

Background: FSP27 contributes to unilocular lipid droplet formation in adipocytes.
Results: Adipocyte-specific FSP27 disruption in mice produced small white adipose mass, hepatosteatosis, and insulin resistance upon high-fat diet feeding.
Conclusion: Adipose FSP27 plays a critical role in minimizing ectopic fat accumulation.
Significance: This mouse model is useful for understanding the significance of fat storage in adipose tissue.
Keywords: Adipose Tissue, Diabetes, Hepatocyte, Insulin Resistance, Lipid, Mouse
Abstract
White adipose tissue (WAT) functions as an energy reservoir where excess circulating fatty acids are transported to WAT, converted to triglycerides, and stored as unilocular lipid droplets. Fat-specific protein 27 (FSP27, CIDEC in humans) is a lipid-coating protein highly expressed in mature white adipocytes that contributes to unilocular lipid droplet formation. However, the influence of FSP27 in adipose tissue on whole-body energy homeostasis remains unclear. Mice with adipocyte-specific disruption of the Fsp27 gene (Fsp27ΔAd) were generated using an aP2-Cre transgene with the Cre/LoxP system. Upon high-fat diet feeding, Fsp27ΔAd mice were resistant to weight gain. In the small WAT of these mice, small adipocytes containing multilocular lipid droplets were dispersed. The expression levels of the genes associated with mitochondrial abundance and brown adipocyte identity were increased, and basal lipolytic activities were significantly augmented in adipocytes isolated from Fsp27ΔAd mice compared with the Fsp27F/F counterparts. The impaired fat-storing function in Fsp27ΔAd adipocytes and the resultant lipid overflow from WAT led to marked hepatosteatosis, dyslipidemia, and systemic insulin resistance in high-fat diet-treated Fsp27ΔAd mice. These results demonstrate a critical role for FSP27 in the storage of excess fat in WAT with minimizing ectopic fat accumulation that causes insulin-resistant diabetes and non-alcoholic fatty liver disease. This mouse model may be useful for understanding the significance of fat-storing properties of white adipocytes and the role of local FSP27 in whole-body metabolism and estimating the pathogenesis of human partial lipodystrophy caused by CIDEC mutations.
Introduction
White adipose tissue (WAT)4 functions as an energy reservoir. Excess circulating fatty acids (FA) are taken into WAT, converted to triglycerides (TG), and stored as unilocular lipid droplets. Lipid droplets in adipocytes are coated with several proteins, such as perilipin 1 (PLIN1) and fat-specific protein 27 (FSP27), to protect lipid droplets from various lipolytic stimuli (1). When energy demand exceeds energy supply, FA are released from WAT into the circulation by accelerated lipolysis, taken into peripheral tissues, and utilized as an efficient energy source. Therefore, WAT plays an essential role in the control of lipid fluxes and the maintenance of energy/nutrient homeostasis.
Dysfunction of WAT causes serious metabolic disruption in energy/nutrient homeostasis and may result in several pathological conditions. For example, obesity is defined by excessive lipid accumulation in adipocytes due to an imbalance between calorie intake and energy consumption. Hypertrophied adipocytes lead to local inflammation and ischemia of WAT and inhibit the production of adipokines, such as adiponectin, resulting in systemic insulin resistance (IR) (2, 3). Obesity and IR often coexist and are closely linked to the pathogenesis of type 2 diabetes, non-alcoholic fatty liver disease/steatohepatitis (NAFLD/NASH), dyslipidemia, atherosclerosis, and cancer. Lipodystrophy is another type of adipocyte dysfunction that is characterized by the reductions in mature adipocyte number. Impaired fat storage capacity in WAT and the ensuing lipid overflow from WAT may enhance fat deposition in extra-adipose tissues, such as liver and muscle, leading to disruption of insulin action in these organs (4, 5). It is intriguing that lipodystrophy causes similar metabolic consequences to obesity, i.e. systemic IR, type 2 diabetes, dyslipidemia, and NAFLD/NASH (4–6).
Fsp27, also known as cell death-inducing DNA fragmentation factor-α-like effector c (CIDEC) in humans, is exclusively expressed in WAT and has an essential role in the formation of unilocular lipid droplets in adipocytes in cooperation with perilipin 1 (7–10). Forced expression of FSP27 in 3T3-L1 adipocytes causes formation of unilocular lipid droplets (8, 9). Knockdown of FSP27 expression in well differentiated adipocytes enhances lipolysis and increases the number of mitochondria and the expression of genes associated with brown adipocyte identity (8–11), resulting in formation of small multilocular lipid droplets. Disruption of the gene encoding FSP27 protected mice from obesity, hepatosteatosis, and IR induced by high-fat diet (HFD) that was likely associated with increased energy expenditure (11, 12). However, FSP27 can be induced in steatotic livers and forced expression of FSP27 in primary hepatocytes caused storage of lipid droplets mainly due to depressed mitochondrial β-oxidation activity and TG turnover (13). Acute disruption of FSP27 in ob/ob mouse livers using an adenovirus expressing a short hairpin RNA of Fsp27 moderated hepatosteatosis (13). These findings imply the possible contribution of FSP27 disruption in extra-adipose tissues on whole-body metabolism in Fsp27-null mice. However, the effect of adipose FSP27 disruption on overall lipid/glucose metabolism has not been precisely defined.
To examine the effect of Fsp27 disruption in adipocytes on whole-body energy homeostasis, mice with adipocyte-specific targeted disruption of the Fsp27 gene (Fsp27ΔAd) were generated using an aP2-Cre transgene with the Cre/LoxP system and treated with HFD. The Fsp27ΔAd mice were resistant to HFD-induced weight gain but demonstrated marked hepatosteatosis, high levels of serum TG and non-esterified FA (NEFA), and IR. This was likely a consequence of enhanced basal lipolytic activities and the inability to hold excess fat in Fsp27ΔAd WAT and the resultant lipid overflow from WAT upon HFD. These findings reveal a crucial role for FSP27 in white adipocytes under dietary fat overload to store excess lipids in WAT and prevent ectopic fat deposition, which is one of the key determinants of systemic IR development.
EXPERIMENTAL PROCEDURES
Generation of Fsp27ΔAd Mice and Treatment
The Fsp27-floxed mouse line (Fsp27F/F) was generated as presented in Fig. 1A. In brief, a 11.5-kbp DNA fragment containing the mouse Fsp27 gene was subcloned into PL253 vector. The first loxP site was introduced in the first intron of the Fsp27 gene. A neomycin (Neo) cassette flanked by two FRT sites and one loxP site was inserted into the third intron of the Fsp27 gene using the PL451 vector that contains this cassette. The final targeting vector was purified and electroporated into C57BL/6J x 129/SvJae hybrid ES cells (14). After screening with G418, the correctly targeted ES clones were confirmed by Southern blotting (Fig. 1B) and injected into blastocysts obtained from C57BL/6N mice. For Southern blotting, genomic DNA was isolated from a tail biopsy and digested with SspI. Digested DNA fragments were run on a 0.4% agarose gel overnight and transferred to a nylon membrane. The probe was amplified by PCR with primer set 5′-AAAGCCACTTGCTTCAAGC-3′ and 5′-CCAGGGCTAAGTGTTAGTCC-3′. 32P-Labeled (PerkinElmer Life Sciences) probes were prepared with Ready-To-Go DNA Labeling Beads (−dCTP) (GE Healthcare). Signals were detected using Strom 840 (GE Healthcare). The offspring of Fsp27-targeted chimeric mice crossed with wild-type C57BL/6N mice. After that, the mice (Flox-Neo) were crossed to β-actin-driven Flp transgenic mice used to remove the Neo cassette to produce the Fsp27-floxed allele, and the allele was confirmed by PCR analysis (Fig. 1C). To separately detect Fsp27-flox and flox-neo genes, PCR was carried out with primer sets 5′-AAAGTCGACGGTTCCTTTCTGATGTCAGC-3′, 5′-GCTGGACGTAAACTCCTCTTCAGACC-3′, and 5′-AAAGCGGCCGCAAGCAAAATTAGAAGGGGGTAGG-3′. The PCR produced for wild-type, Fsp27-flox, and flox-neo genes were 594, 639, and 307 bp, respectively. The mice used in this study were backcrossed to C57BL/6N for six generations. To generate mice in which Fsp27 gene is deleted specifically in adipose tissue, the Cre/LoxP system was used. Homozygous Fsp27F/F mice were crossed with transgenic mice expressing Cre recombinase under the control of aP2 promoter (15), and subsequently Fsp27ΔAd and Fsp27F/F mice were interbred. Mice were housed in a specific pathogen-free environment controlled for temperature and light (25 °C, 12-h light/dark cycle) and maintained with NIH31 regular chow (10% kcal from fat) and tap water ad libitum. Male mice were used for the experiments in the present study unless specifically stated. To induce obesity, high-fat diet F3282 (60% kcal from fat; 5.49 metabolizable kcal/g, BioServ, Frenchtown, NJ) was given from 8 weeks of age. Mouse body weight (BW) was measured every week. Daily food intake under the regular housing condition was assessed by maintaining each mouse into the individual cage for 3 days before killing. At the indicated time point, mice were killed after measurement of BW and body compositions using Echo3-in-1 nuclear magnetic resonance (NMR) analyzer (Echo Medical Systems, Houston TX) and after a 6-h fasting period. Blood was collected using Serum Separator Tubes (BD Biosciences) and centrifuged for 10 min at 8000 rpm at 4 °C to obtain serum. The WAT, interscapular brown adipose tissue (BAT), liver, and kidney were weighed immediately after isolation. Tissues were divided into two parts; one part was immediately frozen in liquid nitrogen, and the other was soaked into 10% neutral formalin for histological evaluation. All animal studies were carried out in accordance with Institute of Laboratory Animal Resources guidelines and approved by the National Cancer Institute Animal Care and Use Committee.
