

Abstract
Background
Gene amplification is a frequent manifestation of genomic instability that plays a role in tumour progression and development of drug resistance. It is manifested cytogenetically as extrachromosomal double minutes (DMs) or intrachromosomal homogeneously staining regions (HSRs). To better understand the molecular mechanism by which HSRs and DMs are formed and how they relate to the development of methotrexate (MTX) resistance, we used two model systems of MTX-resistant HT-29 colon cancer cell lines harbouring amplified DHFR primarily in (i) HSRs and (ii) DMs.
Results
In DM-containing cells, we found increased expression of non-homologous end joining (NHEJ) proteins. Depletion or inhibition of DNA-PKcs, a key NHEJ protein, caused decreased DHFR amplification, disappearance of DMs, increased formation of micronuclei or nuclear buds, which correlated with the elimination of DHFR, and increased sensitivity to MTX. These findings indicate for the first time that NHEJ plays a specific role in DM formation, and that increased MTX sensitivity of DM-containing cells depleted of DNA-PKcs results from DHFR elimination. Conversely, in HSR-containing cells, we found no significant change in the expression of NHEJ proteins. Depletion of DNA-PKcs had no effect on DHFR amplification and resulted in only a modest increase in sensitivity to MTX. Interestingly, both DM-containing and HSR-containing cells exhibited decreased proliferation upon DNA-PKcs depletion.
Conclusions
We demonstrate a novel specific role for NHEJ in the formation of DMs, but not HSRs, in MTX-resistant cells, and that NHEJ may be targeted for the treatment of MTX-resistant colon cancer.
Keywords: Cancer: colon, Genetics, Molecular genetics, Oncology
Introduction
The amplification of oncogenes or drug resistance genes plays a pivotal role in malignant transformation. Cytogenetic studies have identified two major topographical structures of amplified DNA segments: extrachromosomal DNA (double minutes, DMs) and intrachromosomal DNA (homogeneously staining regions, HSRs).1 2 However, the underlying molecular mechanism for their formation remains unclear. A large body of evidence points to free double-strand DNA breaks (DSBs) as key intermediates in the process that leads to gene amplification. The Saccharomyces cerevisiae I-SceI endonuclease system and DSB-inducing agents have been proven to be crucial in providing support for the role of DSBs in initiating gene amplification.3 4 In addition, increased frequency of gene amplification in Chinese hamster cells treated with γ-rays, hypoxia or clastogenic drugs supports a correlation between DSBs and gene amplification.5 6
Non-homologous end joining (NHEJ), one of the major DSB repair mechanisms, can restore the original sequence at the break or generate chromosomal aberrations7 by ligation of the DNA ends. This process often results in the loss of nucleotides, rendering NHEJ prone to errors.8 The key proteins involved in NHEJ include DNA-PKcs, KU70 and KU86, among which DNA-PKcs has been shown to be the central player. NHEJ-deficient cells are characterised by increased sensitivity to DNA-damaging agents, chromosomal instability, gene amplification and predisposition to cancer.6 9 10 Previous reports have also shown that cells lacking DNA-PKcs are radiosensitive and defective in their ability to repair DSBs.11–13 Conversely, increased level of DNA-PKcs was observed in adriamycin-resistant cells.14 Adriamycin-resistant cells are known to exhibit MDR1 amplification, raising the possibility that the highly expressed DNA-PKcs may contribute to gene amplification in drug-resistant cells. There is evidence that NHEJ is involved in junction formation between amplicon microhomologies during gene amplification.15 16 However, the role of NHEJ in the formation of DMs and HSRs relative to drug resistance in cancer cells remains to be investigated.
Gene dosage depends on factors that regulate both gene amplification and gene elimination. Micronuclei (MNs) are derived from chromosomal fragments or whole chromosomes that lag behind during anaphase and nuclear division.17 Nuclear buds (NBUDs) are characterised by the same morphology as MNs, with the exception that they are connected to the nucleus by a stalk of nucleoplasmic material. Previous studies have shown that MNs can be formed via a budding process following exposure to γ-irradiation.18 On the other hand, amplified DNA can be eliminated by DNA synthesis inhibitors such as hydroxyurea.19
In this study, we used methotrexate (MTX)-resistant HT-29 human colon cancer cells to study the mechanism involved in the formation of DMs and HSRs relative to MTX resistance. We show evidence that NHEJ is differentially involved in the formation of DMs and HSRs and in the resistance of cancer to MTX.
Methods
Cell lines and cell culture
HT-29 colon cancer cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and were authenticated by the Beijing Microread Genetics (Beijing, China) using short tandem repeat analysis in 2011. DM-containing and HSR-containing cells were generated by continuous culture of parental HT-29 cells in dulbecco's modified eagle medium DMEM containing high glucose (Gibco BRL, Gaithersburg, Maryland, USA) and supplemented with MTX (Calbiochem Biochemicals, Darmstadt, Germany). All cell lines were maintained in the presence of 15% fetal calf serum (Gibco BRL). The DNA-PK inhibitor, NU7026 (Sigma-Aldrich Co. LLC, Missouri, USA), was added to the medium at a final concentration of 10 μM for 5 days.
Antibodies
The antibodies used and their sources are as follows: DHFR mouse monoclonal antibody was from Abnova, Taipei, Taiwan; KU86 goat polyclonal and DNA-PKcs rabbit polyclonal antibodies were from Santa Cruz Biotechnology Inc., Texas, USA; KU70 mouse monoclonal antibody was from Abcam, Cambridge, UK; phospho-Ser139 H2AX mouse monoclonal antibody was from Millipore, Massachusetts, USA; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mouse monoclonal antibody was from Kang Chen Bio-tech, Shanghai, China and the CF488 goat anti-mouse IgG (H1L) was from Biotium, California, USA.
