

Background: Peptidoglycan metabolites regulate AmpR-mediated control of ampC β-lactamase expression.
Results: AmpR binds DNA as a tetramer and interacts with up to four copies of its repressor UDP-MurNAc-pentapeptide via its d-Ala-d-Ala motif.
Conclusion: d-Ala-d-Ala recognition supports 1,6-anhydroMurNAc-pentapeptide as an AmpR activator, probably through competitive binding with UDP-MurNAc-pentapeptide.
Significance: Understanding how peptidoglycan metabolites modulate AmpR provides insight into how β-lactam classes differentially induce ampC expression.
Keywords: Crystal Structure, Mass Spectrometry (MS), Peptidoglycan, Small Angle X-ray Scattering (SAXS), Transcription, AmpC beta-Lactamase, AmpR, LysR-type Transcriptional Regulator
Abstract
Inducible expression of chromosomal AmpC β-lactamase is a major cause of β-lactam antibiotic resistance in the Gram-negative bacteria Pseudomonas aeruginosa and Enterobacteriaceae. AmpC expression is induced by the LysR-type transcriptional regulator (LTTR) AmpR, which activates ampC expression in response to changes in peptidoglycan (PG) metabolite levels that occur during exposure to β-lactams. Under normal conditions, AmpR represses ampC transcription by binding the PG precursor UDP-N-acetylmuramic acid (MurNAc)-pentapeptide. When exposed to β-lactams, however, PG catabolites (1,6-anhydroMurNAc-peptides) accumulate in the cytosol, which have been proposed to competitively displace UDP-MurNAc-pentapeptide from AmpR and convert it into an activator of ampC transcription. Here we describe the molecular interactions between AmpR (from Citrobacter freundii), its DNA operator, and repressor UDP-MurNAc-pentapeptide. Non-denaturing mass spectrometry revealed AmpR to be a homotetramer that is stabilized by DNA containing the T-N11-A LTTR binding motif and revealed that it can bind four repressor molecules in an apparently stepwise manner. A crystal structure of the AmpR effector-binding domain bound to UDP-MurNAc-pentapeptide revealed that the terminal d-Ala-d-Ala motif of the repressor forms the primary contacts with the protein. This observation suggests that 1,6-anhydroMurNAc-pentapeptide may convert AmpR into an activator of ampC transcription more effectively than 1,6-anhydroMurNAc-tripeptide (which lacks the d-Ala-d-Ala motif). Finally, small angle x-ray scattering demonstrates that the AmpR·DNA complex adopts a flat conformation similar to the LTTR protein AphB and undergoes only a slight conformational change when binding UDP-MurNAc-pentapeptide. Modeling the AmpR·DNA tetramer bound to UDP-MurNAc-pentapeptide predicts that the UDP-MurNAc moiety of the repressor participates in modulating AmpR function.
Introduction
Induction of chromosomal AmpC β-lactamase by the LysR-type transcriptional regulator (LTTR)4 AmpR is a mechanism of β-lactam antibiotic resistance that is common to many enterobacteria as well as Pseudomonas aeruginosa and other nonfermenting Gram-negative bacilli. AmpC enzymes are of considerable medical concern because they confer resistance to a broad range of β-lactams, and their activity is not sufficiently suppressed with clinically available β-lactamase inhibitors (1). The genes encoding AmpC and AmpR form a divergent operon (ampR-ampC), which allows AmpR to bind to overlapping promoters within the intergenic region of the operon to control the transcription of both genes (2). Moreover, AmpR has also been found to exert transcriptional control over an additional chromosomal class D β-lactamase gene as well as numerous virulence-associated genes in P. aeruginosa (3, 4). Accordingly, there has been an increased interest to better understand how AmpR regulates gene expression.
AmpR controls transcription of AmpC by binding to metabolites of the Gram-negative peptidoglycan (PG) recycling pathway (Fig. 1) (5, 6). Despite its critical structural role, PG is a dynamic macromolecule that is continuously turned over and recycled during microbial growth (5–8). Turnover is carried out by periplasmic autolysins that excise GlcNAc-β(1→4)-1,6-anhydroMurNAc-peptide fragments from the PG sacculus (9), and the fragments are transported to the cytosol by the AmpG permease (10) for recycling. In the cytosol, the non-reducing GlcNAc residue is removed from the fragments by the glycosidase NagZ (11–13) to yield a series of 1,6-anhydroMurNAc-tripeptides, -tetrapeptides, and -pentapeptides. The amidase AmpD cleaves the amide bond linking the remaining sugar, 1,6-anhydroMurNAc, to the peptide stems (14, 15), and the resulting pool of monosaccharide and peptide catabolites are subsequently used to build UDP-MurNAc-pentapeptide, an essential anabolite of cell wall synthesis (5). In the absence of β-lactams, UDP-MurNAc-pentapeptide binds to AmpR and causes the repression of ampC transcription (16). In contrast, disruption of PG metabolism by β-lactams increases the cytosolic concentration of 1,6-anhydroMurNAc-peptides, which appears to overwhelm endogenous AmpD activity and allows either the 1,6-anhydroMurNAc-tripeptide (16, 17) or -pentapeptide species (7, 18) to bind to AmpR. It has been proposed that these 1,6-anhydroMurNAc species competitively displace UDP-MurNAc-pentapeptide from AmpR, thus converting AmpR into an activator of ampC transcription (16, 19). The expressed AmpC is then exported to the periplasm, where it hydrolyzes the β-lactam to re-establish normal cell wall homeostasis.
FIGURE 1.
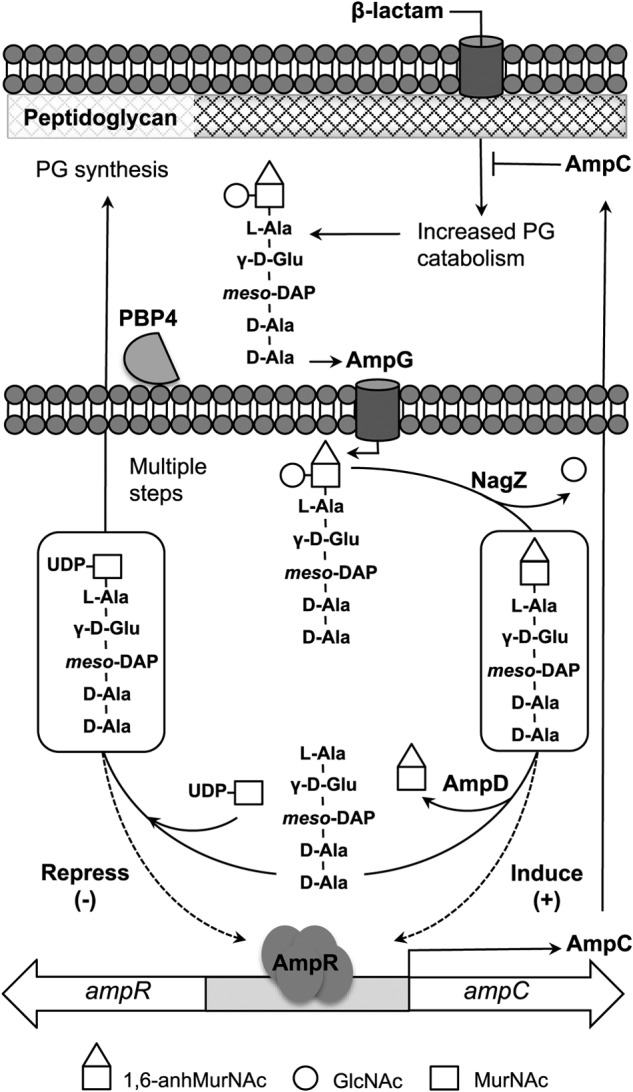
Simplified schematic of the PG recycling pathway (see Refs. 5 and 6 for details). GlcNAc-1,6-anhydroMurNAc-peptides (only the pentapeptide species is shown) are excised from the PG layer and then transported to the cytosol via the AmpG permease, where glycosidase NagZ removes GlcNAc to yield 1,6-anhydroMurNAc-pentapeptide (or -tripeptide species), which have been implicated as activators of AmpR. The amidase activity of AmpD separates the stem peptide from 1,6-anhydroMurNAc, which depletes the cytosolic levels of the AmpR activator ligands, and the products are used to generate the AmpR repressor molecule UDP-MurNAc-pentapeptide (via multiple steps), which is eventually reincorporated into the PG layer.
Some β-lactams are weak inducers of ampC transcription (e.g. ceftazidime) and are thus effective against bacteria harboring the ampR-ampC system, although they are susceptible to AmpC (1). However, spontaneous inactivation by mutations within ampD occurs frequently and causes accumulation of cytosolic 1,6-anhydroMurNAc-peptides and production of AmpC to levels that confer resistance even to these β-lactams (14, 20–23). Inactivation of dacB, which encodes penicillin-binding protein 4 (PBP4), also causes AmpR-dependent AmpC hyperproduction and high level β-lactam antibiotic resistance (24). Loss of PBP4 has recently been found to be the leading cause of single-step mutational resistance to β-lactams in P. aeruginosa (25). Interestingly, PBP4 removes the terminal d-Ala residue from PG catabolites prior to being transported to the cytosol for recycling (26), suggesting that the terminal d-Ala residue may be important for AmpC activation. Although occurring less frequently than ampD or dacB null mutations, mutations in ampR have also been found to drive constitutive high level AmpC production (21, 25, 27, 28).
LTTR proteins, including AmpR, function as oligomers, typically as homotetramers, to regulate gene transcription in response to effector molecule binding (29, 30). Most LTTRs repress transcription in the absence of an effector molecule and only initiate transcription upon binding an activator ligand. AmpR, however, is controlled by both a repressor ligand (UDP-MurNAc-pentapeptide) and an activator ligand (1,6-anhydroMurNAc-tripeptide or -pentapeptide), providing an additional level of metabolite-controlled regulation over AmpC expression (16, 18).