FIGURE 1.
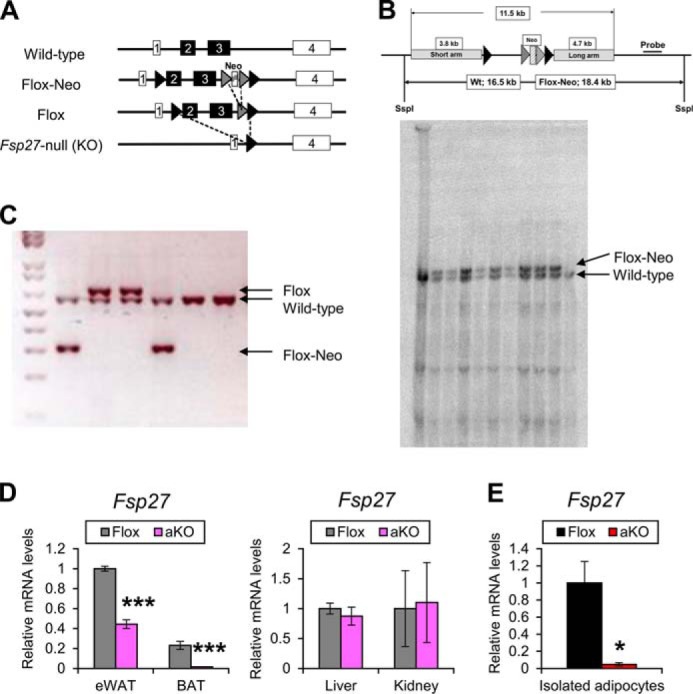
Generation of Fsp27ΔAd mice. A, strategies to generate Fsp27F/F mice. B, Southern blotting for Fsp27F/F gene detection. C, PCR for Fsp27-flox and flox-neo genes. D, Fsp27 expression levels in eWAT, BAT, liver, and kidney of 11-week-old Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice fed a normal diet (n = 4–5/group). E, Fsp27 expression levels in adipocytes isolated from WAT of aKO and Flox mice treated with HFD for 20 weeks (n = 3/group). Data were analyzed using the two-tailed Student's t test. *, p < 0.05; ***, p < 0.001 compared with Flox mice. All values represent the mean ± S.E.
Quantitative Polymerase Chain Reaction (qPCR) Analysis
RNA was extracted using TRIzol reagent (Invitrogen). qPCR was performed using cDNA generated from 1 μg of total RNA with the SuperScript II Reverse Transcriptase kit (Invitrogen). The primer sequences designed by qPrimerDepot were listed in Table 1. qPCR reactions were carried out using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA) in an ABI Prism 7900HT Sequence Detection System. Values were quantified using the comparative CT method and normalized to 18 S ribosomal RNA (16).
TABLE 1.
Primer pairs used for qPCR
F, forward; R, reverse.
| Gene | Primer | Primer Sequence (5′-3′) |
|---|---|---|
| 18 S | F | ATTGGAGCTGGAATTACCGC |
| R | CGGCTACCACATCCAAGGAA | |
| Acadl | F | TCTTGCGATCAGCTCTTTCA |
| R | GGTACATGTGGGAGTACCCG | |
| Acadm | F | AGCTCTAGACGAAGCCACGA |
| R | GCGAGCAGAAATGAAACTCC | |
| Acads | F | AGCTGGACAGGGAGCATCT |
| R | ACTCAGCTCCTCTGGCACAT | |
| Acaca | F | TGGTGCAGAGGTACCGAAGTG |
| R | CGTAGTGGCCGTTCTGAAACT | |
| Acsl1 | F | CTTCCAACCAACACCCTCAT |
| R | ACCATCAGTGGTACCCGCTA | |
| Apoa4 | F | ACCCAGCTAAGCAACAATGC |
| R | TAGCATCCCCAAGTTTGTCC | |
| Apob | F | AAGCACCTCCGAAAGTACGTG |
| R | CTCCAGCTCTACCTTACAGTTGA | |
| Ccl2 | F | CCTGCTGTTCACAGTTGCC |
| R | ATTGGGATCATCTTGCTGGT | |
| Cd36 | F | GCGACATGATTAATGGCACA |
| R | CCTGCAAATGTCAGAGGAAA | |
| Cd137 | F | CCCCCACATATTCAAGCAAC |
| R | TAGCCTCCTCCTCCTCCTTC | |
| Ces3 | F | TGGTATTTGGTGTCCCATCA |
| R | GCTTGGGCGATACTCAAACT | |
| Cidea | F | CAGTGATTTAAGAGACGCGG |
| R | TCTGCAATCCCATGAATGTC | |
| Cideb | F | GACCCTTCCGTGTCTGTGAT |
| R | ACGTAGCAGCAAGGTCTCCA | |
| Cox4i2 | F | GGCTGCGTCTTCTTCTTCAT |
| R | CGTCAGGGTGACAACCTTCT | |
| Cox5b | F | CGTCCATCAGCAACAAGAGA |
| R | ATCGCTGACTCTCGCCTTT | |
| Cox8b | F | TGCGAAGTTCACAGTGGTTC |
| R | AGCCAACGACTATGGCTGAG | |
| Cpt1a | F | GATGAACTTCTTCTTCCAGGAGTGC |
| R | ATGGCAGAGGCTCACCAAGC | |
| Creb3l3 | F | ACGGAAGGTGGATGTGAGAA |
| R | GTTGGATCTCCAGCAAAACC | |
| Cytc | F | TGTGCTACACGGAGGAAGAAGC |
| R | TCAGGGTTAGGGTATGGTTTTGG | |
| Dgat1 | F | GACGGCTACTGGGATCTGA |
| R | TCACCACACACCAATTCAGG | |
| Dgat2 | F | CGCAGCGAAAACAAGAATAA |
| R | GAAGATGTCTTGGAGGGCTG | |
| Dio2 | F | ATGCTGACCTCAGAAGGGCT |
| R | ACACTGGAATTGGGAGCATC | |
| Ebf2 | F | CTTCAGACCCGGTCATCATC |
| R | CCTCATGTCCCTTGGGTTTC | |
| Elovl6 | F | CCAATGGATGCAGGAAAACT |
| R | AACTTGGCTCGCTTGTTCAT | |
| Emr1 | F | GGATGTACAGATGGGGGATG |
| R | CATAAGCTGGGCAAGTGGTA | |
| Fabp1 | F | TGCAGAGCCAGGAGAACTTT |
| R | GATTTCTGACACCCCCTTGA | |
| Fasn | F | AAGTTGCCCGAGTCAGAGAACC |
| R | ATCCATAGAGCCCAGCCTTCCATC | |
| Fatp5 | F | CCAGTGTGCTGATTGTGGAT |
| R | GGCCAAGGTAGAAGCAGTGA | |
| Foxc2 | F | AGTGCGGATTTGTAACCAGG |
| R | ACAGTTGGGCAAGACGAAAC | |
| Fsp27 | F | GCCACGCGGTATTGCCAGGA |
| R | GGGTCTCCCGGCTGGGCTTA | |
| Fsp27b | F | GTGACCACAGCTTGGGTCGGA |
| R | GGGTCTCCCGGCTGGGCTTA | |
| Lipe | F | CCTCCAAGCAGGGCAAAGA |
| R | GCGTAAATCCATGCTGTGTGA | |
| Lxra | F | TGAGAGCATCACCTTCCTCA |
| R | TGGAGAACTCAAAGATGGGG | |
| Mttp | F | AGGTGCTGGGGGTCAGTT |
| R | GGCAAAAGCCCTGGTCTCTT | |
| Nrf1 | F | TCTATCCGAAAGAGACAGCAGACAC |
| R | TTGGGTTTGGAGGGTGAGATG | |
| Plin1 | F | TGAAGCAGGGCCACTCTC |
| R | GACACCACCTGCATGGCT | |
| Plin2 | F | CCCGTATTTGAGATCCGTGT |
| R | CAATTTGTGGCTCCAGCTTC | |
| Plin3 | F | TCCAAGCTGGACTTGGTAGG |
| R | GCGCATCTGTACCATTGCT | |
| Pnpla2 | F | CCACTCACATCTACGGAGCC |
| R | TAATGTTGGCACCTGCTTCA | |
| Pnpla3 | F | CTGTCCTACATCATGCTGCC |
| R | TCCTGGATATCAGGGAGCC | |
| Ppara | F | GCGGCCCCATACAGGAGAGCAG |
| R | CTAACCTTGGGCCACACCTTGACT | |
| Pparg1 | F | GGGGCCCGTCGCACTCA |
| R | TGGTTCACCGCTTCTTTCA | |
| Pparg2 | F | TCTGGGAGATTCTCCTGTTGA |
| R | GGTGGGCCAGAATGGCATCT | |
| Ppargc1a | F | TGTAGCGACCAATCGGAAAT |
| R | TGAGGACCGCTAGCAAGTTT | |
| Ppargc1b | F | GCTCTCGTCCTTCTTCCTCA |
| R | GAGGTCAAGCTCTGGCAAGT | |
| Scd1 | F | ACGCCGACCCTCACAATTC |
| R | CAGTTTTCCGCCCTTCTCTTT | |