Stable shRNA transfection
The shRNA sequences were inserted into the plasmid vector pSUPER.retro.puro (Oligoengine, Washington, USA) to construct the recombinant plasmid. The shRNA sequences for DNA-PKcs were as follows: shDNA-PKcs-SiR+: 5′-GATCCCCAAACTACCTGTTCTGGCAGGATTCAAGAGATCCTGCCAGAACAGGTAGTTTTTTTA-3′; shDNA-PKcs-SiR–: 5′-AGCTTAAAAAAAACTACCTGTTCTGGCAGGATCTCTTGAATCCTGCCAGAACAGGTATTTGGG-3′. MTX-resistant HT-29 cells were transfected with recombinant plasmid pSUPER-DNA-PKcs and control vector pSUPER-control using Lipofectamine 2000 (Invitrogen, California, USA), according to the manufacturer's protocol. Cells were plated at a density of 3×105 cells/well in 6-well plates and grown until 80% confluence. Cells were then transfected with 4 μg of plasmid per well. Puromycin was added to medium to a final concentration 0.5 μg/mL (HSR-containing cells) or 0.3 μg/mL (DM-containing cells) at 24 h post-transfection to select stable clones.
Real-time PCR
Genomic DNA was extracted using QIAmp DNA Mini Kit (Qiagen, Dusseldorf, Germany), following manufacturer's protocol. Real-time PCR was performed using the Light Cycler 480 SYBR Green Kit (Roche Applied Science, Mannheim, Germany). The DNA primers were as follows: DHFR: 5′-ATTTTGTTCAGTGCCTACCACA-3′ and 5′-GCCTGAATGATATCTACAAGCTG-3′, RAD1: 5′-TGTCAGTTGCGTGTCTTCAT-3′ and 5′-AGACAGTAAAACTCCCATCA-3′, PLK2: 5′-ACCCTATGGGACTCCTCTTT-3′ and 5′-GTATGCCTTAGCCTGTTCTG-3′. ZFYVE16: 5′-AGGAAGCAACCACCACAAC-3′ and 5′-CAGCACCACCAACAGATACA-3′, MSH3: 5′-TGTCTGGTGTTTCGCCTGAT-3′ and 5′- TTAGCCAATAACCGCTCTAC-3′, CCNH: 5′-GTATTGCAGCACTGATTATGTCC-3′ and 5′-TCATGAAAATAGCCATAGGTGA-3′, GLRX: 5′-CCCACATTGTAGGGAATCAT-3′ and 5′-CCCACAGTCTATTCGTAGCA-3′, CAST: 5′-TTGACTCCATAGCCAACCTT-3′ and 5′-GTCACTTTTCCCAGAATCCG-3′, ACTN: 5′-ACCGCGAGAAGATGACCCAG-3′ and 5′-TTAATGTCACGCACGATTTAAA-3′.
Western blot analysis
Cells were homogenised in lysis buffer supplemented with protease inhibitors and PhosSTOP phosphatase inhibitor (Roche, Basel, Switzerland). Samples were centrifuged at 14 000 rpm for 40 min at 4°C. The supernatants were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore) and subjected to immunoblotting. GAPDH was used for normalisation.
Immunofluorescence
Cells on coverslips were cultured in serum-free medium for 24 h to achieve cell-cycle synchronisation, and then were washed with phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde for 10 min. Fixed cells were treated with blocking buffer (2.5% bovine serum albumin, 0.2 M glycine, 0.1% Triton X-100) at 37°C for 30 min, followed by incubation with phosphoSer139-H2AX antibody overnight at 4°C. Slides were then washed with PBS and incubated with the CF488 goat anti-mouse IgG secondary antibody at 37°C for 30 min. Following incubation, slides were washed with PBS, and chromosomes were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). Images were obtained using a Leica DM5000B microscope (Leica Microsystems, Solms, Germany).
Fluorescence In Situ Hybridization
Metaphase spreads from Colcemid (Sigma-Aldrich Co. LLC)-arrested cells were prepared according to standard cytogenetic methods. The BAC clones, PR11-90A9, PR11-27F9, PR11-42H4 and PR11-957J15 (BAC PAC Resources Center, California, USA), were extracted using Genopure Plasmid Midi Kit (Roche) according to the manufacturer's protocol and labelled with Cy3-dUTP, Cy5-dUTP or Green-dUTP using a BioPrime DNA Labelling System Kit (Invitrogen). The template of whole chromosome 5 painting probe, kindly provided by Dr Hong Chen (Institute of Basical Medical Sciences, Peking Union Medical College), was labelled with fluorescein isothiocyanate-deoxyuridine triphosphate (FITC-dUTP). The slides with interphase or metaphase spreads were treated with RNase for 40 min at 37°C and then washed in 2× saline sodium citrate (SSC) for 3 min. After dehydration through an ethanol series, the slides were incubated with pepsin for 15 min at 37°C and washed in 1× PBS for 5 min. The cells were fixed in 1% paraformaldehyde for 10 min, then washed in 1× PBS for 5 min. Following dehydration, the slides were treated in 70% formamide for 3 min at 75°C and subsequently washed twice in prechilled 2× SSC for 3 min. After dehydration, the cells were hybridised with labelled probes, mounted with rubber cement and incubated overnight at 37°C. The slides were then immersed in 44°C prewarmed 50% formamide for 15 min and subsequently washed twice in 2× SSC for 3 min. Following dehydration, the slides were counterstained with DAPI and mounted. The images were obtained using a fluorescent microscope equipped with the MetaMorph Imaging System (Universal Imaging Corporation).
Proliferation assay
Cells were seeded into 96-well plates at a density of 800 cells/well. CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Wisconsin, USA) was used to measure the cell viability during a 5-day time course according to the manufacturer's protocol. The absorbance at 490 nm was measured daily using a microplate reader (Tecan Austria GmbH, Grödig Austria). Three independent experiments (n=3) were performed.
Drug sensitivity assay
Cells were seeded into 96-well plates at a density of 4500 cells/well and incubated for 72–96 h in the presence of MTX. Cell viability was measured using CellTiter 96 AQueous One Solution Cell Proliferation Assay as described above. The absorbance at 490 nm was measured using a microplate reader (Tacan) to calculate IC50 values (n=3).