AmpR is a 32-kDa protein, that like other LTTRs, has a well conserved N-terminal DNA-binding domain, containing a winged helix-turn-helix motif, and a C-terminal effector-binding domain (EBD) (31). It recognizes and binds to a 38-base pair (bp) region within the overlapping promoters of the ampR-ampC operon, shown by Lindquist et al. (32) to be protected by AmpR in DNase footprinting assays (Fig. 2). This region includes a canonical LTTR high affinity T-N11-A motif (29, 30) and, according to the “sliding dimer” model of LTTR proteins (29, 30), may also include two additional low affinity binding sites for AmpR that directly control ampC gene expression as described below (Fig. 2). When LTTR proteins tetramerize, the N-terminal DNA-binding domains of the monomers work together in pairs to bind DNA. Whereas one pair of DNA-binding domains binds with high affinity to the T-N11-A motif (29), the other pair is thought to be mobile and interacts with at least two low affinity sites that are close to the RNA polymerase recognition site (−35 site) of the regulated gene (33–35). According to the sliding dimer model, interaction of the mobile pair of DNA binding domains with these sites causes DNA bending to occur, and when the pair binds to a low affinity site near the −35 site, it shields the site from the RNA polymerase and suppresses transcription. However, when bound to an appropriate effector ligand, conformational changes in the LTTR protein prompt the domains to “slide” upstream to an alternate low affinity site, relaxing the DNA bend and exposing the −35 site to the polymerase to permit transcription (29, 30).
FIGURE 2.
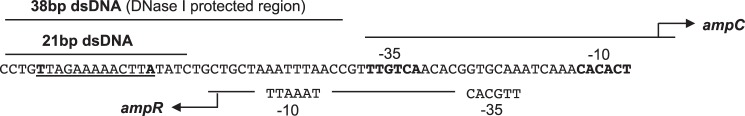
The intergenic region of the ampR-ampC operon of C. freundii containing the high affinity LTTR binding motif T-N11-A (underlined) and the promoter regions of the divergently transcribed ampR and ampC genes (32), which according to the sliding dimer model of LTTR proteins (29, 30) are predicted to contain low affinity binding sites for AmpR. The sequences of the 21- and 38-bp DNA that were bound to AmpR in this study are as indicated, with the latter region corresponding to the DNase-protected region identified by Lindquist et al. (32).
Although a number of crystal structures of full-length LTTRs have been determined, including one bound to its natural activator ligand (36), none have been determined in complex with a natural repressor molecule. Nor have any full-length LTTR complexes with DNA been structurally studied, thus the interactions with promoter elements and associated conformational changes that underpin the switch from gene repression to activation remain poorly understood.
Here we provide new structural insights into the archetypal AmpR protein from Citrobacter freundii to further our understanding of how this LTTR protein modulates AmpC β-lactamase gene expression. Using non-denaturing mass spectrometry, we demonstrate that AmpR is a tetramer that possesses two independent DNA binding sites that have high affinity for the LTTR T-N11-A DNA binding motif. We find that AmpR is able to bind up to four molecules of UDP-MurNAc-pentapeptide, the natural repressor ligand of this protein. To investigate how AmpR binds UDP-MurNAc-pentapeptide, we determined the crystal structure of the EBD of AmpR bound to the metabolite, which is the first structure of an LTTR bound to a natural repressor ligand. This structure reveals that the terminal d-Ala-d-Ala motif of UDP-MurNAc-pentapeptide plays a central role in the interaction of the ligand with AmpR. Finally, small angle x-ray scattering (SAXS) studies of the full-length AmpR tetramer bound to DNA indicate that it adopts a conformation remarkably similar to AphB, an LTTR protein involved in virulence gene regulation in Vibrio cholera, and that the complex undergoes a subtle conformational change upon binding UDP-MurNAc-pentapeptide. Modeling the repressor-bound AmpR·DNA complex (based on our X-ray data and the AphB structure) suggests that whereas the pentapeptide of the repressor and activator ligands are involved in binding AmpR, it is likely the UDP-MurNAc and 1,6-anhydroMurNAc moieties of the molecules that modulate AmpR function at the quaternary level and control its ability to control transcription.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification of the AmpR EBD
Escherichia coli BL21 (DE3) GOLD HTE cells harboring pMB400 (31) were grown to mid-log phase (A600 = 0.5) at 37 °C, with shaking, in 500-ml volumes of LB media supplemented with 35 μg/ml kanamycin. Expression of the AmpR EBD was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h at 28 °C with shaking. Cells were pelleted by centrifugation and stored at −80 °C. Pellets were thawed in 20 ml of ice-cold lysis buffer (1 m NaCl, 20 mm Tris-HCl (pH 8.0), and 1 μm PMSF) and lysed using a French pressure cell press (Aminco). The lysate was clarified by centrifugation and loaded onto a nickel-nitrilotriacetic acid-agarose column that had been pre-equilibrated with binding buffer (1 m NaCl and 20 mm Tris-HCl (pH 8.0)). The column was washed with 10 column volumes of binding buffer, followed by 2 column volumes of binding buffer supplemented with 25 mm imidazole (pH 7.4) and 50 mm imidazole (pH 7.4), respectively. The AmpR EBD protein was eluted from the column using binding buffer supplemented with 250 mm imidazole (pH 7.4). The eluate was dialyzed overnight against 300 mm NaCl and 20 mm MES (pH 6.0). Chromatographic steps were performed using AKTA FPLC. All centrifugation, chromatography, and dialysis steps were conducted at 4 °C.
Protein Expression, Refolding, and Purification of Full-length AmpR
The full-length ampR gene (GenBankTM ID GU322411.3) was PCR-amplified from C. freundii ATCC8090 genomic DNA using oligonucleotide primers 5′-AGATATACATATGACGCGTAGCTATATCCCTC-3′ and 5′-GATATAACTAGTTTTGTGCAGCACCCCGGTC-3′ and ligated into a modified pET-28 vector using NdeI and SpeI restriction sites to generate plasmid pMB-AmpR. E. coli BL21 (DE3) GOLD HTE cells transformed with plasmid pMB-AmpR were grown in four 500-ml volumes of LB media supplemented with 35 μg/ml kanamycin to an A600 of 1.0 at 28 °C. AmpR expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 16 h at 28 °C with shaking. Cells from four 500-ml cultures were combined together to yield two 1-liter volumes of culture, pelleted by centrifugation, resuspended in lysis buffer (50 mm Tris-HCl (pH 7.0) and 1 μm PMSF), and lysed using a French pressure cell press (Aminco). The lysate was then centrifuged at 12,000 rpm for 1 h, and the pellets from each combined 1-liter preparation were reconstituted in 4 ml of resuspension buffer (RB) (500 mm KCl, 10% glycerol, 10 mm Tris-HCl (pH 7.0), and 1 mm DTT) with 0.1% Triton X-100. Resuspended pellets were centrifuged again at 12,000 rpm for 20 min at 4 °C, supernatant was discarded, and pellets were washed with RB containing 0.5% Triton X-100. Subsequently, the centrifugation and wash steps were repeated using RB and then in RB containing 10 mm guanidine hydrochloride, in RB containing 50 mm guanidine hydrochloride, and finally in a final reconstitution buffer of RB only. The resuspended pellets were then sonicated (Branson Sonicator) in 2 × 15-s bursts, each at 50% duty cycle at a power setting of 5. The sonicated pellet resuspensions were then combined and denatured in 16 ml of denaturation buffer (2 m guanidine hydrochloride, 10% glycerol, 10 mm Tris-HCl (pH 7.0), and 1 mm DTT) at room temperature for 1 h, with vortexing every 10 min.
The denatured pellet was centrifuged for 40 min at 12,000 rpm at 4 °C, and the supernatant was poured into a syringe with a stopcock. Denatured protein was slowly dripped dropwise into 350 ml of refolding buffer (300 mm KCl, 10% glycerol, 10 mm Tris-HCl (pH 7.0), and 1 mm DTT) at 4 °C with slow stirring. Refolded AmpR was centrifuged for 40 min at 3700 rpm at 4 °C to pellet insoluble protein and then vacuum-filtered through a 0.22-μm cellulose membrane and concentrated to a final concentration of 0.7 mg/ml prior to being loaded on to a size exclusion gel filtration column (Superdex 200) pre-equilibrated in gel filtration buffer (300 mm KCl, 10% glycerol, 10 mm Tris (pH 7.0), and 1 mm DTT) at 4 °C. Gel filtration fractions containing purified AmpR were pooled together in the presence of 50 mm NDSB-195 to prevent protein aggregation before further concentration and biophysical analyses.
Preparation of Double-stranded DNA (dsDNA)
Pairs of oligonucleotides were purchased from AlphaDNA (Montreal, Canada) or Integrated DNA Technologies. Set 1 (21 bp) had only the T-N11-A motif: sense strand, 5′-CCTGTTAGAAAAACTTATATC-3′ (6388.2 Da); antisense strand, 5′-GATATAAGTTTTTCTAACAGG-3′ (6459.3 Da). Set 2 (38 bp) contained the T-N11-A motif and the additional region shown to be protected during DNase footprinting (32): sense strand, 5′-CCTGTTAGAAAAACTTATATCTGCTGCTAAATTTAACC-3′ (11,594.6 Da); antisense strand, 5′-GGTTAAATTTAGCAGCAGATATAAGTTTTTCTAACAGG-3′ (11,754.7 Da). Equimolar amounts of each pair of sense and antisense DNA oligonucleotides were added to buffer containing 50 mm NaCl, 10 mm Tris-HCl (pH 7.9), 10 mm MgCl2, 1 mm DTT (NEBuffer 2, New England Biolabs) and annealed by heating for 3 min at 98 °C, with a slow cool for 1 h to room temperature.