| Srebf1 | F | GGAGCCATGGATTGCACATT |
| R | GCTTCCAGAGAGGAGGCCAG | |
| Tfam | F | GGAATGTGGAGCGTGCTAAAAG |
| R | CAGACAAGACTGATAGACGAGGGG | |
| Tmem | F | GCTCACCCTCAAGTTCAAGC |
| R | ATGGTGCATTTCAAGAAGCC | |
| Tnf | F | CCACCACGCTCTTCTGTCTAC |
| R | AGGGTCTGGGCCATAGAACT | |
| Ucp1 | F | TCTCTGCCAGGACAGTACCC |
| R | AGAAGCCACAAACCCTTTGA |
Measurement of Biochemical Parameters
Serum lipids were measured using kits purchased from Wako (Wako Chemicals USA, Inc., Richmond, VA). Serum levels of insulin and alanine aminotransferase were determined using ultrasensitive mouse insulin the ELISA kit (Crystal Chem Inc., Downers Grove, IL) and the alanine aminotransferase assay kit (Catachem, Bridgeport, CT), respectively. Hepatic TG contents were assayed as described previously (16).
Histological Analysis
Formalin-fixed tissues were dehydrated by graded ethanol and xylene and embedded in paraffin. Sections (4 μm thick) were stained with H&E (17). At least three discontinuous sections were evaluated in the same samples. Immunohistochemical staining against F4/80 was performed as described previously (3).
Immunoblot Analysis
Immunoblot analysis was carried out as described elsewhere (18). Briefly, ∼40 mg of epididymal WAT (eWAT) or liver was homogenized in radioimmune precipitation assay buffer containing a proteinase inhibitor mixture. The homogenates were centrifuged at 18,000 × g for 5 min at 4 °C, and cytosolic extracts (20 μg of protein) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% bovine serum albumin or skim milk and incubated overnight with primary antibodies against hormone-sensitive lipase (HSL; Cell Signaling Technology, Inc., Danvers, MA, #4107, 1:1000 dilution), phosphorylated HSL (Cell Signaling, #4139, 1:1000 dilution), adipose TG lipase (ATGL; Cell Signaling, #2439, 1:1000 dilution), and phosphorylated ATGL (Abcam, Cambridge, MA, #135093, 1:1000 dilution). The antibody against β-actin (Abcam, #8227, 1:10000 dilution) was used as the loading control. After washing, the blots were incubated with peroxidase-conjugated goat anti-rabbit IgG (Cell Signaling, #7074, 1:3000 dilution) and scanned.
Assessment of Glucose Metabolism
For glucose tolerance tests, mice were injected intraperitoneally with 1 g/kg BW of glucose dissolved in 0.9% sterile saline (K·D Medical, Columbia, MD) using a 27-gauge needle after 18 h of fasting. For the insulin tolerance test, mice were injected intraperitoneally with 1 unit/kg BW of insulin (Humalin R; Lilly) in the same manner after a 6-h fasting. Blood glucose concentrations at 0, 15, 30, 60, 90, and 120 min after the injection were measured using Contour blood glucose meter (Bayer HealthCare LLC, Mishawaka, IN). Hyperinsulinemic-euglycemic clamp assay was performed described previously (3). Primed continuous infusion of d-[3-3H]glucose was used: 2.5-μCi bolus, 0.05 μCi/min during the basal condition and 0.1 μCi/min during the clamp study. Humalin R was infused as a bolus of 18 milliunits/kg over a period of 3 min followed by continuous insulin infusion at the rate of 2.5 milliunits/kg/min to raise plasma insulin concentration to 4 ng/ml.
Total Energy Expenditure
Total free-living energy expenditure was calculated from the caloric intake, change in BW, and change in body composition using an energy balance technique (19). Mice were caged individually and fed HFD (5.49 kcal/g diet) for 8 weeks using Rodent CAFÉ feeders (OYC Americas, Inc., Andover, MA). Food intake was measured twice a week. Body composition was measured at weeks 2 and 8 of feeding. In the body composition, caloric equivalents (fat mass, 9.4 kcal/g; fat-free mass, 1.0 kcal/g) were used in calculations. The increase in body kcal content was subtracted from the total metabolizable energy intake during weeks 3–8, yielding the total energy expenditure, which was divided by the experiment duration.
Indirect Calorimetry
Indirect calorimetry was performed on 8-week-old mice before administration of HFD and on the same mice after 8 weeks on HFD using a 12-chamber Environment Controlled CLAMS (Columbus Instruments, Columbus, OH). After an 48-h acclimatization, mice were monitored for 24 h at 22 °C and then for the following 24 h at thermoneutrality (30 °C) for recording data. To measure thermogenic capacity of HFD-fed mice, CL316243, a selective β3-adrenoreceptor agonist (100 μg/kg in saline, intraperitoneally, Sigma) was administered to mice adapted to 30 °C for 24 h. During testing, food and water were provided ad libitum.