Statistical analysis
At least three independent experiments were performed for each variable. Analysis of variance (ANOVA) was used to analyse differences in mean variables in control and DNA-PKcs knockdown clones. Ridit test was used to assess differences in the γH2AX signals. χ2 analysis was used to assess differences in total MNs and NBUDs as well as signal-positive MNs and NBUDs formation rates. p<0.05 was considered statistically significant.
Results
Increased DSBs and levels of NHEJ-associated proteins are associated with the development of DM-containing MTX-resistant HT-29 cells
We have generated two HT-29 cell lines that are resistant to MTX, which selects cells that harbour amplified DHFR: HT-29/10−5 mol/L MTX, which exhibits amplified DHFR primarily in the form of HSRs, and HT-29/10−4 mol/L, which exhibits amplified DHFR primarily in the form of DMs. Generation of these lines has been reported previously.20–22 In this study, we initially sought to examine the existence of DSBs in these cells through analysis of γH2AX foci by immunocytochemistry. Cells were categorised into four different groups based on the number of γH2AX foci in the nucleus: no foci, 1–15 foci, 16–30 foci and >30 foci (figure 1A). Using the Ridit test, we found that γH2AX foci formation was significantly increased in both the HSR-containing cells (HT-29/10−5 mol/L MTX) (p<0.05) and the DM-containing cells (HT-29/10−4 mol/L MTX) (p<0.05) compared with parental cells (figure 1B). This indicates that MTX-induced formation of DMs and HSRs in HT-29 cells is associated with increased DSBs.
Figure 1.

Levels of double-strand DNA breaks and non-homologous end joining (NHEJ)-associated proteins in methotrexate (MTX)-resistant cancer cells. (A) IF assay for γH2AX foci in HT-29 parental cells, and homogeneously staining region (HSR)-containing and double minute (DM)-containing MTX-resistant cells (red, γH2AX; blue, 4′,6′-diamidino-2-phenylindole). Cells were grouped into four categories according to the number of γH2AX foci: no foci, 0–15 foci, 16–30 foci and >30 foci. (B) The proportion of cells in each γH2AXfoci category is shown. Ridit test was used to assess statistical significance (*p<0.05). (C) Representative immunoblots of NHEJ-associated proteins (KU70, KU86 and DNA-PKcs) in HT-29 parental, HSR-containing and DM-containing HT-29 cells. GAPDH was used as loading control. Right panel shows densitometric scanning of three independent blots from three separate experiments. Values are means±SD.
KU70, KU86 and DNA-PKcs are key proteins involved in NHEJ. Thus, we examined whether expression of these proteins is altered in DM-containing and HSR-containing HT-29 cells. By immunoblot analysis, we found that HSR-containing cells showed no noticeable change in levels of these proteins (figure 1C). However, the DM-containing cells showed significant increase in DNA-PKcs expression (figure 1C, right panel). KU70 and KU86 expression was also increased but did not reach significance. This suggests that the NHEJ contributes to the development of DM-containing but not HSR-containing MTX-resistant HT-29 cells.
Inhibition of NHEJ significantly decreases gene amplification in DM-containing MTX-resistant HT-29 cells
As described above, we found that increased DSBs as well as increased levels of the NHEJ-associated proteins, particularly DNA-PKcs, are associated with the development of DM-containing MTX-resistant HT-29 cells that exhibit DHFR amplification. We then tested whether inhibition of NHEJ results in decreased gene amplification. To do so, we depleted DNA-PKcs from DM-containing MTX-resistant cells by shRNA transfection and assessed gene amplification by real-time PCR and fluorescence in situ hybridization (FISH) analyses. As shown in figure 2A, we developed two clones of DM-containing MTX-resistant cells that were efficiently depleted of DNA-PKcs following stable transfection with DNA-PKcs shRNA (figure 2A). We then examined whether these cells have impaired NHEJ repair pathway through analysis of their γH2AX foci levels. We found that DNA-PKcs-depleted cells have significantly (p<0.05, Ridit test) increased levels of DSBs (figure 2B), indicating that depletion of DNA-PKcs caused impairment of the NHEJ repair pathway in these cells. To examine the effect of DNA-PKcs depletion on gene amplification, real-time PCR for DHFR was performed. As shown in figure 2C, there was a significant (p<0.001) decrease in DHFR amplification in DNA-PKcs-depleted cells compared with control cells. Consistent with these data, we found that the level of DHFR protein was also significantly decreased in cells depleted of DNA-PKcs (figure 2D). In addition, FISH analysis using metaphase spreads revealed a dramatic decrease in DHFR amplification in cells depleted of DNA-PKcs (figure 2E, red). Interestingly, DHFR amplification (red) in the DNA-PKcs knockdown cells was lost in DMs. Instead, DHFR amplification occurred in HSRs on chromosome 5 (figure 2E, 2nd and 3rd panels) as indicated by colocalisation of DHFR amplification with centromere staining (green) of chromosome 5. We also analysed at least 50 karyotypes from each of the control and DNA-PKcs-depleted cells and further found that HSRs became the major cytogenetic manifestation of the amplified DHFR in DNA-PKcs knockdown cells (figure 2F). These findings support the notion that DNA-PKcs is important in the development of DMs and suggest that NHEJ contributes to DHFR amplification through the formation of DMs.
Figure 2.