Binding dsDNA to AmpR
dsDNA was added to purified AmpR in a molar excess of 2:1 to the purified AmpR protein. Concentrated AmpR·dsDNA complex was purified by gel filtration (Superdex 200) in a column pre-equilibrated in gel filtration buffer (300 mm KCl, 10% glycerol, 10 mm Tris (pH 7.0), and 1 mm DTT) at 4 °C. Purified AmpR·dsDNA fractions were pooled together and stored at 4 °C.
Binding and Purification of AmpR·dsDNA with UDP-MurNAc-pentapeptide
A molar excess of UDP-MurNAc-pentapeptide (BaCWAN, University of Warwick) was added to the purified AmpR·21 bp dsDNA complex, left to bind at room temperature for 30 min, and then reloaded on to a Superdex 200 gel filtration column pre-equilibrated in gel filtration buffer at room temperature. Purified AmpR·21-bp dsDNA·UDP-MurNAc-pentapeptide gel filtration fractions were pooled together and concentrated for analysis by dynamic light scattering, small angle x-ray scattering and mass spectrometry or stored at 4 °C for up to 5 days.
Non-denaturing Mass Spectrometry
After refolding and purification of full-length AmpR, the pure protein was digested with trypsin and analyzed by MALDI mass spectrometry to confirm its identification. Protein from precipitates and gel slices was processed as reported (37). These spectra were acquired on a prototype MALDI tandem quadrupole/time-of-flight (QqTOF) instrument (38). Intact AmpR and the AmpR·dsDNA complexes were desalted into 0.3–0.5 m ammonium acetate buffer (Aldrich, 99.99%) with 1 mm DTT (Aldrich, 99%), using “waterbugs” (39, 40). Samples were diluted to about 2 μm and inserted into a New Objective PicoTipTM nanospray capillary attached to an electrospray time-of-flight mass spectrometer built for these kinds of experiments (41). The declustering voltage was adjusted to give the best resolution (42) but can also be used to test the relative stability of large non-covalent complexes (examples in Refs. 40 and 41). The UDP-MurNac-pentapeptide was lost during dialysis into volatile buffer. Therefore, the AmpR·dsDNA complex in 250 mm ammonium acetate and 1 mm DTT was mixed with increasing concentrations of repressor UDP-MurNAc-pentapeptide (dissolved in water) at 0, 5, 10, and 50 μm, keeping the final concentration of AmpR·dsDNA complex fixed at 2 μm. The mixtures were then held on ice for 1 h before the mass spectrometry experiments. All data were analyzed using TOFMA, software developed with the instruments, in the Department of Physics and Astronomy at the University of Manitoba.
Crystallization of the AmpR EBD Bound to UDP-MurNAc-pentapeptide and X-ray Data Collection
Purified wild-type AmpR EBD was concentrated to 4.5 mg/ml using an Amicon Ultra Centricon spin cartridge (Merck Millipore) and co-crystallized with 5 mm UDP-MurNAc-pentapeptide. X-ray diffraction data were obtained from a single crystal formed in a solution of 10% PEG 3350, 9% glycerol, and 100 mm MES, pH 6.2, via hanging drop vapor diffusion at room temperature. Crystals were flash-cooled in liquid N2 after replacing the mother liquor in the drop with a cryosolution consisting of 35% PEG 4000 and 15% glycerol.
X-ray diffraction data were collected from a single crystal (held at 100 K in an N2 gas stream) using beamline 08ID-1 at the Canadian Light Source (Saskatoon, Canada). The x-ray data were indexed using Mosflm (43) and then scaled and averaged using SCALA (44). The AmpR EBD·UDP-MurNAc-pentapeptide complex structure was determined by molecular replacement using PHASER (PHENIX package) (45) and a structure of the AmpR EBD (PDB entry 3KOS) as the search model (from which all solvent and ligands had been removed). The molecular replacement model was rebuilt using phenix.autobuild (45). The restraint file for UDP-MurNAc-pentapeptide was generated using PHENIX eLBOW (45), and the ligand was manually fit into its electron density in the complex. Further refinement and model building were carried out using phenix.refine (45) and Coot (46).
Dynamic Light Scattering
The dynamic light scattering profile for AmpR·21-bp dsDNA with and without UDP-MurNAc-pentapeptide was measured using the Zetasizer Nano S system (Malvern Instruments Ltd., Malvern, UK) as described earlier (47). AmpR·21-bp dsDNA was filtered through a 0.1-μm filter via centrifugation and diluted in 300 mm KCl, 10% glycerol, 10 mm Tris-HCl (pH 7.0), and 1 mm DTT to final concentrations ranging from 2.9 to 4.1 mg/ml. Similarly, AmpR·21-bp dsDNA·UDP-MurNAc-pentapeptide complexes were diluted within 0.8–2.3 mg/ml. For each measurement, the protein was allowed to equilibrate for 4 min at 20 °C prior to dynamic light scattering measurements, after which multiple records of the dynamic light scattering profile were collected for data analysis.
Small-angle X-ray Scattering
SAXS data for the AmpR·21-bp dsDNA complex and for AmpR·21-bp dsDNA·UDP-MurNAc-pentapeptide complex were collected at multiple concentrations (3, 4, 4.6, and 4.8 mg/ml and 0.8, 1.0, 1.4, and 1.8 mg/ml, respectively) using a Rigaku three-pinhole camera (S-MAX3000) equipped with a MicroMax+002 microfocus sealed tube and confocal max-flux optics operating at 40 W, as described earlier (47). The system was also equipped with a 200-mm multiwire two-dimensional detector for data collection. SAXS data were collected at a wavelength of 1.54 Å (λ) with a scattering vector (q) of 0.008 ≤ q ≤ 0.26 Å−1 (q = (4π sinθ)/λ, where 2θ is the scattering angle) with 3 h of exposure for each concentration. Raw data were processed and merged together using the program PRIMUS (48) to obtain a single output file (represented graphically in the inset to Fig. 8B), which was further processed using GNOM (49) to obtain the radius of gyration (Rg) and maximum particle dimension (Dmax) for both complexes as described previously (50, 51). Multiple ab initio models for the AmpR·21-bp dsDNA complex and for the AmpR·21-bp dsDNA·UDP-MurNAc-pentapeptide complex were built using the program DAMMIF (52) with enforced particle symmetry of P22. These models were then rotated and averaged using the program DAMAVER (53) to obtain a representative model.
FIGURE 8.

Low resolution solution structures of the AmpR·21-bp dsDNA complex determined by SAXS. A, hydrodynamic radii (Rh) determined for the AmpR·21-bp dsDNA complex (blue) and AmpR·21-bp dsDNA·UDP-MurNAc-pentapeptide complex (red) at various protein concentrations. SAXS data were collected using an in-house instrument (Rigaku) at multiple concentrations, from which pair distribution functions (p(r)) for the AmpR·21-bp dsDNA complex (blue) and AmpR·21-bp dsDNA·UDP-MurNAc-pentapeptide complex (red, scaled data to AmpR·DNA p(r) data) were calculated using GNOM (49) from merged data revealing that AmpR undergoes only a slight conformational change upon binding the repressor (B). Guinier plots for AmpR complexes are shown in B (inset). Ab initio modeling of the unliganded AmpR·21-bp dsDNA complex (C) and UDP-MurNAc-pentapeptide-bound complex (D) using DAMMIF (52) with a particle symmetry of P22 suggests that repressor binding causes a slight conformational change in AmpR, most notably demonstrating an absence of the central surface pocket observed in the unliganded AmpR·DNA complex. E, fitting the structure of the AphB (N100E) variant on to the ab initio SAXS model of the AmpR·DNA complex indicates that the central cavity in the AphB tetramer is consistent with the indent in the SAXS model. 21-bp dsDNA fragments are placed within the ab initio model such that the α3-recognition helices of AphB (N100E) (colored in red) fit into the major groove of the dsDNA. Error bars, S.D.
The program HYDROPRO (54) was employed to calculate hydrodynamic properties, such as the Rh, Rg, and Dmax for each ab initio model as described earlier (47, 50). The partial specific volume of 0.738 ml/g for AmpR was calculated using SEDNTERP from its amino acid sequence, whereas that for DNA was considered to be 0.57 ml/g (55). By using Equation 1 from Dzananovic et al. (56) and considering the molecular mass of tetramer for protein (130.8 kDa) and dimer of DNA (25.7 kDa), the partial specific volume of 0.710 ml/g was obtained. Buffer density (1.03 g/ml) and viscosity (1.276 centipoises) were also calculated using the program SEDNTERP (57) and were used as the input parameters to run HYDROPRO.
RESULTS AND DISCUSSION
C. freundii AmpR Is a Tetramer in Solution with Two Active DNA-binding Domains
C. freundii AmpR was overexpressed as inclusion bodies in E. coli BL21 (DE3) GOLD HTE cells. Following denaturation of inclusion bodies in 2 m guanidine hydrochloride, the protein could be refolded and purified using an approach we significantly modified from that originally described by Bishop et al. (58) (see “Experimental Procedures”). Refolded AmpR was obtained as a single peak of ∼130 kDa by gel filtration, indicating that the protein is a tetramer in solution. The purified full-length protein was only stable at low concentrations (<1 mg/ml). The poor stability and solubility of LTTR proteins (30) is often associated with their DNA-binding domains, which have been suggested to oligomerize in solution in the absence of DNA, thus contributing to their aggregation and precipitation (59, 60). However, binding AmpR to 21- or 38-bp dsDNA fragments of the ampR-ampC intergenic promoter, containing the T-N11-A motif only or the complete region that is protected by AmpR during DNase footprinting, respectively (Fig. 2), greatly stabilized the protein and allowed it to be concentrated for biophysical studies.