Isolation of White Adipocytes from Mice
Epididymal and inguinal fat pads were immediately isolated, minced by scissors, and incubated in Krebs-Ringer bicarbonate HEPES buffer (120 mm NaCl, 30 mm HEPES, 10 mm NaHCO3, 4 mm KH2PO4, 1 mm MgSO4 and 1 mm CaCl2, pH 7.4) containing FA-free bovine serum albumin (20 mg/ml, Sigma) and collagenase I (2 mg/ml, Sigma) with gentle shaking (150 rpm) at 37 °C for 2 h. Subsequently, the floating layer was filtered through nylon mesh (125-μm pore size, Component Supply Co., Fort Meade, FL) into 60 ml of Separatory (Corning, Corning, NY). The floating cell layer was washed twice with Krebs-Ringer bicarbonate HEPES buffer to eliminate collagenase. Isolated adipocytes was resuspended with Krebs-Ringer bicarbonate HEPES buffer at a density of 10% (v/v) and immediately subjected to ex vivo lipolysis assay. RNA was extracted from isolated adipocytes using QIAzol and the RNeasy Mini kit (Qiagen, Valencia, CA).
Ex Vivo Lipolysis Assay
Freshly isolated adipocytes were used for this assay. Two-hundred μl of Krebs-Ringer bicarbonate HEPES buffer with and without CL316243 were prepared in 5-ml round-bottom glass tube in a 37 °C water bath, and 100 μl of 10% adipocyte suspension was added to each tube and incubated with gentle shaking (100 rpm) at 37 °C for 30 min and 2 h. The infranatant in each tube was collected, and glycerol concentration was determined using Free Glycerol Reagent (Sigma).
Statistical Analysis
Data were expressed as the mean ± S.E., and statistical analyses were performed using the two-tailed Student's t test. A p value of <0.05 was considered to be statistically significant.
RESULTS
Generation of Fsp27ΔAd Mice
To generate mice in which Fsp27 gene is disrupted specifically in adipose tissue, the Cre/LoxP system was used. Generation of the Fsp27F/F is shown in Fig. 1, A–C. Homozygous Fsp27F/F mice were crossed with transgenic mice expressing Cre recombinase under the control of adipose-specific aP2 promoter (15). Because aP2 and FSP27 are expressed in the late stage of adipocyte maturation (8, 9, 11, 12), this strategy was expected to lead to recombination of the Fsp27 allele after completing normal differentiation from pre-adipocytes to functional adipocytes, i.e. Fsp27 disruption in mature adipocytes. qPCR analysis revealed significant decreases in the Fsp27 mRNA levels in eWAT and BAT, but not in other tissues such as liver and kidney, in 11-week-old Fsp27ΔAd mice (Fig. 1D). The Fsp27 mRNA levels were further decreased in eWAT of 28-week-old Fsp27ΔAd mice (∼20% of Fsp27F/F mice) (Fig. 2A). Furthermore, Fsp27 mRNA levels in white adipocytes isolated from Fsp27ΔAd mice were <5% that of those from Fsp27F/F mice (Fig. 1E). This mouse line was considered to be a useful model to determine the role of adipose FSP27 in whole-body metabolism.
FIGURE 2.
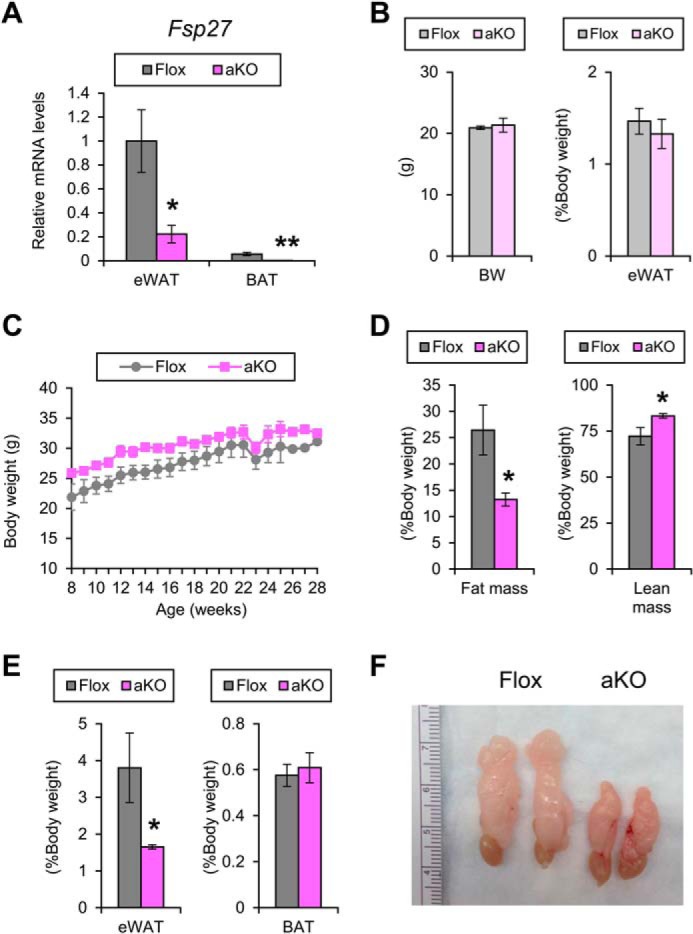
Small WAT mass in Fsp27ΔAd mice. A, Fsp27 mRNA levels in the eWAT and BAT of 28-week-old Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice fed a normal diet. B, body weight and eWAT weight of 11-week-old aKO and Flox mice fed a normal diet. C, body weight changes of aKO and Flox mice fed a normal diet. D, E, and F, body composition measured by NMR (D), weight of eWAT and interscapular BAT (E), and gross appearance of eWAT (F) in 28-week-old aKO and Flox mice fed a normal diet. Data were analyzed using the two-tailed Student's t test. *, p < 0.05; **, p < 0.01, compared with Flox mice. All values represent the mean ± S.E. n = 3–4/group.
Emergence of Small Adipocytes Containing Multilocular Lipid Droplets in WAT of Fsp27ΔAd Mice
At the age of 11 weeks, Fsp27ΔAd and Fsp27F/F mice fed a chow diet had similar BW and eWAT weight (Fig. 2B) and exhibited similar body weight gain up to 28 weeks of age (Fig. 2C). Food intake measured at the age of 9 and 28 weeks was also similar between the groups (4.1 ± 0.4 g/day versus 4.0 ± 0.5 g/day, p = 0.65 and 2.7 ± 1.1 g/day versus 3.3 ± 0.6 g/day, p = 0.43 in Fsp27F/F and Fsp27ΔAd mice, respectively). However, at the age of 28 weeks, fat mass/BW ratio, as determined by use of NMR imaging, and eWAT weight were significantly decreased, and lean mass/BW ratio was increased in Fsp27ΔAd mice (Fig. 2, D and E). The color of eWAT appeared slightly darker in the Fsp27ΔAd mice than that of Fsp27F/F mice (Fig. 2F). In accordance with smaller eWAT mass in the Fsp27ΔAd mice, microscopic examination revealed a unique mosaic adipocyte distribution, and small adipocytes containing multilocular lipid droplets were dispersed among typical adipocytes having unilocular lipid droplets (Fig. 3A). The percentage of the small adipocytes relative to total adipocytes was 76 ± 4.5% in eWAT of Fsp27ΔAd mice (n = 3). These bizarre small adipocytes were also detected in other WAT, such as subcutaneous adipose tissue and mesentery (Fig. 4), and resembled adipocytes found in Fsp27-null mice (11, 12). Apparent infiltration of F4/80-positive cells was not seen around adipocytes in the eWAT of Fsp27ΔAd mice, and qPCR analysis demonstrated no increases in the expression of genes associated with inflammation, such as the EGF-like module containing mucin-like hormone receptor-like sequence 1 (Emr1, encoding F4/80), tumor necrosis factor α (Tnf), and chemokine (C-C motif) ligand 2 (Ccl2) (Fig. 3B). There were no significant differences in BAT weight (Fig. 2E). Microscopically, adipocytes with small lipid droplets were seen in the BAT of both mouse lines, but the number of these cells appeared to be fewer, and the size of lipid droplets looked bigger in Fsp27ΔAd mice (Fig. 3A). No histopathological abnormalities were obvious in the liver, kidney, quadriceps, and pancreas of Fsp27ΔAd mice (Fig. 4). These results indicate that adipocyte-specific Fsp27 disruption reduces WAT mass and induces multilocularization of lipid droplets in white adipocytes.