Inhibition of non-homologous end joining decreases gene amplification in double minute (DM)-containing methotrexate (MTX)-resistant cells. (A) Immunoblot and densitometric scanning of DNA-PKcs in DM-containing cells: control and two DNA-PKcs-depleted clones (shDNA-PKcs1 and shDNA-PKcs2). GAPDH was used as loading control. (B) The proportion of DM-containing cells (with and without DNA-PKcs depletion) in each γH2AX foci category is shown. Ridit test, *p<0.05. (C) Amplification of DHFR in DM-containing cells (with and without DNA-PKcs depletion) was examined by real-time PCR; ***p<0.001 by ANOVA. (D) Immunoblot for DHFR in DM-containing cells. GAPDH was used as a loading control. (E) FISH analysis of metaphase spreads of DM-containing cells using BACs containing human DHFR or centromere of chromosome 5 as probes (red, DHFR; green, centromere of chromosome 5; blue, 4′,6′-diamidino-2-phenylindole). (F) Quantification of the proportion of DM-containing cells in each category according to the cytogenetic manifestations of amplified DHFR is shown. (G) Real-time PCR analysis of RAD1, PLK2, ZFYVE16, MSH3, CCNH, GLRX and CAST amplification in DM-containing control and DNA-PKcs knockdown cells.
We then investigated whether inhibition of NHEJ by depleting DNA-PKcs generally reduces the incidence of cytogenetically manifested gene amplifications in MTX-resistant cells. By comparative genomic hybridization (CGH) array, we found that ZFYVE16, MSH3, CCNH, GLRX and CAST colocalised with DHFR within the same amplicon on chromosome 5 in HSR-containing cells but only ZFYVE16 and MSH3 showed similar colocalisation in DM-containing cells. RAD1 and PLK2 are not amplified on chromosome 5 during the development of MTX resistance, and thus, were used as negative controls (data not shown). Consistent with this finding, FISH analysis showed that ZFYVE16 and MSH3 colocalised with DHFR within the same DM in DM-containing cells (see online supplementary figure S1A). We then evaluated the DNA levels of ZFYVE16, MSH3, CCNH, GLRX and CAST, which are cytogenetically manifested gene amplifications in MTX-resistant cells. As with DHFR (figure 2C), levels of ZFYVE16 and MSH3 were decreased (p<0.001) in the DNA-PKcs knockdown cells as measured by real-time PCR (figure 2G). Conversely, levels of RAD1 and PLK2 as well as CCNH, GLRX and CAST were not affected. Thus, our results suggest that inhibition of NHEJ by knocking down DNA-PKcs reduces gene amplification in DMs of MTX-resistant cells.
Inhibition of NHEJ has no effect on gene amplification in HSR-containing MTX-resistant cells
There was no noticeable change in levels of NHEJ-associated proteins (KU70, KU86 and DNA-PKcs) in HSR-containing cells (figure 1C). Therefore, we speculated that NHEJ is not involved in gene amplification in these cells. To investigate this possibility, we used two clones of HSR-containing MTX-resistant cells that were efficiently depleted of DNA-PKcs (particularly clone 2) following stable transfection with DNA-PKcs shRNA (figure 3A). Analysis for γH2AX foci showed that DNA-PKcs-depleted cells (clone 2) had significantly (p<0.05 by the Ridit test) increased level of unrepaired DSBs compared with control (figure 3B). This indicates that depletion of DNA-PKcs also causes impairment of the NHEJ pathway in HSR-containing cells. However, real-time PCR analysis showed that depletion of DNA-PKcs does not affect DHFR amplification in HSR-containing cells (figure 3C). Immunoblot analysis further showed no difference in DHFR protein level between control and DNA-PKcs-depleted cells (figure 3D). As expected, the cytogenetic manifestation of DHFR amplification was HSRs on chromosome 5 in both control and DNA-PKcs-depleted cells (figure 3E). Therefore, DNA-PKcs depletion has no significant effect on DHFR amplification in HSR-containing cells.
Figure 3.

Inhibition of non-homologous end joining has no effect on gene amplification in homogeneously staining region (HSR)-containing methotrexate (MTX)-resistant cells. (A) Immunoblot analysis showing decreased DNA-PKcs level in DNA-PKcs-depleted clones. GAPDH was used as a loading control. (B) The proportion of HSR-containing cells (with and without DNA-PKcs depletion) in each γH2AX foci category is shown. Ridit test, *p<0.05. (C) Amplification of DHFR in HSR-containing cells (with and without DNA-PKcs depletion) was examined by real-time PCR; p<0.05 by ANOVA. (D) Immunoblot for DHFR in HSR-containing cells. GAPDH was used as a loading control. (E) FISH analysis of metaphase spreads of HSR-containing cells using BACs containing human DHFR or centromere of chromosome 5 as probes (red, DHFR; green, centromere of chromosome 5; blue, 4′,6′-diamidino-2-phenylindole). (F) Real-time PCR analysis of RAD1, PLK2, ZFYVE16, MSH3, CCNH, GLRX and CAST amplification in HSR-containing control and DNA-PKcs knockdown cells.
The DNA levels of RAD1, PLK2, ZFYVE16, MSH3, CCNH, GLRX and CAST were then assessed. We found that there were no significant alternations in the levels of these genes in DNA-PKcs knockdown cells compared with control (figure 3F). FISH analysis showed that, as in DMs, ZFYVE16 and MSH3 colocalised with DHFR within the same HSRs (see online supplementary figure S1B). Taken together, these further indicate that inhibition of NHEJ by depletion of DNA-PKcs does not affect gene amplification in HSR-containing cells.
Previously, breakage–fusion–bridge (B/F/B) cycles have been associated with low-copy gene amplification in human cancer cells.23 Consistent with this observation, we found that HSR-containing MTX-resistant cells exhibited mitotically unstable dicentric chromosomes (see online supplementary figure S2A) and nucleoplasmic bridges (see online supplementary figure S2B), which are typical structural features of B/F/B cycles. This suggests that B/F/B cycles may be associated with gene amplification during the formation of HSRs. However, since inhibition of NHEJ by DNA-PKcs depletion has no effect on gene amplification in HSR-containing cells, it is conceivable that NHEJ does not contribute to the end-to-end fusion of sister chromatids during B/F/B cycles.