Nanospray mass spectrometry (MS) was used to measure the mass of the components in the AmpR·dsDNA complexes to investigate the oligomeric state of the folded protein and to determine the stoichiometry of AmpR complexed with either the 21- or 38-bp dsDNA. The measured mass of the AmpR monomer was 32,725 Da, identical to the predicted mass of the recombinant protein (Fig. 3, D and E), and our MS analysis also revealed transient ions consistent with a tetramer (Fig. 4). The AmpR tetramer was considerably more stable when bound to DNA, and the spectra indicated that the difference in mass of the most abundant ions could be accounted for by the mass of two molecules of dsDNA bound to one AmpR tetramer (Fig. 5). For the AmpR·21-bp dsDNA complex, the observed mass of 156,916 Da (Fig. 5A) agreed well with that expected for a tetramer bound to two copies of dsDNA, calculated to be 156,590 Da. Similarly, for the AmpR·38-bp dsDNA complex (Fig. 5B), the observed mass of 177,825 Da agrees with the calculated 177,596 Da. Both spectra show a less abundant species that is probably from a dimeric form of AmpR. Also present in the sample, but only seen at low declustering voltage, were ions from the DNA (Fig. 3, A and B), which meant that some dissociation was occurring in the mass spectrometer.
FIGURE 3.

A series of nanospray spectra from the same AmpR·21-bp dsDNA sample in the same capillary where only the declustering voltage was changed, thus changing the internal energy of the ions. These spectra show all of the components of the AmpR·21-bp dsDNA complex. The full width-half-maximum indicated at the arrows is a measurement of error (see Ref. 42). A, at low voltage, there are the major ions of the complex and monoisotopic resolution of free dsDNA and ssDNA; B, an expansion of the spectrum at low m/z and deconvolution of the major labeled ions (inset). The mass of each oligonucleotide comprising the dsDNA was 6388.2 and 6459.3 Da, respectively, with dsDNA expected at 12,847.5 Da. Ions flanking the 6+ and 8+ of the dsDNA ions are the 3+ and 4+ ions of the sense and antisense oligonucleotides, at monoisotopic resolution. A similar experiment with the AmpR·38-bp dsDNA complex verified the mass of the 38-bp dsDNA to be 23,350 Da (data not shown); C, under our control conditions (200 V), there is only one major ion envelope (see Fig. 6A); D, at maximum voltage, a new ion envelope from the protein monomer is present at low m/z. E, expansion of the low m/z region and deconvolution of the main ions (inset) give a protein with a mass of 32,725 Da and a small amount at 32,824 Da, indicating some retention of the initial Met residue. From the published amino acid sequence, the expected mass of AmpR is 32,523 Da (Swissprot P12529). The AmpR protein in our study has four extra vector-derived amino acids, TSGS, on the C terminus. The addition of that extra mass gives an expected value of 32,855 Da, or 32,724 Da if the initial Met is processed off.
FIGURE 4.
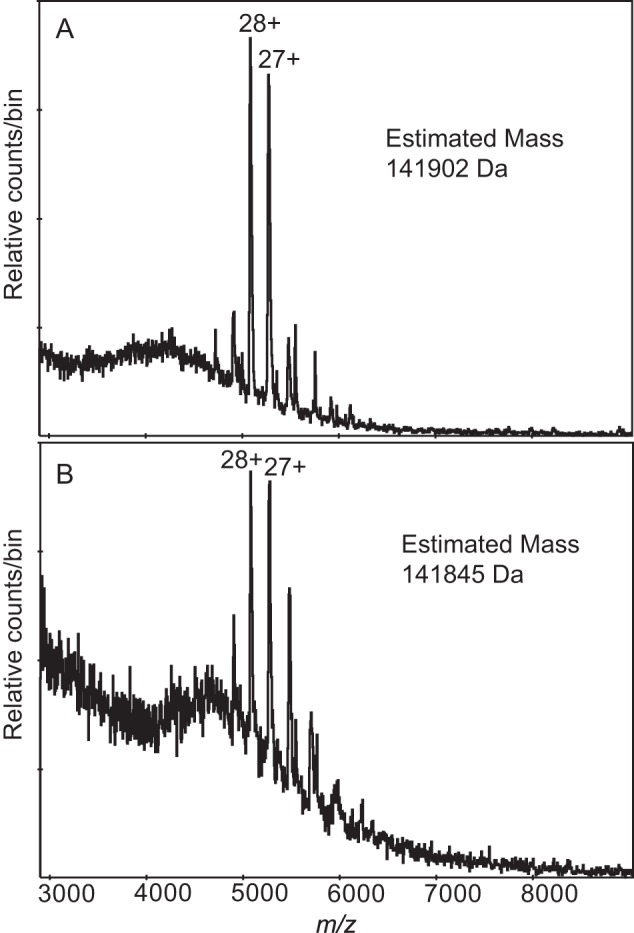
The major non-salt ions from two nanospray spectra showing the ions expected for an AmpR tetramer. A, the beginning acquisition of the purified protein prepared without dsDNA and buffer-exchanged into 0.3 m ammonium acetate, 1 mm DTT. Within 5 min, this ion envelope was replaced by a series best explained by aggregation; B, the beginning acquisition of the AmpR·21-bp dsDNA complex showing the same ions. Within 5 min, this ion envelope was replaced by the one shown in Fig. 5A. Although the observation was transient, it happened frequently, always at the very beginning of the experiment when there might be residual salts in the new capillary being used for the test. Mass was manually determined from the m/z values of the labeled ions.
FIGURE 5.
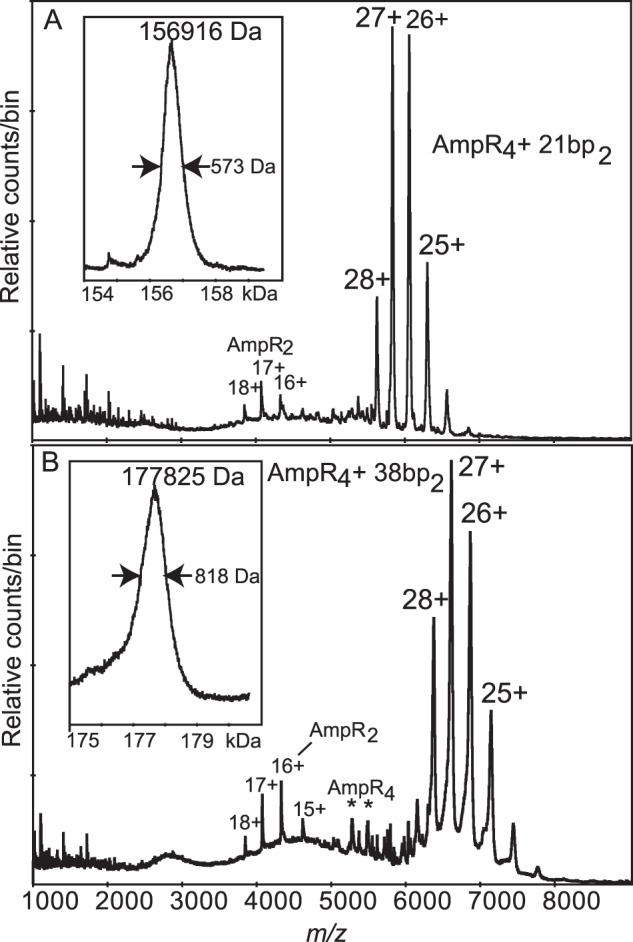
Nanospray spectra of the complexes of AmpR protein with 21- and 38-bp dsDNA. Insets, deconvolutions of the major labeled ions. A, with the 21-bp dsDNA containing only the T-N11-A motif, there is one major charge envelope, the 24+ to 28+ ions of a species of mass 156,916 Da (full-width-half-maximum, 573 Da). A minor series, 16+ to 18+, belongs to a species of mass 68,987 Da (full-width-half-maximum, 22 Da). The complex was in 100 mm ammonium acetate buffer, and the declustering voltage was 200 V. B, with the 38-bp dsDNA containing both the T-N11-A motif and two putative low affinity binding sites, as with 21-bp dsDNA, there is one major ion envelope with almost the same charge states but shifted to higher m/z with measured mass 177,825 Da (full-width-half-maximum, 818 Da). Also present in low abundance are ions at m/z 4000 (as in A) and unresolved ions in the m/z 5000 region, some of which, marked with an asterisk, may be from the tetramer (see Fig. 4). The complex was in 50 mm ammonium acetate, 0.5 mm DTT, and the declustering voltage was 200 V.
If each AmpR tetramer was able to bind both the T-N11-A motif and a putative low affinity site present in the 38-bp fragment of dsDNA at the same time, then that complex would have been smaller, only 154,246 Da. However, the same 4:2 binding stoichiometry of AmpR monomer to dsDNA was observed with both the 38- and 21-bp dsDNA. We speculate that each pair of DNA-binding domains of tetrameric AmpR preferentially binds the high affinity T-N11-A site on the DNA molecules in our experiments rather than binding in tandem to the high and low affinity binding sites, as would occur in vivo.
The AmpR·dsDNA Complex Binds up to Four UDP-MurNAc-pentapeptide Repressor Molecules
Non-denaturing mass spectra collected from mixtures of 2 μm AmpR·21-bp dsDNA complex and increasing amounts of UDP-MurNAc-pentapeptide (up to 50 μm) revealed four repressor-binding sites (Fig. 6). At 5 μm repressor (Fig. 6B), the most intense ions correspond to those of the unliganded AmpR·21-bp dsDNA complex, having charge states ranging from 24+ to 28+. However, an additional series of ions appeared with the same charge states but with an m/z ratio equivalent to the complex plus one repressor molecule (Fig. 6B). At 10 and 50 μm concentrations of UDP-MurNAc-pentapeptide (Fig. 6, C and D), additional series of ions appear and increase in intensity while the abundance of the unliganded species decreases. At a 50 μm concentration of UDP-MurNAc-pentapeptide, there are five ions at each charge state, with a difference in m/z between each series corresponding to the expected m/z for the repressor molecule (mass 1193 Da). These MS results provide a direct determination of the binding stoichiometry of UDP-MurNAc-pentapeptide to the AmpR·DNA complex. The data reveal that the tetramer can bind four molecules of UDP-MurNAc-pentapeptide (Fig. 6D), although full saturation was not possible because the high concentration of the repressor and associated sodium overloaded the detector on the mass spectrometer. The observed binding stoichiometry suggests that the effector-binding domain of each monomer in the AmpR tetramer binds one molecule of repressor. This proposed binding model is supported by our crystal structure of the AmpR EBD bound to UDP-MurNAc-pentapeptide as described below.