FIGURE 3.
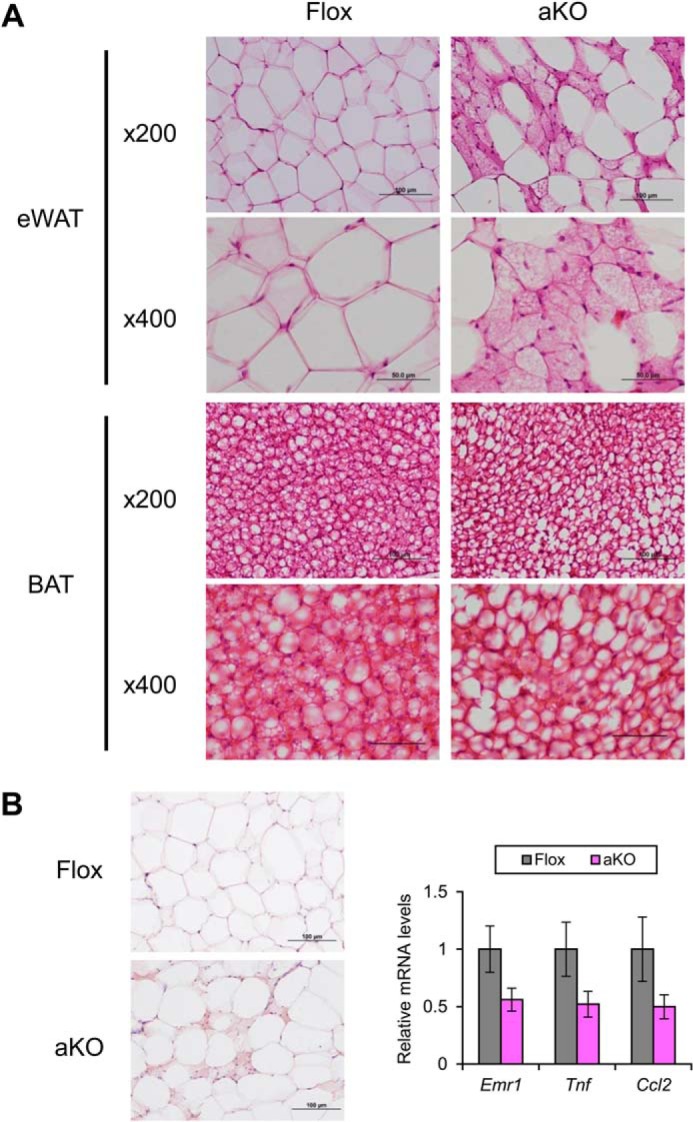
Emergence of small adipocytes containing multilocular lipid droplets in Fsp27ΔAd mice fed a normal diet. A, histological appearance of eWAT and BAT in 28-week-old Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice stained with H&E. Scale bars: 100 μm (×200 magnification) and 50 μm (×400 magnification), respectively. B, immunohistochemical staining of eWAT against F4/80 and qPCR analysis of genes associated with inflammation. Scale bars: 100 μm. All values represent the mean ± S.E. n = 3–4/group.
FIGURE 4.
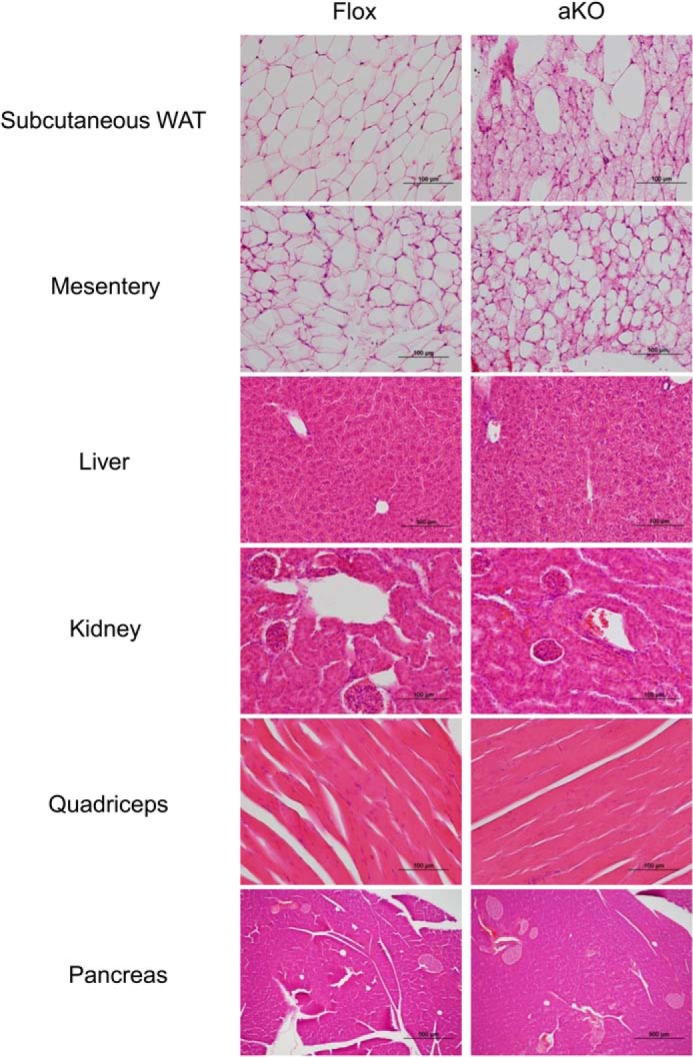
Histological findings of other tissues in 28-week-old Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice fed a normal diet. Scale bars: 100 μm (other than pancreas) and 500 μm (pancreas), respectively.
Protection from HFD-induced Obesity in Fsp27ΔAd Mice
To investigate the role of adipose FSP27 in HFD-induced obesity, male Fsp27ΔAd and Fsp27F/F mice were treated with HFD for 17 weeks. At the starting point of HFD treatment (8 weeks of age), there were no significant differences in BW between the genotypes (Fig. 5A). The BW was gradually increased in Fsp27F/F mice upon HFD feeding. However, in Fsp27ΔAd mice, increases in BW plateaued around 8 weeks after starting the HFD. Consequently, significant differences in BW appeared between the two groups from 9 weeks after commencing the HFD (Fig. 5A). The mean changes in BW at the end point were 210 ± 9.8% and 170 ± 9.3% in Fsp27F/F and Fsp27ΔAd mice (p < 0.0001), respectively. NMR measurement at 17 weeks of HFD feeding revealed significant decreases in fat mass and fat mass/BW ratio in the Fsp27ΔAd mice (Fig. 5B), but there were no differences in the absolute lean mass between Fsp27ΔAd and Fsp27F/F mice (28 ± 2.9 g versus 27 ± 1.5 g, p = 0.34). In agreement with the NMR results, gross appearance after necropsy revealed smaller eWAT compared with their counterparts (Fig. 5, C and D). The color of eWAT in HFD-treated Fsp27ΔAd mice was less transparent and darker than that of Fsp27F/F mice (Fig. 5C). In another cohort of 30-week HFD-fed mice, the weight of other WAT deposits, such as inguinal, retroperitoneal, perirenal, and mesenteric WAT, was also significantly smaller in the Fsp27ΔAd mice (Fig. 5E). The BAT weight was greater in the Fsp27ΔAd mice after a 17-week HFD treatment but not after a 30-week treatment (Fig. 5, D and E).
FIGURE 5.