Inhibition of NHEJ eliminates the amplified DHFR through MNs and NBUDs in DM-containing MTX-resistant cells
To determine whether the decrease in gene amplification resulting from inhibition of DNA-PKcs can be attributed to elimination via MNs and NBUDs, DM-containing cells were treated with NU7026, a specific inhibitor of DNA-PK, and harvested after 1, 3 and 5 days of treatment. FISH analysis of metaphase spreads using BAC containing the DHFR revealed that the DHFR copy number in the 3-day and 5-day treatments was significantly (p<0.05) reduced compared with dimethyl sulfoxide (DMSO)-treated control group (figure 4A). We then counted and classified at least 50 interphase nuclei by FISH analysis. The nuclei were categorised into nuclei with (i) no MNs or NBUDs, (ii) MNs or NBUDs with DHFR signal and (iii) MNs or NBUDs without DHFR signal (figure 4B). The percentages of nuclei belonging to each category are shown in figure 4C. Determination of the formation of MNs and NBUDs by x2 analysis showed a significant (p<0.05) increase in the transient 3-day and 5-day treatment groups compared with the dimethyl sulfoxide (DMSO)-treated control group. Furthermore, the number of MNs and NBUDs with DHFR was also increased compared with the control group. Since the formation of MNs and NBUDs corresponds to amplified DNA elimination, our data further suggest that inhibition of DNA-PKcs promotes the elimination of amplified DHFR via increases in MNs and NBUDs in DM-containing cells. Interestingly, we observed that stable depletion of DNA-PKcs resulted in a significant (p<0.01) decrease in the formation of MNs and NBUDs (see online supplementary figure S3).
Figure 4.

Inhibition of non-homologous end joining causes elimination of amplified DHFR in double minute (DM)-containing cells via micronuclei (MNs) and nuclear buds (NBUDs). (A) Quantification of DHFR amplification in the presence of the DNA-PKcs inhibitor, NU7026, for 1, 3 and 5 days. DMSO treatment was used as control. t test, *p<0.05. (B) FISH analyses of interphase nuclei using BACs containing human DHFR or centromere of chromosome 5 as probes. The MNs and NBUDs were grouped into two categories: with DHFR signal (top panel) and without DHFR signal (bottom panel). Red, DHFR; green, centromere of chromosome 5; blue, 4′,6′-diamidino-2-phenylindole. (C) The proportion of nuclei without or with MNs and NBUDs (with or without DHFR) in the presence of NU7026 for 1, 3 and 5 days are shown. DMSO treatment was used as control.
Inhibition of NHEJ decreases MTX resistance and proliferation in MTX-resistant cells
As DHFR amplification is the main mechanism that underlies MTX resistance, we examined the effect of DNA-PKcs depletion on the MTX sensitivity of MTX-resistant DM-containing and HSR-containing cells. By MTS assay, we found that DM-containing cells depleted of DNA-PKcs had 4.8-fold to 8.8-fold decrease in IC50 values compared with the control (table 1). However, in HSR-containing cells, depletion of DNA-PKcs resulted only in a onefold to twofold decrease in IC50 values compared with the control (table 1). These results indicate that DNA-PKcs depletion renders higher MTX sensitivity in DM-containing cells compared with HSR-containing cells. We then examined the effect of DNA-PKcs depletion on the proliferation of MTX-resistant DM-containing and HSR-containing cells. As shown in figure 5, DNA-PKcs depletion caused reduced proliferation of both DM-containing (figure 5A) and HSR-containing (figure 5B) cells.
Table 1.
Methotrexate (MTX) sensitivity of double minute (DM)-containing and homogeneously staining region (HSR)-containing cells, with or without DNA-PKcs depletion
| Cell | IC50 (mM) | Fold change |
|---|---|---|
| DM-containing control | 0.049195 | |
| shDNA-PKcs 1 | 0.010174 | 4.8 |
| shDNA-PKcs 2 | 0.005578 | 8.8 |
| HSR-containing control | 0.014116 | |
| shDNA-PKcs 1 | 0.013980 | 1.0 |
| shDNA-PKcs 2 | 0.006982 | 2.0 |
Figure 5.
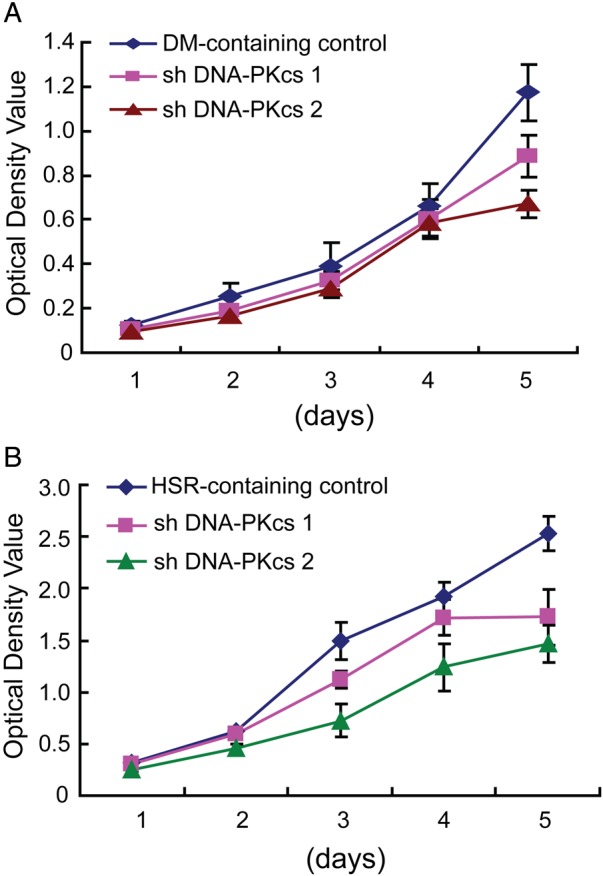
Inhibition of non-homologous end joining decreases proliferation of methotrexate (MTX)-resistant cells. Growth of (A) double minute (DM)-containing and (B) homogeneously staining region (HSR)-containing cells, with and without DNA-PKcs depletion, over 5 days.