FIGURE 6.
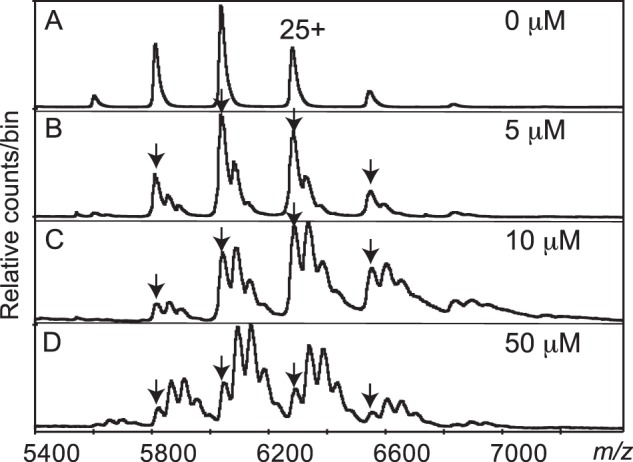
Parts of four nanospray spectra of the AmpR·21-bp dsDNA complex mixed with increasing amounts of UDP-MurNAc-pentapeptide. Arrows, ions at the same m/z as those in the control. Spacing between the adducts can be explained by sequential additions of the repressor molecule, with up to four present. A, control, no repressor; B, 5 μm repressor; C, 10 μm repressor; D, 50 μm repressor. High ion counts at low m/z from the free repressor molecules, and fragments thereof caused serious interference at higher concentrations of repressor. All samples were in 125 mm ammonium acetate, 0.5 mm DTT, and all spectra were at 200 V declustering voltage.
It is well known that AmpR is able to “tune” ampC expression in response to various levels of activator ligand, such as the stepwise increase in AmpC production that occurs when the three AmpD homologues are systematically deleted from P. aeruginosa (61). Based on this observation, we suggest that four UDP-MurNAc-pentapeptide molecules are needed to most efficiently repress AmpR, and as the concentration of the activator ligand increases during β-lactam exposure, AmpR-mediated ampC transcription increases in a stepwise manner until all four UDP-MurNAc-pentapeptide molecules have been displaced by activator ligand, at which point AmpR drives ampC transcription at its maximal rate.
AmpR Recognizes and Binds the Peptide Motif of UDP-MurNAc-pentapeptide
Although we were unable to crystallize full-length AmpR, its EBD (residues 94–289) could be crystallized in complex with UDP-MurNAc-pentapeptide. The structure of the complex was determined by molecular replacement using the unliganded AmpR EBD (PDB entry 3KOS) as a search model (31) and refined to 2.15 Å (see Table 1 for crystallographic and refinement statistics).
TABLE 1.
Crystallographic and refinement statistics for the AmpR EBD from C. freundii bound to UDP-MurNAc-pentapeptide
| Data collection | |
| Space group | P 21 21 2 |
| Unit cell (Å) | a = 62.7, b = 183.6, c = 197.81 |
| α = β = γ = 90° | |
| Wavelength (Å) | 0.9795 |
| Resolution range (Å) | 59.3–2.15 (2.27–2.15)a |
| Total observations | 495,866 (63,842) |
| Unique reflections | 121,101 (17,581) |
| Ι/σΙ | 8.3 (3.0) |
| Completeness (%) | 97.7 (98.0) |
| Rmerge | 0.096 (0.497) |
| Rpim | 0.053 (0.316) |
| Multiplicity | 4.1 (3.6) |
| Refinement statistics | |
| Reflections (work) | 120,936 |
| Reflections (test) | 2722 |
| Total atoms refined | 26,109 |
| Rwork (Rfree) | 0.20 (0.25) |
| Mean B-values (Å2) | |
| Protein | 36.03 |
| Ligands | 37.34 |
| Solvent | 39.87 |
| Root mean square deviations | |
| Bond lengths (Å)/angles (degrees) | 0.013/1.46 |
| Ramachandran plot | |
| Favored/allowed (%) | 96.9/3.1 |
a Values in parentheses refer to the high resolution shell.
The AmpR EBD is known to dimerize in solution (31), and it crystallized in space group P21212 when bound to UDP-MurNAc-pentapeptide, with four homodimers present within the asymmetric unit. Each monomer of the AmpR EBD dimer is composed of two subdomains that adopt α/β-Rossman-like folds and are connected to each other by a hinge region (Fig. 7A) (31). The effector-binding site is located between the two subdomains of each monomer, and electron density maps clearly revealed that the effector-binding site of each monomer was occupied by the pentapeptide motif of one UDP-MurNAc-pentapeptide molecule (Fig. 7, A–C). The UDP-MurNAc portion of each ligand (Fig. 7B) was disordered in all copies of the AmpR EBD monomer with the exception of four monomers where the MurNac sugar could be modeled into density.
FIGURE 7.

A, the crystal structure of the AmpR EBD dimer (monomers indicated by purple and gray schematics) bound to UDP-MurNAc-pentapeptide (stick format; oxygen atoms are indicated in red, nitrogen atoms in blue, and carbon atoms in yellow) refined to 2.15 Å resolution. The hinge regions connecting each subunit within a monomer are colored magenta. The UDP moiety of the repressor was disordered and not visible in electron density maps, as were some of the MurNAc sugars. B, chemical schematic of UDP-MurNAc-pentapeptide (the UDP moiety not visible in the electron density maps is colored red). C, σ-A-weighted 2Fo − Fc density map contoured at 1.0σ showing the MurNAc-pentapeptide portion of the repressor bound within the AmpR effector-binding pocket. Hydrogen-bonding interactions between residues forming the binding site and the pentapeptide stem of the repressor are shown by dashed magenta lines. Waters are shown as red spheres. D, superposition of AmpR EBD monomers comprising the asymmetric unit of the AmpR EBD·UDP-MurNAc-pentapeptide crystal structure (gray) with the AmpR EBD bound to MES (PDB entry 3KOS; shown in cyan) and the inactivated variant AmpR EBD (T103V/S221A/Y264F) (PDB entry 3KOT; shown in pink). UDP-MurNAc-pentapeptide and MES molecules are represented by sticks, with carbon atoms indicated in yellow and pink, respectively, and oxygen and nitrogen atoms in red and blue. Regions with relative shifts in loop/residue position are circled in orange.
It was striking to observe that the terminal d-Ala-d-Ala motif of the pentapeptide plays a primary role in interacting with the effector-binding site of AmpR (Fig. 7C). Buried deep within the binding pocket, the carboxylate on the terminal d-Ala group acts as an acceptor of three hydrogen bonds from the side chain hydroxyl groups of Thr-103 (2.6 Å), Ser-221 (2.7 Å), and Tyr-264 (2.5 Å), which together form the base of the effector binding pocket. The backbone oxygen of the penultimate d-Ala residue forms a 3.0-Å hydrogen bond with the amide backbone of Tyr-192. Three additional hydrogen-bonding contacts are made between the pentapeptide and AmpR residues at the entrance of the effector-binding pocket; the indole ring nitrogen of Trp-152 participates in a 2.8-Å hydrogen bond with the side chain carboxylate group of the d-Glu within the pentapeptide. The carbonyl group of the meso-DAP side chain forms a hydrogen bond within 2.8 and 3.0 Å of the side chain groups of Arg-193 and Asn-132. Six water molecules are also present within the effector-binding pocket and form hydrogen-bonding interactions with either the bound repressor or residues of the pocket (Fig. 7C).
Previously, in an attempt to predict how both 1,6-anhydroMurNAc-tripeptide and -pentapeptide species appear to behave as AmpR inducers, we speculated that the sugar moiety, 1,6-anhydroMurNAc, played a key role in converting AmpR into an activator of ampC transcription by binding within the effector binding pocket (31) because the 1,6-anhydroMurNAc sugar is common to both the 1,6-anhydroMurNAc-tripeptide and -pentapeptide species (16, 18). However, our crystal structure of the AmpR EBD bound to UDP-MurNAc-pentapeptide repressor now shows that it is the pentapeptide stem of the repressor that is solely responsible for binding to the AmpR EBD. This binding mode implies that 1,6-anhydroMurNAc-pentapeptide (18) or possibly free pentapeptide (as suggested by Park and Uehara) (7) bind in an identical fashion to UDP-MurNAc-pentapeptide to outcompete the repressor in a concentration-dependent manner and convert AmpR into an activator of ampC transcription. Although the terminal d-Ala-d-Ala motif contributes the majority of the interactions of the ligand to the AmpR EBD, additional interactions between the d-Glu and meso-DAP of the repressor and Arg-193, Asn-132, and Trp-152 on the surface of the AmpR EBD ligand binding pocket suggest that 1,6-anhydroMurNAc-tripeptide could also displace UDP-MurNAc-pentapeptide from AmpR as observed by Jacobs et al. (16). However, these binding interactions of the tripeptide species would probably be weaker than those of 1,6-anhydroMurNAc-pentapeptide or free pentapeptide that contain the d-Ala-d-Ala motif. Moreover, involvement of the d-Ala-d-Ala motif in AmpR regulation is entirely consistent with the observed AmpC hyperproduction that occurs upon loss of PBP4 (24) because this protein has a dd-peptidase activity that removes the terminal d-Ala residue from PG catabolites (26). Thus, our findings support a scenario where loss of PBP4 causes 1,6-anhydroMurNAc-pentapeptide (or free pentapeptide) to accumulate and drive AmpR to hyperproduce AmpC. Finally, as pointed out previously by Park and Uehara (7), it is interesting to note that the strongest inducers of AmpR-mediated ampC transcription, such as cefoxitin and imipenem, are those that generate pentapeptide-containing PG fragments (7, 18).