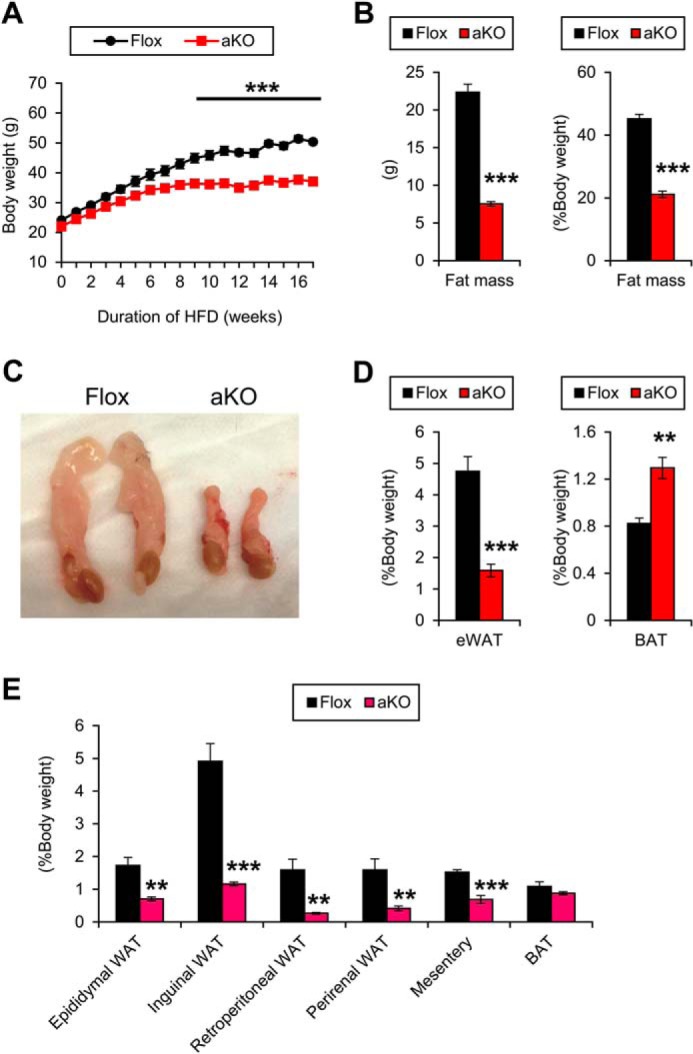
Protection from HFD-induced obesity in Fsp27ΔAd mice. A, body weight changes of Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice during 17-week HFD feeding. B, body composition measured by NMR after 17-week HFD feeding. C, gross appearance of eWAT. D, Weight of eWAT and interscapular BAT. E, weight of other white adipose deposits in aKO and Flox mice after a 30-week HFD treatment. Data were analyzed using the two-tailed Student's t test. **, p < 0.01; ***, p < 0.001 compared with Flox mice. All values demonstrate the mean ± S.E. n = 6–8/group.
Microscopic evaluation of eWAT and mesentery detected emergence of a lot of small adipocytes containing multilocular lipid droplets showing a mosaic appearance of adipocyte arrangement in Fsp27ΔAd mice (Fig. 6A). The percentage of the small adipocytes relative to total adipocytes was 79 ± 2.8% and 85 ± 3.8% in eWAT and mesentery, respectively (n = 4–7). Although typical unilocular adipocytes were bigger in HFD-fed Fsp27F/F mice compared with normal diet-fed Fsp27F/F mice, the size of atypical adipocytes with multilocular lipid droplets in Fsp27ΔAd mice was similar between normal diet and HFD feeding (Figs. 3A, 4, and 6A), suggesting an inability to enlarge the size of adipocytes and lipid droplets by fat overload in Fsp27-disrupted adipocytes. The appearance frequency of F4/80-positive cells around adipocytes was lesser, and qPCR analysis revealed significant reductions in inflammation-related genes in the eWAT of HFD-treated Fsp27ΔAd mice (Fig. 6B). Similar morphological changes in adipocytes were also found in subcutaneous and perirenal WATs (Fig. 7). These results clearly demonstrate that Fsp27ΔAd mice are protected from HFD-induced BW gain and have small WAT that contains small adipocytes with multilocular fat droplets.
FIGURE 6.
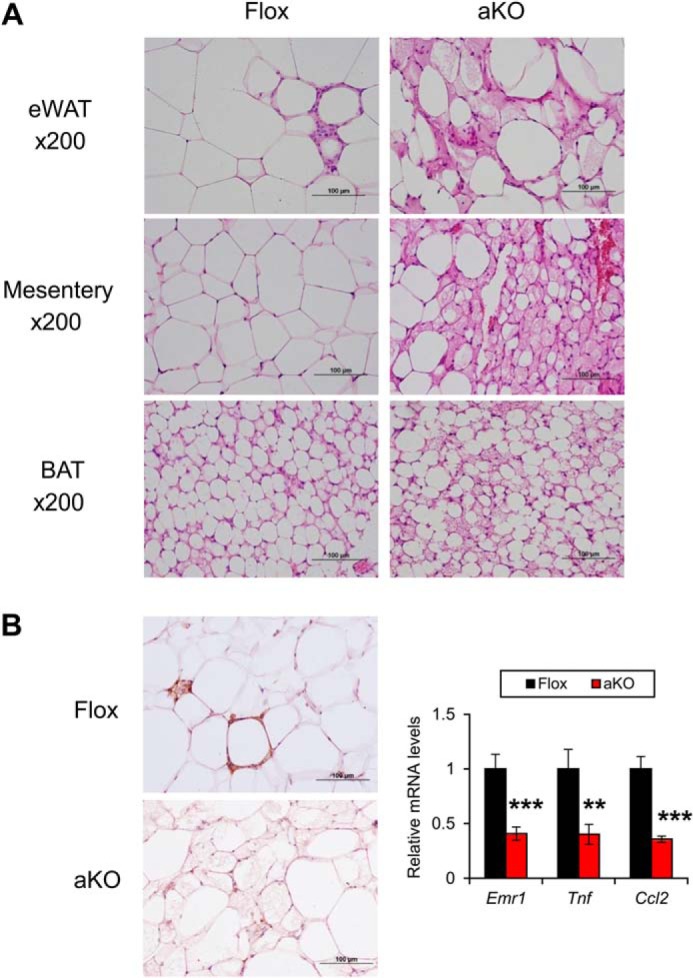
Emergence of small adipocytes containing multilocular lipid droplets in Fsp27ΔAd mice treated with HFD for 17 weeks. A, histological appearance of eWAT, mesentery, and BAT of 17-week HFD-treated Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice stained with H&E. B, immunohistochemical staining of eWAT against F4/80 and qPCR analysis of genes associated with inflammation. Scale bars: 100 μm. Data were analyzed using the two-tailed Student's t test. **, p < 0.01; ***, p < 0.001 compared with Flox mice. All values demonstrate the mean ± S.E. n = 6–8/group.
FIGURE 7.
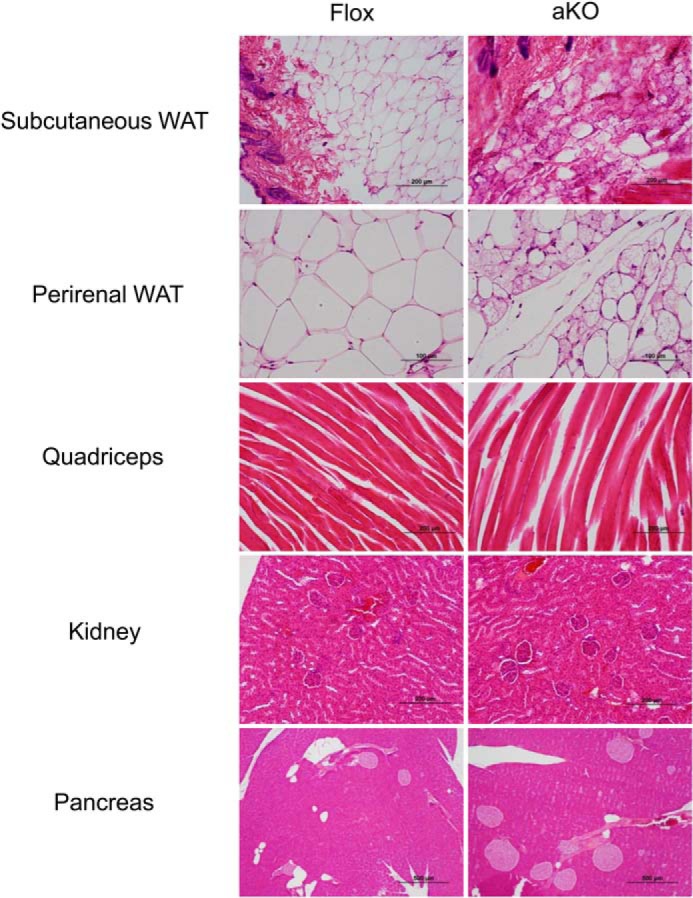
Histological findings of other tissues in Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice treated with HFD for 17 weeks. Scale bars: 100 μm (perirenal WAT), 200 μm (subcutaneous WAT, quadriceps, and kidney), and 500 μm (pancreas), respectively.