Discussion
Cytogenetic studies have identified two types of gene amplification, intrachromosomal HSRs and extrachromosomal DMs. Although these two genetic structures have been suggested to be associated with increased DSBs and subsequent NHEJ repair process, the precise mechanism underlying the formation of HSRs and DMs remains to be investigated. Using a method that has been described previously,20–22 we have generated two HT-29 colon cancer cell lines that are resistant to different concentrations of MTX: (i) a 10−5 mol/L resistant line that harbours amplified DHFR primarily in HSRs and (ii) a 10−4 mol/L resistant line that harbours amplified DHFR primarily in DMs. These two cell lines serve as model systems that provided us a unique opportunity to study the molecular mechanism by which HSRs and DMs are formed and how they relate to the development of dose-dependent MTX resistance.
Initially, we examined whether MTX causes DSBs and whether the two MTX-resistant cell lines exhibit altered levels of NHEJ-associated proteins. We found that γH2AX foci formation increased in both the HSR-containing and DM-containing cells, indicating that MTX-induced formation of DHFR-harbouring DMs and HSRs, at least in HT-29 cells, is associated with increased DSBs and genomic instability. Our data are consistent with previous reports by Lorico et al24 that MTX causes single-strand DNA breaks (SSBs) and DSBs, and Coquelle et al5 proposed that MTX induces the formation of fragile sites and triggers gene amplification. We also found that HSR-containing cells showed no significant change in levels of the key NHEJ proteins, but DM-containing cells showed an increase in expression of the major NHEJ protein, DNA-PKcs, suggesting that NHEJ contributes to the development of DM-containing but not HSR-containing MTX-resistant HT-29 colon cancer cells. This may indicate differential roles of NHEJ at different stages of MTX resistance, which cytogenetically manifests as HSRs and DMs.
To investigate the differential roles of NHEJ on amplification of genes in MTX-resistant cancer cells harbouring HSRs or DMs, we analysed the effect of depleting DNA-PKcs in these cells. We demonstrated that DHFR amplification was decreased in DM-containing cells and that the predominant cytogenetic manifestation changed from DMs to HSRs, whereas no change was observed in HSR-containing cells, suggesting that NHEJ contributes to the formation of DMs but not HSRs. Indeed, depletion of DNA-PKcs specifically reduced the amplification of MSH3 and ZFYVE16, which colocalise with DHFR in DM-containing cells.
B/F/B cycles, which are assumed to be the primary mechanism for the formation of HSRs,5 23 25–29 are triggered by a DSB. Thus, our observation that dicentric chromosome 5 and nucleoplasmic bridges exist in HSR-containing cells suggests that DSBs generated by MTX may lead to enhanced occurrence of B/F/B cycles and subsequently, the formation of HSRs harbouring DHFR. However, alternative mechanisms for HSR formation remain to be investigated such as the potential involvement of HR-dependent pathways30–33 when DNA-PKcs is compromised.
Different mechanisms have also been implicated in the formation of extrachromosomal DMs. For example, breakage and recombination at stalled replication forks can generate circular DNA.34 35 DNA segments can also be excised from the chromosome (looping out mechanism) during G1 or G2 and circularise giving rise to extrachromosomal elements.25 36–38 In addition, genomic fragments resulting from chromosome shattering (chromothripsis) can be assembled into DMs.39 40 In all these cases, DSBs are produced and must be repaired to generate circular elements. Previously, Storlazzi et al38 found nucleotide microhomologies in amplicon junctions in DMs that have been shown to be used in the fusion of amplicon ends.41 This suggests that DNA circularisation in DMs is mediated by an abnormal NHEJ repair pathway. In the current study, we have demonstrated increased expression of NHEJ proteins in DM-containing cells, and that depletion of DNA-PKcs in these cells caused a decrease in DHFR amplification and disappearance of DMs. These findings indicate a crucial role for NHEJ in the formation of DMs in MTX-resistant cells. Thus, while previous DNA sequencing analysis has implicated NHEJ in the formation of DMs,38 41 in the current study, we describe for the first time the involvement of the NHEJ pathway in the formation of DMs. We propose that NHEJ, which is known to play a critical role in DNA repair,42 is involved in the later stage (ie, formation of DMs) but not in the early stage (ie, formation of HSRs) of gene amplification.
Previous reports have shown that DMs may undergo selective elimination from cells if DNA damage in DMs is induced by low concentrations of a replication inhibitor such as hydroxyurea or gemcitabine hydrochloride (GEM) or by low-dose radiation therapy.19 43–45 Here, we demonstrate for the first time that NU7026, a DNA-PK inhibitor, can inhibit DSB repair in chromosomes and DMs and can contribute to a transient increase in the formation of NBUDs or MNs which correlates with amplified DNA elimination. Interestingly, we observed that stable depletion of DNA-PKcs in DM-containing cells resulted in decreased number of NBUDs and MNs. This may reflect decreased number of DMs in these cells compared with NU7026-treated cells and thus, have decreased demand for the formation of NBUDs and MNs to eliminate DMs. Nonetheless, our data support the requirement for DNA-PKcs and thus, NHEJ in the formation of DMs.
In DM-containing cells, we also found that depletion of DNA-PKcs resulted in considerably increased sensitivity to MTX. This is consistent with previous reports indicating that inhibition of DNA-PKcs can confer increased sensitivity to DNA-damaging agents.46 47 Conversely, HSR-containing cells showed only a modest increase in sensitivity to MTX following DNA-PKcs depletion. Interestingly, both DM-containing and HSR-containing cells exhibited decreased proliferation upon DNA-PKcs depletion. This is supported by data presented by An et al48 showing decreased proliferation of DM-containing cells with silenced DNA-PKcs. We presume that in DM-containing cells, increased sensitivity to MTX following inhibition or depletion of DNA-PKcs is a result of DHFR elimination and reduced proliferation. However, the fact that DNA-PKcs depletion has no effect on DHFR dosage in HSR-containing cells suggests the differential involvement of NHEJ in MTX-resistant cancers. Together, we provide insight on the potential for NHEJ as a target in the development of personalised medicine.
