI and two (22%) showed ischemic lesions on the MRI, one of them associated with acute right motor deficit and one asymptomatic.
Most of the children with DS without epilepsy (70%) had mild mental retardation and only 30% had moderate to severe cognitive disability.
Only 22% of the patients with DS and epilepsy had mild mental retardation, the other 78% had moderate to severe cognitive impairment. The children with DS that associate epilepsy seem to have more significant cognitive impairment (6,9,13).
After the seizures control, all the patients had slight/mild improvement in the cognitive/ laguage area, even if the degree of mental retardation was the same, although it is very difficult to assess corectly neurodevelopment in this population due to the associated comorbidities and to the lack of specific scales It appears to be a significant association between early recognition of IS and early initiation of therapy, the delay between the diagnostic and therapy influencing directly the negative impact on neurodevelopment and autistic behavior of DS patients (7,9).
A bias in the results interpretation may be due to the heterogenicity of the two groups including different proportion of children of different ages and also to the low and different number of analysed subjects. Further studies including higher number of children with DS+/- epilepsy, and homogenous groups are needed. Prospective studies including formal cognition testing before starting antiepileptic medication and after seizures control are to be developed, to check if early diagnosis and treatment of epileptic seizures, especially IS, in DS, will improve cognitive outcome, as some of the studies in the literature suggested (5-7,9, 14-16).
Only four (44%) children with DS and epilepsy associated cardiac malformation (three with no hemodinamic influence and one with atrial and ventricular septal defect), one (11%) of them presenting also hypothyroidism which might have additionally negative impact on cognition. ❑
CONCLUSIONS
In our cohort, 23% of DS cases had epilepsy, more than cited in the literature (between 1.4-17%, according to the different studies. Most patients with DS had epileptic spasms, with a good response to ACTH associated or not with TPM or VGB. The two patients with generalized seizures had spectacular response to LEV and therefore we advice the use of LEV as a first line of treatment in DS patients associating generalized seizures; this medication being proved very effective in DS patients with myoclonic epilepsy with late onset in the literature studies.
All 39 patients had mental retardation of different degrees of severity. The patients with DS and epilepsy had a slight improvement of the cognitive and language abilities after achieving seizures control.
We strongly recommend early diagnosis and treatment of epilepsy in children with DS for a better outcome, this requiring a better knowledge of the association of the two disorders.
The epilepsy in patients with DS, as epilepsies in other chromosomal abnormalities syndromes, requires a systematic semiological delineation, an early recognition, an adequate treatment adaptated to the global management plan. A better characterization of epileptic phenotypes will be a valuable aid in the progress of understanding the molecular mechanisms that lie beneath these syndromes, thus hopefully better treatments could become available, with improved quality of life as result.
CONFLICT OF INTEREST
none declared.
FINANCIAL SUPPORT
none declared.
References
- 1.Bull MJ and the Committee on Genetics. Clinical report - Health supervision for children with Down syndrome. Pediatrics. 2011;128:393–406. doi: 10.1542/peds.2011-1605. [DOI] [PubMed] [Google Scholar]
- 2.Thiel RJ, Fowkes SW. Down syndrome and epilepsy: a nutritional connection? Med Hypotheses. 2004;62:35–44. doi: 10.1016/s0306-9877(03)00294-9. [DOI] [PubMed] [Google Scholar]
- 3.Sangani M, Shahid A, Amina S, et al. Improvement of myoclonic epilepsy in Down syndrome treated with levetiracetam. Epileptic Disord. 2010;12:151–4. doi: 10.1684/epd.2010.0306. [DOI] [PubMed] [Google Scholar]
- 4.Ferlazzo E, Adjien CK, Guerrini R, et al. Lennox-Gastaut syndrome with late- onset and prominent reflex seizures in trisomy 21 patients. Epilepsia. 2009;50:1587–95. doi: 10.1111/j.1528-1167.2008.01944.x. [DOI] [PubMed] [Google Scholar]
- 5.Hamouda HB, Mnasri H, Ghanmi S, et al. West syndrome associated with Down syndrome: case report and literature review. Pediatria Polska. 2014;89:203–6. [Google Scholar]
- 6.Ulate-Campos A, Nascimento A, Ortez C. Down's syndrome and epilepsy. Rev Med Int Sindr Down. 2014;18:3–8. [Google Scholar]
- 7.Verotti A, Cusmai R, Nicita F, et al. Electroclinical features and long-term outcome of cryptogenic epilepsy in children with Down syndrome. J Pediatr. 2013;163:1754–8. doi: 10.1016/j.jpeds.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg-Stern H, Strawsburg RH, Patterson B, et al. Seizure frequency and characteristics in children with Down syndrome. Brain Dev. 2001;23:375–8. doi: 10.1016/s0387-7604(01)00239-x. [DOI] [PubMed] [Google Scholar]
- 9.Arya R, Kabra M, Gulati S. Epilepsy in children with Down syndrome. Epileptic Disord. 2011;13:1–7. doi: 10.1684/epd.2011.0415. [DOI] [PubMed] [Google Scholar]
- 10.Cortez MA, Shen L, Wu Y, et al. Infantile spasms and Down Syndrome: a new animal model. Pediatr Res. 2009;65:499–503. doi: 10.1203/PDR.0b013e31819d9076. [DOI] [PubMed] [Google Scholar]
- 11.Nabbout R, Melki I, Gerbaka B, et al. Infantile Spasms in Down Syndrome: good response to a short course of vigabatrin. Epilepsia. 2001;42:1580–3. doi: 10.1046/j.1528-1157.2001.13501.x. [DOI] [PubMed] [Google Scholar]
- 12.Scorza CA, Scorza FA, Arida RM, et al. Sudden unexpected death in people with Down syndrome and epilepsy: another piece in this complicated puzzle. Clinics (Sao Paulo) 2011;66:719–20. doi: 10.1590/S1807-59322011000500001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vignoli A, Zambrelli E, Chiesa V, et al. Epilepsy in adult patients with Down syndrome: a clinical-video EEG study. Epileptic Disord. 2011;13:125–32. doi: 10.1684/epd.2011.0426. [DOI] [PubMed] [Google Scholar]
- 14.Chaanine A, Hugonenq C, Lena G, et al. Les complications neurologiques liées à la trisomie 21. Arch Pediatr. 2008;15:388–96. doi: 10.1016/j.arcped.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Smigielska-Kuzia J, Sobaniec W, Kuak W, et al. Clinical and EEG features of epilepsy in children and adolescents in Down syndrome. J Child Neurol. 2009;24:416–20. doi: 10.1177/0883073808324542. [DOI] [PubMed] [Google Scholar]
- 16.Bahi-Buisson N, Ville D, Eisermann M, et al. L'épilepsie dans les aberrations chromosomiques. Arch Pediatr. 2005;12:449–58. doi: 10.1016/j.arcped.2004.12.016. [DOI] [PubMed] [Google Scholar]