Increased Energy Expenditure in Fsp27ΔAd Mice
To study the mechanism of resistance to HFD-induced obesity in Fsp27ΔAd mice, energy balance was analyzed in a separate cohort of individually caged Fsp27ΔAd and Fsp27F/F mice during 8 weeks of high fat feeding. At the initiation of the experiment, Fsp27ΔAd and Fsp27F/F mice had similar body weights, body compositions, and food intake. Energy expenditure and respiratory exchange ratio (RER), as detected by indirect calorimetry, were also comparable between the groups (data not shown). During 8 weeks of HFD treatment, both groups consumed similar amounts of diet; however, Fsp27ΔAd gained significantly less fat mass and more lean mass compared with controls (Table 2). Total energy expenditure calculated using the energy balance technique (19) during the last 6 weeks of HFD feeding was 12% higher in Fsp27ΔAd compared with Fsp27F/F mice (15.1 ± 0.5 versus 13.5 ± 0.3, p = 0.045). Indirect calorimetry performed on mice fed a HFD for 8 weeks revealed significantly increased resting and total whole-body oxygen consumption (adjusted to body weight0.75), reduced RER, and increased activity in Fsp27ΔAd mice (Table 2).
TABLE 2.
Comparison of body composition and metabolic parameters between Fsp27ΔAd and Fsp27F/F mice after 8 weeks of HFD feeding
VO2, whole-body oxygen consumption rate; TEE, total energy expenditure. Indirect calorimetry was performed at thermoneutral temperature. Data are expressed as the mean ± S.E., and statistical analyses were performed by means of the Student's t test.
| Measurements | Fsp27F/F (n = 4) | Fsp27ΔAd (n = 5) |
|---|---|---|
| Body composition by NMR analyzer | ||
| Body mass (g) | 36.2 ± 2.5 | 33.4 ± 2.8 |
| Fat mass (g) | 11.6 ± 1.7 | 5.6 ± 0.8a |
| Lean mass (g) | 23.8 ± 0.8 | 26.7 ± 2.0 |
| Fat mass/body mass (%) | 31.5 ± 2.8 | 16.5 ± 1.2b |
| Lean mass/body mass (%) | 66.3 ± 2.7 | 80.2 ± 1.3b |
| Metabolic parameters by indirect calorimetry | ||
| Resting | ||
| VO2 (ml/g0.75/h) | 3.21 ± 0.09 | 3.85 ± 0.11b |
| RER | 0.85 ± 0.00 | 0.80 ± 0.00c |
| REE (kcal/h) | 0.23 ± 0.01 | 0.26 ± 0.02 |
| Total | ||
| VO2 (ml/g0.75/h) | 3.98 ± 0.08 | 4.91 ± 0.13c |
| RER | 0.86 ± 0.00 | 0.82 ± 0.01b |
| TEE (kcal/h) | 0.29 ± 0.02 | 0.33 ± 0.02 |
| Total activity (beam break/min) | 175 ± 4.46 | 236 ± 19.58a |
| Ambulatory activity (beam brake/min) | 69.0 ± 4.01 | 115 ± 8.79b |
| Total food intake (kcal/day) | 11.9 ± 0.90 | 11.5 ± 1.50 |
| TEE (kcal/mouse) | 7.00 ± 0.38 | 8.00 ± 0.54 |
a p < 0.05 compared with Fsp27F/F mice.
b p < 0.01, compared with Fsp27F/F mice.
c p < 0.001 compared with Fsp27F/F mice.
To test whether loss of FSP27 in adipose tissue affected thermogenic capacity, mice were treated with an acute dose of the selective β3-adrenergic agonist, CL316243. Serum NEFA concentrations were similarly increased after CL316243 injection in HFD-treated Fsp27ΔAd and Fsp27F/F mice (Fig. 8A). Both groups showed comparable reduction in RERs in response to CL316243, indicating stimulating of lipid oxidation (Fig. 8B); however, Fsp27ΔAd mice had a significantly higher increase in oxygen consumption, which is consistent with increased thermogenic capacity (Fig. 8B). Taken together, these data indicate that the Fsp27ΔAd mice are resistant to HFD-induced obesity due to increased energy expenditure rather than reduced caloric intake.
FIGURE 8.
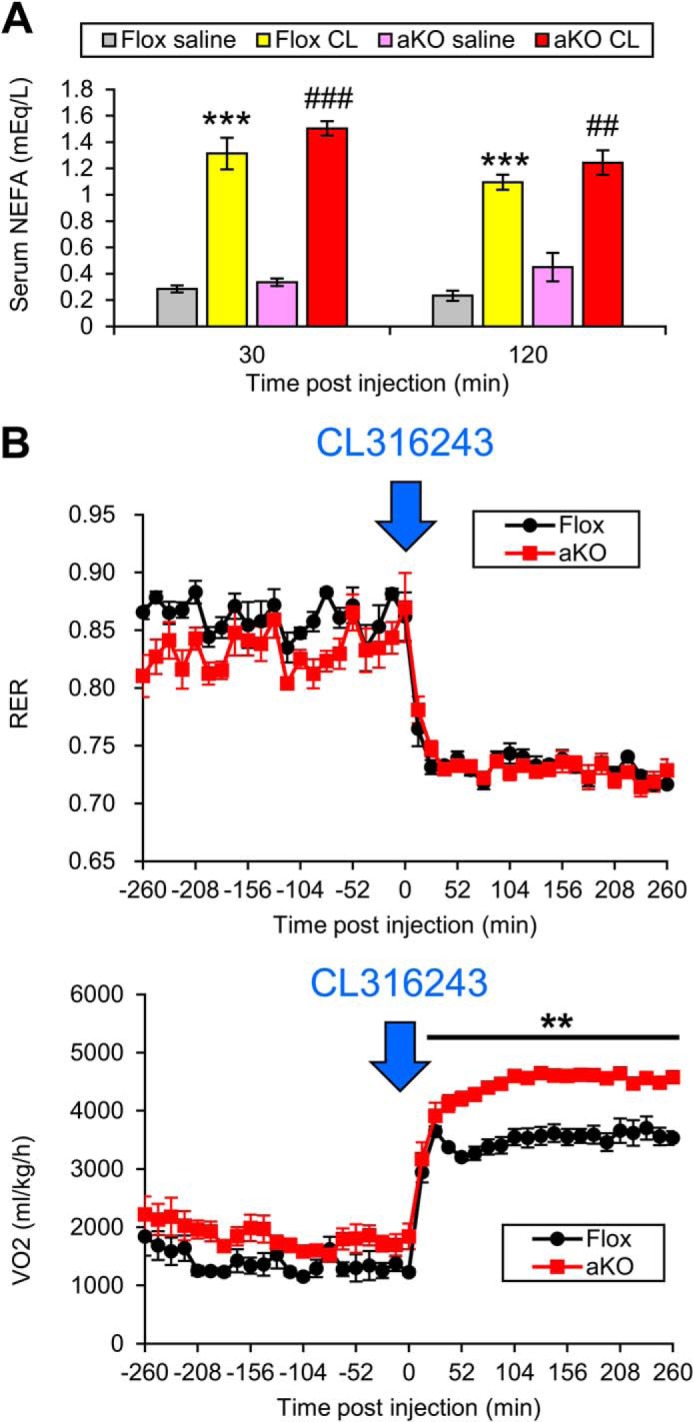
Changes in serum NFEA and oxygen consumption after injection of a selective β3-adrenergic agonist CL316243. A, CL316243 (CL, 100 μg/kg in saline) or saline was administered intraperitoneally to HFD-treated Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice, and serum NEFA concentrations were measured 30 min and 2 h after the injection. Data were analyzed using the two-tailed Student's t test. ***, p < 0.001 compared with saline-injected Flox mice. ##, p < 0.01; ###, p < 0.001 compared with saline-injected aKO mice. Data were analyzed using the two-tailed Student's t test. All values represent the mean ± S.E. n = 5–7/group. B, CL316243 (100 μg/kg in saline) was injected intraperitoneally to HFD-treated Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice, and the changes in RER and whole-body oxygen consumption (VO2) were monitored. All values represent the mean ± S.E. n = 4–5/group. Data were analyzed using the two-tailed Student's t test. **, p < 0.01 compared with Flox mice.