Supplementary Material
Acknowledgments
This work was supported in part by the International Science and Technology Cooperation Program of China (2013DFA31610 to SF); Program for Changjiang Scholars and Innovative Research Team in University (IRT1230 to YJ); New Century Support Program for the Excellent Scholar, Ministry of Education of China (NCET-11-0954 to YY, NCET-13-0758 to CZ); CIHR and NSERC to K-YL. and National Natural Science Foundation of China (81372784 to SF, 81101646 to XM).
Footnotes
Contributors: Our manuscript presents original research. Neither the submitted manuscript nor any similar manuscript, in whole or in part, is under consideration, in press, published or reported elsewhere.
Competing interests: None.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Chatterjee S, Martinez-Lopez W, Grigorova M, Darroudi F, Obe G, Natarajan AT. Comparison of AluI-induced frequencies of dicentrics and translocations in human lymphocytes by chromosome painting. Mutagenesis 1999;14:283–6. [DOI] [PubMed] [Google Scholar]
- 2.Natarajan AT, Boei JJ. Formation of chromosome aberrations: insights from FISH. Mutat Res 2003;544:299–304. [DOI] [PubMed] [Google Scholar]
- 3.Monteilhet C, Perrin A, Thierry A, Colleaux L, Dujon B. Purification and characterization of the in vitro activity of I-Sce I, a novel and highly specific endonuclease encoded by a group I intron. Nucleic Acids Res 1990;18:1407–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuo MT, Vyas RC, Jiang LX, Hittelman WN. Chromosome breakage at a major fragile site associated with P-glycoprotein gene amplification in multidrug-resistant CHO cells. Mol Cell Biol 1994;14:5202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell 1997;89:215–25. [DOI] [PubMed] [Google Scholar]
- 6.Mondello C, Guasconi V, Giulotto E, Nuzzo F. Gamma-ray and hydrogen peroxide induction of gene amplification in hamster cells deficient in DNA double strand break repair. DNA Repair (Amst) 2002;1:483–93. [DOI] [PubMed] [Google Scholar]
- 7.Chakraborty S, Stark JM, Sun CL, Modi H, Chen W, O'Connor TR, Forman SJ, Bhatia S, Bhatia R. Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage-fusion-bridge cycles mediated by increased nonhomologous end joining. Blood 2012;119:6187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albertson DG. Gene amplification in cancer. Trends Genet 2006;22:447–55. [DOI] [PubMed] [Google Scholar]
- 9.Mc CB. Chromosome organization and genic expression. Cold Spring Harb Symp Quant Biol 1951;16:13–47. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu N. Extrachromosomal double minutes and chromosomal homogeneously staining regions as probes for chromosome research. Cytogenet Genome Res 2009;124:312–26. [DOI] [PubMed] [Google Scholar]
- 11.Pipiras E, Coquelle A, Bieth A, Debatisse M. Interstitial deletions and intrachromosomal amplification initiated from a double-strand break targeted to a mammalian chromosome. EMBO J 1998;17:325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mondello C, Rebuzzini P, Dolzan M, Edmonson S, Taccioli GE, Giulotto E. Increased gene amplification in immortal rodent cells deficient for the DNA-dependent protein kinase catalytic subunit. Cancer Res 2001;61:4520–5. [PubMed] [Google Scholar]
- 13.Shen H, Schultz M, Kruh GD, Tew KD. Increased expression of DNA-dependent protein kinase confers resistance to adriamycin. Biochim Biophys Acta 1998;1381:131–8. [DOI] [PubMed] [Google Scholar]
- 14.Ader I, Muller C, Bonnet J, Favre G, Cohen-Jonathan E, Salles B, Toulas C. The radioprotective effect of the 24 kDa FGF-2 isoform in HeLa cells is related to an increased expression and activity of the DNA dependent protein kinase (DNA-PK) catalytic subunit. Oncogene 2002;21:6471–9. [DOI] [PubMed] [Google Scholar]
- 15.Cahill D, Connor B, Carney JP. Mechanisms of eukaryotic DNA double strand break repair. Front Biosci 2006;11:1958–76. [DOI] [PubMed] [Google Scholar]
- 16.Okuno Y, Hahn PJ, Gilbert DM. Structure of a palindromic amplicon junction implicates microhomology-mediated end joining as a mechanism of sister chromatid fusion during gene amplification. Nucleic Acids Res 2004;32:749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fenech M. The in vitro micronucleus technique. Mutat Res 2000;455:81–95. [DOI] [PubMed] [Google Scholar]
- 18.Haaf T, Raderschall E, Reddy G, Ward DC, Radding CM, Golub EI. Sequestration of mammalian Rad51-recombination protein into micronuclei. J Cell Biol 1999;144:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu N, Misaka N, Utani K. Nonselective DNA damage induced by a replication inhibitor results in the selective elimination of extrachromosomal double minutes from human cancer cells. Genes Chromosomes Cancer 2007;46:865–74. [DOI] [PubMed] [Google Scholar]
- 20.Singer MJ, Mesner LD, Friedman CL, Trask BJ, Hamlin JL. Amplification of the human dihydrofolate reductase gene via double minutes is initiated by chromosome breaks. Proc Natl Acad Sci USA 2000;97:7921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morales C, Garcia MJ, Ribas M, Miro R, Munoz M, Caldas C, Peinado MA. Dihydrofolate reductase amplification and sensitization to methotrexate of methotrexate-resistant colon cancer cells. Mol Cancer Ther 2009;8:424–32. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz-Herrera A, Smirnova A, Khouriauli L, Nergadze SG, Mondello C, Giulotto E. Gene amplification in human cells knocked down for RAD54. Genome Integr 2011;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo AW, Sabatier L, Fouladi B, Pottier G, Ricoul M, Murnane JP. DNA amplification by breakage/fusion/bridge cycles initiated by spontaneous telomere loss in a human cancer cell line. Neoplasia 2002;4:531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorico A, Toffoli G, Boiocchi M, Erba E, Broggini M, Rappa G, D'Incalci M. Accumulation of DNA strand breaks in cells exposed to methotrexate or N10-propargyl-5,8-dideazafolic acid. Cancer Res 1988;48:2036–41. [PubMed] [Google Scholar]