A general examination of the repressor-bound AmpR EBD monomers revealed little change in the relative orientation of secondary structural elements between each monomer within the asymmetric unit. However, superposition of a crystal structure of the AmpR EBD monomer bound to the buffer molecule MES (PDB entry 3KOS) or a variant of the AmpR EBD (T103V/S221A/Y264F) that is unable to bind MES (PDB entry 3KOT) with the AmpR·UDP-MurNAc-pentapeptide complex revealed that the hydrogen bonds with γ-d-Glu and meso-DAP residues in the pentapeptide tether Trp-152 and Arg-193 such that they point toward the center of the interdomain cleft (Fig. 7D). Similarly to the terminal d-Ala stem of the repressor, MES forms hydrogen-bonding interactions at the base of the binding pocket with Thr-103, Ser-221, and Tyr-264. However, in the absence of hydrogen-bonding interactions at the opening of the binding cleft, Trp-152 and Arg-193 and the loop regions they reside on exhibit more flexibility in the wild-type AmpR EBD·MES complex and the unliganded AmpR variant relative to the UDP-MurNAc-pentapeptide-bound structure (Fig. 7D). This change might be involved in modulating AmpR function because it has been suggested that conformational changes in the EBD upon effector molecule binding may trigger larger reorganization of the LTTR homotetramer (62).
The AmpR·21-bp dsDNA Complex Adopts a Flat Conformation That Undergoes Only a Slight Conformational Change upon Binding of UDP-MurNAc-pentapeptide
Given that we identified a stable AmpR·21-bp dsDNA quaternary complex by non-denaturing MS, we decided to examine the effects of repressor ligand binding on the conformation of this complex using SAXS measurements. Prior to SAXS data collection, dynamic light scattering was used to confirm that the complex remained monodisperse in the presence and absence of UDP-MurNAc-pentapeptide over a protein concentration range of 0.8–4.1 mg/ml (Fig. 8A and Table 2). Analysis of the SAXS data collected from the complex with and without UDP-MurNAc-pentapeptide using GNOM (49) yielded a radius of gyration (Rg) of 4.40 ± 0.32 nm for the AmpR·DNA complex and Rg of 4.63 ± 0.10 nm for the AmpR·DNA·repressor complex (Table 2).
TABLE 2.
Measurements and statistics from SAXS and DLS analysis of the AmpR·21 bp dsDNA complex with and without repressor UDP-MurNAc-pentapeptide
| AmpR·DNA complex |
AmpR·DNA·UDP-MurNAc-pentapeptide |
|||
|---|---|---|---|---|
| Experimental value | HYDROPRO-DAMMIFa | Experimental value | HYDROPRO-DAMMIF | |
| Rh (nm)b | 4.95 ± 0.01 | 5.10 ± 0.03 | 5.18 ± 0.01 | 5.05 ± 0.02 |
| Rg (nm)c | 4.20 ± 0.10 | 4.30 ± 0.20 | ||
| Rg (nm)d | 4.40 ± 0.32 | 4.45 ± 0.05 | 4.63 ± 0.10 | 4.72 ± 0.08 |
| Dmax (nm)d | 12.5 | 13.4 ± 0.30 | 13.8 | 15.0 ± 0.28 |
| Chi | 0.85 | 0.72 | ||
| NSDe | 0.53 ± 0.18 | 0.68 ± 0.11 | ||
a Data calculated from ab initio models using HYDROPRO.
b Experimentally determined using dynamic light scattering.
c Experimentally determined from SAXS data using Guinier analysis.
d Experimentally determined from SAXS data from p(r) analysis by GNOM.
e Normalized spatial discrepancy.
The maximal particle dimension (Dmax) of the repressor-free AmpR·21-bp dsDNA complex was calculated from the pairwise distribution (p(r)) function as 12.5 nm, which only increased slightly to 13.8 nm upon binding UDP-MurNAc-pentapeptide (Fig. 8B). Using the program DAMMIF (52), ab initio models of the AmpR·DNA complex alone and bound to UDP-MurNAc-pentapeptide were calculated and were in excellent agreement with the experimental SAXS data (Table 2). Interestingly, a representative ab initio model for the AmpR·21-bp dsDNA complex possesses a large central cavity, which is flanked at opposite ends by two smaller cavities present along a diagonal of the tetramer, which are absent in the ab initio model of the complex bound to UDP-MurNAc-pentapeptide, suggesting that the surface indents may become occupied by repressor ligands (Fig. 8, C and D).
Only slight conformational changes appear to occur in AmpR upon binding UDP-MurNAc-pentapeptide according to our SAXS data. The prevailing model of LTTR activation proposes that small conformational changes in the EBD of LTTR proteins translate to larger conformational changes in their linker helix and DNA binding domains to trigger transcription (63). TsaR, the only full-length LTTR crystallized with its native inducer ligand did not demonstrate significant conformational changes relative to the unliganded protein (36). In the case of the DntR EBD, however, it was observed that binding of its inducer (salicylate) triggered a noticeable conformational change within the protein relative to other LTTRs bound to their respective ligands (62). We did observe a small conformational change within the crystal structure of the AmpR EBD when bound to UDP-MurNAc-pentapeptide (Fig. 7D), and although our SAXS data do not suggest that this translates into large changes at the quaternary level, it is possible that the full scope of conformational changes within full-length AmpR may only become apparent when it is bound to one contiguous strand of dsDNA rather than to separate fragments. This would be technically challenging to achieve, however, due to preferential binding of the DNA-binding domains to the high affinity T-N11-A motif when presented with a molar excess of promoter DNA in vitro, a scenario that may be common to other tetrameric LTTR proteins.
The SAXS-based Solution Structure of the AmpR·21-bp dsDNA Complex Closely Resembles the Structure of V. cholera Virulence Regulator AphB
By individually fitting all known crystal structures of full-length LTTR proteins into the SAXS-based model of the AmpR·21-bp dsDNA complex, it became clear that the shape of the AmpR·21-bp dsDNA complex most closely resembles that of the tetrameric LTTR AphB, a protein that regulates virulence factors in V. cholerae (64). A transcriptional activator ligand for AphB has not been identified; however, the protein is known to respond to intracellular pH and oxygen concentrations (64). Unlike other full-length tetrameric LTTRs that have been structurally characterized (36, 65, 66), AphB adopts a distinctively flat shape, with a linear arrangement of its DNA-binding domains (64). Although AphB and a variant of the protein (N100E) both bind DNA in solution, AphB (N100E) crystallized in a conformation where the relative orientations of the α-helices in its DNA-binding domains suggest that this variant can bind a linear stretch of DNA, unlike in the wild-type crystal structure, where the DNA binding domains are not appropriately aligned (64). Thus, AphB (N100E) was used as a basis for modeling the AmpR·21-bp dsDNA complex into the ab initio SAXS envelope. The AphB (N100E) tetramer fit comfortably into the SAXS envelope of the AmpR·21-bp dsDNA complex (Fig. 8E), and interestingly, AphB (N100E) has a somewhat hollow core that is consistent with the central cavity that we observed in our SAXS model of the AmpR·21-bp dsDNA complex (Fig. 8, C and E).
After fitting AphB (N100E) into the SAXS envelope, room remained available within the envelope to also fit two 21-bp dsDNA oligomers onto the DNA-binding domain of AphB (Fig. 8E). The dsDNA was oriented such that the α3-recognition helices of each DNA-binding domain (colored in red, Fig. 8E) fit into the major groove of the dsDNA, as observed in a crystal structure of dsDNA bound to the isolated DNA-binding domain from the LTTR protein BenM (67). Based on the fitting of AphB (N100E) and dsDNA into the SAXS envelope, we built a model of AmpR bound to the dsDNA molecules by superposing our crystal structure of the AmpR EBD homodimer bound to UDP-MurNAc-pentapeptide onto the equivalent EBD dimers of the modeled AphB·dsDNA complex. Interestingly, the superposition revealed that the effector binding site of each of the four AmpR EBD domains was fully accessible to the solvent because the sites reside on the side of the tetramer opposite from the bound length of dsDNA (Fig. 9). Moreover, although the pentapeptide motif of each repressor is bound deep within the effector-binding pocket of each EBD monomer, the superposition suggests that the UDP-MurNAc portion of each bound repressor may project into the central cavity of AmpR, which is consistent with the filling of the central cavity in the ab initio SAXS model of the AmpR·21-bp dsDNA complex that occurred after binding UPD-MurNAc-pentapeptide (Figs. 8 (C and D) and 9).
FIGURE 9.

Modeling of the AmpR·DNA complex bound to UDP-MurNAc-pentapeptide. A, based on the fitting of AphB (N100E) and dsDNA into our SAXS envelopes of the unliganded and UDP-MurNAc-pentapeptide-bound AmpR·DNA complexes, we built a molecular model of AmpR bound to the dsDNA molecules by superposing our crystal structure of the AmpR EBD homodimer bound to UDP-MurNAc-pentapeptide (shown in gold) onto the equivalent EBD dimers of the modeled AphB·DNA complex (shown in gray). A short, continuous fragment of double-stranded DNA (light blue) is shown docked onto the DNA-binding domains of AphB. UDP moieties (carbon atoms are shown in magenta, nitrogen in blue, oxygen in red, and phosphorus in yellow) were manually modeled on MurNAc-pentapeptide portions of the repressor that were visible in the electron density maps (green). B, a close-up of the central cavity predicted to exist within the AmpR tetramer showing how the UDP-MurNAc moieties of the bound repressor ligands may become positioned to interact with each other or with residues of the DNA-binding domains and/or EBDs to trigger conformational rearrangements in the quaternary structure of AmpR that cause it to bind the operator DNA in a manner that represses gene transcription.