- 25.Toledo F, Buttin G, Debatisse M. The origin of chromosome rearrangements at early stages of AMPD2 gene amplification in Chinese hamster cells. Curr Biol 1993;3:255–64. [DOI] [PubMed] [Google Scholar]
- 26.Ma C, Martin S, Trask B, Hamlin JL. Sister chromatid fusion initiates amplification of the dihydrofolate reductase gene in Chinese hamster cells. Genes Dev 1993;7:605–20. [DOI] [PubMed] [Google Scholar]
- 27.Toledo F, Smith KA, Buttin G, Debatisse M. The evolution of the amplified adenylate deaminase 2 domains in Chinese hamster cells suggests the sequential operation of different mechanisms of DNA amplification. Mutat Res 1992;276:261–73. [DOI] [PubMed] [Google Scholar]
- 28.Debatisse M, Malfoy B. Gene amplification mechanisms. Adv Exp Med Biol 2005;570:343–61. [DOI] [PubMed] [Google Scholar]
- 29.Murnane JP. Telomeres and chromosome instability. DNA Repair (Amst) 2006;5:1082–92. [DOI] [PubMed] [Google Scholar]
- 30.Narayanan V, Lobachev KS. Intrachromosomal gene amplification triggered by hairpin-capped breaks requires homologous recombination and is independent of nonhomologous end-joining. Cell Cycle 2007;6:1814–18. [DOI] [PubMed] [Google Scholar]
- 31.Li YH, Wang X, Pan Y, Lee DH, Chowdhury D, Kimmelman AC. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS ONE 2012;7:e39588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen C, Halbrook J, Nickoloff JA. Interactive competition between homologous recombination and non-homologous end joining. Mol Cancer Res 2003;1:913–20. [PubMed] [Google Scholar]
- 33.Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, Meek K. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol 2011;31:1719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Windle BE, Wahl GM. Molecular dissection of mammalian gene amplification: new mechanistic insights revealed by analyses of very early events. Mutat Res 1992;276:199–224. [DOI] [PubMed] [Google Scholar]
- 35.Nonet GH, Carroll SM, DeRose ML, Wahl GM. Molecular dissection of an extrachromosomal amplicon reveals a circular structure consisting of an imperfect inverted duplication. Genomics 1993;15:543–58. [DOI] [PubMed] [Google Scholar]
- 36.Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M. A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell 1998;2:259–65. [DOI] [PubMed] [Google Scholar]
- 37.Roelofs H, Tasseron-de Jong JG, van der Wal-Aker J, Rodenburg RJ, van Houten GB, van de Putte P, Giphart-Gassler M. Gene amplification in a human osteosarcoma cell line results in the persistence of the original chromosome and the formation of translocation chromosomes. Mutat Res 1992;276:241–60. [DOI] [PubMed] [Google Scholar]
- 38.Storlazzi CT, Fioretos T, Surace C, Lonoce A, Mastrorilli A, Strombeck B, D'Addabbo P, Iacovelli F, Minervini C, Aventin A, Dastugue N, Fonatsch C, Hagemeijer A, Jotterand M, Muhlematter D, Lafage-Pochitaloff M, Nguyen-Khac F, Schoch C, Slovak ML, Smith A, Sole F, Van Roy N, Johansson B, Rocchi M. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet 2006;15:933–42. [DOI] [PubMed] [Google Scholar]
- 39.Kloosterman WP, Koster J, Molenaar JJ. Prevalence and clinical implications of chromothripsis in cancer genomes. Curr Opin Oncol 2014;26:64–72. [DOI] [PubMed] [Google Scholar]
- 40.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011;144:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogt N, Lefevre SH, Apiou F, Dutrillaux AM, Cor A, Leuraud P, Poupon MF, Dutrillaux B, Debatisse M, Malfoy B. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci USA 2004;101:11368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roth DB, Gellert M. New guardians of the genome. Nature 2000;404:823–5. [DOI] [PubMed] [Google Scholar]
- 43.Yu L, Zhao Y, Quan C, Ji W, Zhu J, Huang Y, Guan R, Sun D, Jin Y, Meng X, Zhang C, Yu Y, Bai J, Sun W, Fu S. Gemcitabine eliminates double minute chromosomes from human ovarian cancer cells. PLoS ONE 2013;8:e71988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimizu N. Molecular mechanisms of the origin of micronuclei from extrachromosomal elements. Mutagenesis 2011;26:119–23. [DOI] [PubMed] [Google Scholar]
- 45.Schoenlein PV, Barrett JT, Kulharya A, Dohn MR, Sanchez A, Hou DY, McCoy J. Radiation therapy depletes extrachromosomally amplified drug resistance genes and oncogenes from tumor cells via micronuclear capture of episomes and double minute chromosomes. Int J Radiat Oncol Biol Phys 2003;55:1051–65. [DOI] [PubMed] [Google Scholar]
- 46.Collis SJ, Swartz MJ, Nelson WG, DeWeese TL. Enhanced radiation and chemotherapy-mediated cell killing of human cancer cells by small inhibitory RNA silencing of DNA repair factors. Cancer Res 2003;63:1550–4. [PubMed] [Google Scholar]
- 47.Peng Y, Zhang Q, Nagasawa H, Okayasu R, Liber HL, Bedford JS. Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer Res 2002;62:6400–4. [PubMed] [Google Scholar]
- 48.An J, Yang DY, Xu QZ, Zhang SM, Huo YY, Shang ZF, Wang Y, Wu DC, Zhou PK. DNA-dependent protein kinase catalytic subunit modulates the stability of c-Myc oncoprotein. Mol Cancer 2008;7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.