Proposed Model for the Regulation of AmpR by PG Recycling Intermediates
Ruangprasert et al. (60) recently proposed two structural schemes, I and II, that describe the inactive and active conformations of tetrameric LTTR proteins, respectively. In scheme I (as adopted by the LTTR CbnR), the tetrameric interface is composed of contacts between the DNA-binding domains and linker helices that connect the EBDs to the DNA-binding domains as well as direct interactions between adjacent effector-binding domains, which together result in a “closed” conformation of the protein (60, 65). Scheme II, on the other hand (as observed for the unliganded TsaR, ArgP, and AphB LTTR proteins and a putative transcriptional regulator from P. aeruginosa (PDB entry 2ESN)), presents a more “open” conformation, with an expanded hollow core within the tetramer, and associating solely through the linker helices and DNA-binding domains. Activation of a tetrameric LTTR by inducer binding is thought to trigger a conformational rearrangement of the tetramer, converting it from scheme I to scheme II (60, 64). Because Jacobs et al. (16) found that AmpR is fully active in the absence of PG metabolites, our SAXS envelope for the unliganded, activated AmpR·21-bp dsDNA complex probably mimics the inducer-bound form with a central cavity that corresponds to the “open” tetramer described by scheme II. In comparison, the SAXS model for the repressed AmpR·DNA·UDP-MurNAc-pentapeptide complex does not contain any surface indents, unlike the unliganded, active protein complex. It is possible that when AmpR is repressed by UDP-MurNAc-pentapeptide, the ligand occupies the space within the cavity and thus fills in the indents observed in the unliganded complex. However, binding of the repressor ligand may also trigger conformational changes within the monomers of the tetramer that result in closing of the central core via contacts between the EBDs, thus converting the protein into a form described by scheme I.
Finally, our model of the AmpR·dsDNA complex bound to UDP-MurNAc-pentapeptide (Fig. 9A) predicts that the UDP-MurNAc-moiety of the repressor molecules are oriented such that they could interact with each other as well as with residues comprising the DNA-binding domains and areas of the EBDs that flank the pentapeptide binding pocket (Fig. 9B). Given that the UDP-MurNAc moiety is unique to the repressor UDP-MurNAc-pentapeptide, compared with the putative activator ligands, it is tempting to speculate that the UDP-MurNAc moiety of the repressor may effect a conformational change in the quaternary structure of AmpR that causes the protein to repress ampC transcription (Fig. 9). Competitive binding of 1,6-anhydroMurNAc-pentapeptide and perhaps the tripeptide species would effectively remove the UDP-MurNAc moiety and convert the protein into an activator of ampC transcription. The molecular details underlying these ligand-induced changes await high resolution crystallographic analyses, an aspect we are currently exploring.
Significance
Although UDP-MurNAc-pentapeptide is generally accepted as the PG metabolite responsible for repressing AmpR-mediated gene transcription, the identity of the activator ligand(s) remains controversial (7, 16, 18). In vitro transcription studies by Jacobs et al. (16) showed that AmpR constitutively activates ampC transcription in the absence of any PG metabolites and that the addition of UDP-MurNAc-pentapeptide causes the protein to repress ampC transcription. Notably, they found that the addition of 1,6-anhydroMurNAc-tripeptide could reverse this repression (16). In vivo studies by Dietz et al. (18), however, reported that two of the strongest AmpC inducers (cefoxitin and imipenem) cause 1,6-anhydroMurNAc-pentapeptide to accumulate in the cytosol, not the tripeptide species, and thus argued that the pentapeptide species activates AmpR. Park and Uehara (7) have also stated that, in addition to 1,6-anhydroMurNAc-pentapeptide, large quantities of free pentapeptide accumulate in the cytosol of Gram-negative bacteria when challenged with strong ampC inducers such as cefoxitin and imipenem, suggesting that free pentapeptide could also activate AmpR. Our finding that the terminal d-Ala-d-Ala motif of UDP-MurNAc-pentapeptide forms the primary interactions that bind the repressor to AmpR suggests that 1,6-anhydroMurNAc-pentapeptide and free pentapeptide are more potent activators of AmpR than the tripeptide species, which is a possibility we are actively setting out to confirm by in vitro transcription. Identification of the activator ligands of AmpR has important implications in understanding how AmpR modulates ampC gene expression and why some β-lactams induce AmpC production more potently than others, which could be taken into account when developing next generation β-lactam antibiotics.
Acknowledgments
We thank Veronica Larmour, Vladimir Collado, and Victor Spicer for technical advice and assistance and Shaun Labiuk and the staff of the Canadian Light Source beamline 08B1-1 for assistance with data collection.
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP-97818 and Cystic Fibrosis Canada grants (to B. L. M. and D. J. V.). The Canadian Light Source is supported by Natural Sciences and Engineering Research Council of Canada (NSERC), the National Research Council, the CIHR, and the University of Saskatchewan.
The atomic coordinates and structure factors (code 4WKM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LTTR
- LysR-type transcriptional regulator
- PG
- peptidoglycan
- MurNAc
- N-acetylmuramic acid
- 1,6-anhydroMurNAc
- 1,6-anhydro-N-acetylmuramic acid
- GlcNAc
- N-acetylglucosamine
- PBP4
- penicillin-binding protein 4
- EBD
- effector-binding domain
- SAXS
- small angle x-ray scattering
- RB
- resuspension buffer
- PDB
- Protein Data Bank.
REFERENCES
- 1. Jacoby G. A. (2009) AmpC β-lactamases. Clin. Microbiol. Rev. 22, 161–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Honoré N., Nicolas M. H., Cole S. T. (1986) Inducible cephalosporinase production in clinical isolates of Enterobacter cloacae is controlled by a regulatory gene that has been deleted from Escherichia coli. EMBO J. 5, 3709–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balasubramanian D., Schneper L., Merighi M., Smith R., Narasimhan G., Lory S., Mathee K. (2012) The regulatory repertoire of Pseudomonas aeruginosa AmpC β-lactamase regulator AmpR includes virulence genes. PLoS One 10.1371/journal.pone.0034067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kong K.-F., Jayawardena S. R., Indulkar S. D., Del Puerto A., Koh C.-L., Høiby N., Mathee K. (2005) Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB β-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob. Agents Chemother. 49, 4567–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson J. W., Fisher J. F., Mobashery S. (2013) Bacterial cell-wall recycling. Ann. N.Y. Acad. Sci. 1277, 54–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mark B. L., Vocadlo D. J., Oliver A. (2011) Providing β-lactams a helping hand: targeting the AmpC β-lactamase induction pathway. Future Microbiol. 6, 1415–1427 [DOI] [PubMed] [Google Scholar]
- 7. Park J. T., Uehara T. (2008) How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol. Mol. Biol. Rev. 72, 211–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Typas A., Banzhaf M., Gross C. A., Vollmer W. (2012) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10, 123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vollmer W., Joris B., Charlier P., Foster S. (2008) Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 32, 259–286 [DOI] [PubMed] [Google Scholar]
- 10. Cheng Q., Park J. T. (2002) Substrate specificity of the AmpG permease required for recycling of cell wall anhydro-muropeptides. J. Bacteriol. 184, 6434–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng Q., Li H., Merdek K., Park J. T. (2000) Molecular characterization of the β-N-acetylglucosaminidase of Escherichia coli and its role in cell wall recycling. J. Bacteriol. 182, 4836–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vötsch W., Templin M. F. (2000) Characterization of a β-N-acetylglucosaminidase of Escherichia coli and elucidation of its role in muropeptide recycling and β-lactamase induction. J. Biol. Chem. 275, 39032–39038 [DOI] [PubMed] [Google Scholar]
- 13. Bacik J.-P., Whitworth G. E., Stubbs K. A., Vocadlo D. J., Mark B. L. (2012) Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem. Biol. 19, 1471–1482 [DOI] [PubMed] [Google Scholar]
- 14. Höltje J. V., Kopp U., Ursinus A., Wiedemann B. (1994) The negative regulator of β-lactamase induction AmpD is a N-acetyl-anhydromuramyl-L-alanine amidase. FEMS Microbiol. Lett. 122, 159–164 [DOI] [PubMed] [Google Scholar]
- 15. Jacobs C., Joris B., Jamin M., Klarsov K., Van Beeumen J., Mengin-Lecreulx D., van Heijenoort J., Park J. T., Normark S., Frère J. M. (1995) AmpD, essential for both β-lactamase regulation and cell wall recycling, is a novel cytosolic N-acetylmuramyl-l-alanine amidase. Mol. Microbiol. 15, 553–559 [DOI] [PubMed] [Google Scholar]
- 16. Jacobs C., Frère J. M., Normark S. (1997) Cytosolic intermediates for cell wall biosynthesis and degradation control inducible β-lactam resistance in Gram-negative bacteria. Cell 88, 823–832 [DOI] [PubMed] [Google Scholar]
- 17. Jacobs C., Huang L. J., Bartowsky E., Normark S., Park J. T. (1994) Bacterial cell wall recycling provides cytosolic muropeptides as effectors for β-lactamase induction. EMBO J. 13, 4684–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dietz H., Pfeifle D., Wiedemann B. (1997) The signal molecule for β-lactamase induction in Enterobacter cloacae is the anhydromuramyl-pentapeptide. Antimicrob. Agents Chemother. 41, 2113–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uehara T., Park J. T. (2002) Role of the murein precursor UDP-N-acetylmuramyl-l-Ala-γ-d-Glu-meso-diaminopimelic acid-d-Ala-d-Ala in repression of β-lactamase induction in cell division mutants. J. Bacteriol. 184, 4233–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Juan C., Maciá M. D., Gutiérrez O., Vidal C., Pérez J. L., Oliver A. (2005) Molecular mechanisms of β-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 49, 4733–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaneko K., Okamoto R., Nakano R., Kawakami S., Inoue M. (2005) Gene mutations responsible for overexpression of AmpC β-lactamase in some clinical isolates of Enterobacter cloacae. J. Clin. Microbiol. 43, 2955–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lindberg F., Lindquist S., Normark S. (1987) Inactivation of the ampD gene causes semiconstitutive overproduction of the inducible Citrobacter freundii β-lactamase. J. Bacteriol. 169, 1923–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith E. E., Buckley D. G., Wu Z., Saenphimmachak C., Hoffman L. R., D'Argenio D. A., Miller S. I., Ramsey B. W., Speert D. P., Moskowitz S. M., Burns J. L., Kaul R., Olson M. V. (2006) Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 103, 8487–8492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moya B., Dötsch A., Juan C., Blázquez J., Zamorano L., Haussler S., Oliver A. (2009) β-Lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 10.1371/journal.ppat.1000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cabot G., Ocampo-Sosa A. A., Domínguez M. A., Gago J. F., Juan C., Tubau F., Rodríguez C., Moyà B., Peña C., Martínez-Martínez L., Oliver A. (2012) Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob. Agents Chemother. 56, 6349–6357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korat B., Mottl H., Keck W. (1991) Penicillin-binding protein 4 of Escherichia coli: molecular cloning of the dacB gene, controlled overexpression, and alterations in murein composition. Mol. Microbiol. 5, 675–684 [DOI] [PubMed] [Google Scholar]
- 27. Hanson N. D., Sanders C. C. (1999) Regulation of inducible AmpC β-lactamase expression among Enterobacteriaceae. Curr. Pharm. Des. 5, 881–894 [PubMed] [Google Scholar]
- 28. Kuga A., Okamoto R., Inoue M. (2000) ampR gene mutations that greatly increase class C β-lactamase activity in Enterobacter cloacae. Antimicrob. Agents Chemother. 44, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schell M. A. (1993) Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47, 597–626 [DOI] [PubMed] [Google Scholar]
- 30. Maddocks S. E., Oyston P. C. F. (2008) Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154, 3609–3623 [DOI] [PubMed] [Google Scholar]
- 31. Balcewich M. D., Reeve T. M., Orlikow E. A., Donald L. J., Vocadlo D. J., Mark B. L. (2010) Crystal structure of the AmpR effector binding domain provides insight into the molecular regulation of inducible AmpC β-lactamase. J. Mol. Biol. 400, 998–1010 [DOI] [PubMed] [Google Scholar]
- 32. Lindquist S., Lindberg F., Normark S. (1989) Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC β-lactamase gene. J. Bacteriol. 171, 3746–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parsek M. R., Ye R. W., Pun P., Chakrabarty A. M. (1994) Critical nucleotides in the interaction of a LysR-type regulator with its target promoter region. J. Biol. Chem. 269, 11279–11284 [PubMed] [Google Scholar]
- 34. Akakura R., Winans S. C. (2002) Constitutive mutations of the OccR regulatory protein affect DNA bending in response to metabolites released from plant tumors. J. Biol. Chem. 277, 5866–5874 [DOI] [PubMed] [Google Scholar]
- 35. Porrúa O., García-Jaramillo M., Santero E., Govantes F. (2007) The LysR-type regulator AtzR binding site: DNA sequences involved in activation, repression and cyanuric acid-dependent repositioning. Mol. Microbiol. 66, 410–427 [DOI] [PubMed] [Google Scholar]
- 36. Monferrer D., Tralau T., Kertesz M. A., Dix I., Solà M., Usón I. (2010) Structural studies on the full-length LysR-type regulator TsaR from Comamonas testosteroni T-2 reveal a novel open conformation of the tetrameric LTTR fold. Mol. Microbiol. 75, 1199–1214 [DOI] [PubMed] [Google Scholar]
- 37. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 38. Loboda A. V., Krutchinsky A. N., Bromirski M., Ens W., Standing K. G. (2000) A tandem quadrupole/time-of-flight mass spectrometer with a matrix-assisted laser desorption/ionization source: design and performance. Rapid Commun. Mass Spectrom. 14, 1047–1057 [DOI] [PubMed] [Google Scholar]
- 39. Orr A., Ivanova V. S., Bonner W. M. (1995) “Waterbug” dialysis. BioTechniques 19, 204–206 [PubMed] [Google Scholar]
- 40. Donald L. J., Duckworth H. W., Standing K. G. (2006) in Cell Biology: A Laboratory Handbook (Celis J., ed) pp. 457–464, 3rd Ed., Elsevier Science Inc., San Diego [Google Scholar]
- 41. Kozlovski V. I., Donald L. J., Collado V. M., Spicer V., Loboda A. V., Chernushevich I. V., Ens W., Standing K. G. (2011) A TOF mass spectrometer for the study of noncovalent complexes. Int. J. Mass Spectrom. 10.1016/j.ijms.2011.08.009 [DOI] [Google Scholar]
- 42. Lössl P., Snijder J., Heck A. J. R. (2014) Boundaries of mass resolution in native mass spectrometry. J. Am. Soc. Mass Spectrom. 25, 906–917 [DOI] [PubMed] [Google Scholar]
- 43. Leslie A. G. W., Powell H. R. (2007) Processing diffraction data with mosflm. NATO Sci. Ser. 245, 41–51 [Google Scholar]
- 44. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D. Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 45. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 47. Patel T. R., Morris G. A., Zwolanek D., Keene D. R., Li J., Harding S. E., Koch M., Stetefeld J. (2010) Nano-structure of the laminin γ-1 short arm reveals an extended and curved multidomain assembly. Matrix Biol. 29, 565–572 [DOI] [PubMed] [Google Scholar]
- 48. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 49. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 10.1107/S0021889892001663 [DOI] [Google Scholar]
- 50. Patel T. R., Bernards C., Meier M., McEleney K., Winzor D. J., Koch M., Stetefeld J. (2014) Structural elucidation of full-length nidogen and the laminin-nidogen complex in solution. Matrix Biol. 33, 60–67 [DOI] [PubMed] [Google Scholar]
- 51. Meier M., Patel T. R., Booy E. P., Marushchak O., Okun N., Deo S., Howard R., McEleney K., Harding S. E., Stetefeld J., McKenna S. A. (2013) Binding of G-quadruplexes to the N-terminal recognition domain of the RNA helicase associated with AU-rich element (RHAU). J. Biol. Chem. 288, 35014–35027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 10.1107/S0021889809000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. García De La Torre J., Huertas M. L., Carrasco B. (2000) Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Serdyuk I. N., Zaccai N. R., Zaccai J. (2007) Methods in Molecular Biophysics: Structure, Dynamics, Function, pp. 21–37, Cambridge University Press, Cambridge, UK [Google Scholar]
- 56. Dzananovic E., Patel T. R., Deo S., McEleney K., Stetefeld J., McKenna S. A. (2013) Recognition of viral RNA stem-loops by the tandem double-stranded RNA binding domains of PKR. RNA 19, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laue T. M., Shah B. D., Ridgeway T. M., Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., Horton J. C., eds) pp. 90–125, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 58. Bishop R. E., Weiner J. H. (1993) Overproduction, solubilization, purification and DNA-binding properties of AmpR from Citrobacter freundii. Eur. J. Biochem. 213, 405–412 [DOI] [PubMed] [Google Scholar]
- 59. Ezezika O. C., Haddad S., Neidle E. L., Momany C. (2007) Oligomerization of BenM, a LysR-type transcriptional regulator: structural basis for the aggregation of proteins in this family. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 63, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ruangprasert A., Craven S. H., Neidle E. L., Momany C. (2010) Full-length structures of BenM and two variants reveal different oligomerization schemes for LysR-type transcriptional regulators. J. Mol. Biol. 404, 568–586 [DOI] [PubMed] [Google Scholar]
- 61. Juan C., Moyá B., Pérez J. L., Oliver A. (2006) Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level β-Lactam resistance involves three AmpD homologues. Antimicrob. Agents Chemother. 50, 1780–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Devesse L., Smirnova I., Lönneborg R., Kapp U., Brzezinski P., Leonard G. A., Dian C. (2011) Crystal structures of DntR inducer binding domains in complex with salicylate offer insights into the activation of LysR-type transcriptional regulators. Mol. Microbiol. 81, 354–367 [DOI] [PubMed] [Google Scholar]
- 63. Momany C., Neidle E. L. (2012) Defying stereotypes: the elusive search for a universal model of LysR-type regulation. Mol. Microbiol. 83, 453–456 [DOI] [PubMed] [Google Scholar]
- 64. Taylor J. L., De Silva R. S., Kovacikova G., Lin W., Taylor R. K., Skorupski K., Kull F. J. (2012) The crystal structure of AphB, a virulence gene activator from Vibrio cholerae, reveals residues that influence its response to oxygen and pH. Mol. Microbiol. 83, 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Muraoka S., Okumura R., Ogawa N., Nonaka T., Miyashita K., Senda T. (2003) Crystal structure of a full-length LysR-type transcriptional regulator, CbnR: unusual combination of two subunit forms and molecular bases for causing and changing DNA bend. J. Mol. Biol. 328, 555–566 [DOI] [PubMed] [Google Scholar]
- 66. Zhou X., Lou Z., Fu S., Yang A., Shen H., Li Z., Feng Y., Bartlam M., Wang H., Rao Z. (2010) Crystal structure of ArgP from Mycobacterium tuberculosis confirms two distinct conformations of full-length LysR transcriptional regulators and reveals its function in DNA binding and transcriptional regulation. J. Mol. Biol. 396, 1012–1024 [DOI] [PubMed] [Google Scholar]
- 67. Alanazi A. M., Neidle E. L., Momany C. (2013) The DNA-binding domain of BenM reveals the structural basis for the recognition of a T-N11-A sequence motif by LysR-type transcriptional regulators. Acta Crystallogr. D Biol. Crystallogr. 69, 1995–2007 [DOI] [PubMed] [Google Scholar]