Enhanced Expression of Genes Associated with Mitochondrial Abundance and BAT Identity in WAT of HFD-fed Fsp27ΔAd Mice
To explore the mechanism underlying morphological changes in WAT and increased whole-body energy expenditure found in HFD-fed Fsp27ΔAd mice, the eWAT mRNA levels of the genes associated with lipid metabolism were assessed by qPCR analysis. The mRNA levels of the genes associated with TG synthesis and lipid droplet coating, such as diacylglycerol acyltransferase 1 and 2 (Dgat1 and Dgat2), Plin1, and nuclear receptor peroxisome proliferator-activated receptor (PPAR) γ2 (Pparg2), were increased in HFD-treated Fsp27ΔAd mice (Fig. 9A). Similarly, mRNAs encoding mitochondrion-related genes, such as mitochondrial β-oxidation enzymes (medium-, long-, and short-chain acyl-coenzyme A dehydrogenases (Acadm, Acadl, and Acads, respectively) and respiratory chain components (cytochrome c (Cytc) and cytochrome c oxidase (Cox) 4i2, 5b, and 8b), were markedly elevated in the Fsp27ΔAd mice (Fig. 9B). Normally mitochondria are rich in BAT but not WAT. To examine whether WAT of HFD-treated Fsp27ΔAd mice has BAT-like properties, the mRNAs encoding proteins specifically expressed in BAT were measured. Uncoupling protein 1 (Ucp1), cell death-inducing DNA fragmentation factor-α-like effector a (Cidea), and type 2 iodothyronine deiodinase (Dio2) mRNAs, which are exclusively expressed in BAT, were robustly increased in the Fsp27ΔAd mice (Fig. 9B). Additionally, the mRNAs for transcription factors involved in determination of BAT identity (early B-cell factor 2 (Ebf2) and forkhead box protein C2 (Foxc2)), mitochondriogenesis (PPARγ co-activator 1α (Ppargc1a) and 1β (Ppargc1b)), and regulation of the expression of mitochondrial β-oxidation enzymes (PPARα (Ppara)) were increased in these mice (Fig. 9C). These findings suggest that the properties of WAT are markedly altered in the HFD-treated Fsp27ΔAd mice.
FIGURE 9.
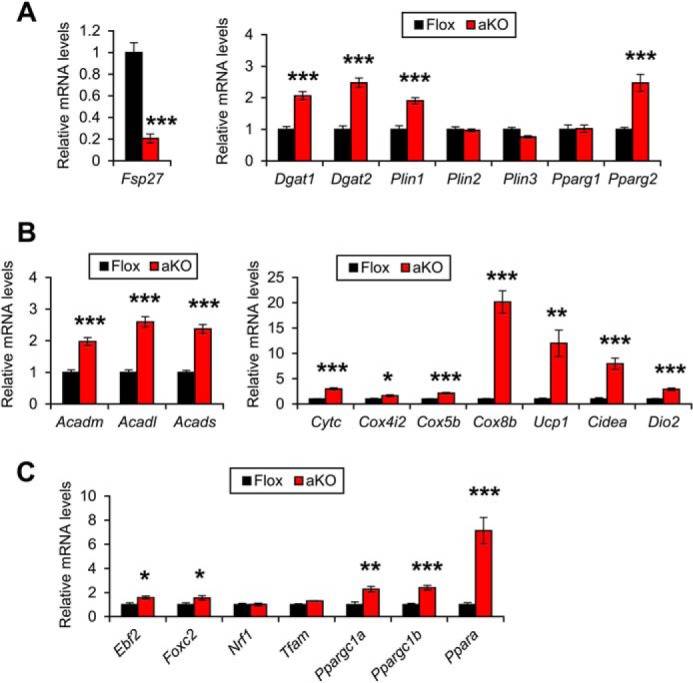
Increased expression of genes associated with mitochondrial abundance and BAT identity in epididymal white adipose tissue of HFD-treated Fsp27ΔAd mice. Expression levels of genes related with lipid droplet formation (A), mitochondrion and BAT identity (B and C) in Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice treated with HFD for 17 weeks. Data were analyzed using the two-tailed Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with Flox mice. All values represent the mean ± S.E. n = 6–8/group.
Hepatic IR in HFD-fed Fsp27ΔAd Mice
Protection from HFD-induced obesity is usually associated with improvement of glucose tolerance and insulin sensitivity. Indeed, Fsp27-null mice showed protection from glucose intolerance and IR induced by HFD (11). Therefore, glucose metabolism was assessed in HFD-treated Fsp27ΔAd mice. Although there were no differences in blood glucose levels in the glucose tolerance test between the two mouse groups, insulin tolerance tests uncovered obvious deterioration of whole-body insulin sensitivity in Fsp27ΔAd mice (Fig. 10A). The area under the insulin tolerance test curve was greater in the Fsp27ΔAd mice, and hyperinsulinemia and increased HOMA index were also detected (Fig. 10, B and C). Protection from HFD-induced BW gain and paradoxical aggravation of whole-body insulin sensitivity were also found in female Fsp27ΔAd mice (Fig. 10, D and E).
FIGURE 10.

Development of insulin resistance in HFD-treated Fsp27ΔAd mice. A, glucose tolerance test (left) and insulin tolerance test (right) in male Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice treated with HFD. These assays were performed 15 and 14 weeks after starting HFD feeding, respectively. B, area under the insulin tolerance test curve. C, serum levels of glucose and insulin and HOMA-IR values. D, body weight changes in female aKO and Flox mice during 17-week HFD feeding. E, glucose tolerance test (left) and insulin tolerance test (right) in female aKO and Flox mice treated with HFD. These assays were performed 15 and 13 weeks after starting HFD treatment, respectively. Data were analyzed using the two-tailed Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with Flox mice. All values represent the mean ± S.E. n = 6–8/group in A–C; n = 5–8/group in D–E.
To explore the precise mechanism of impaired insulin sensitivity, a hyperinsulinemic-euglycemic clamp study was conducted on HFD-fed mice. The glucose infusion rate (GIR) was significantly decreased in the Fsp27ΔAd mice, corroborating the presence of whole-body IR in these mice (Fig. 11A). The whole-body glycolysis rate was decreased in Fsp27ΔAd mice, but the rate of glycogen synthesis was similar to Fsp27F/F mice (Fig. 11E). The rate of glucose disappearance (RD), an indicator of the degree of glucose uptake in skeletal muscle, was also unchanged between the genotypes (Fig. 11B). However, endogenous glucose production during the clamp period was significantly increased in the Fsp27ΔAd mice (Fig. 11C). Reductions in glucose uptake were not detected in skeletal muscles and BAT, whereas glucose uptake in WAT was increased in the Fsp27ΔAd mice (Fig. 11D). These results suggest that liver is a causative organ of systemic IR in the Fsp27ΔAd mice.
FIGURE 11.
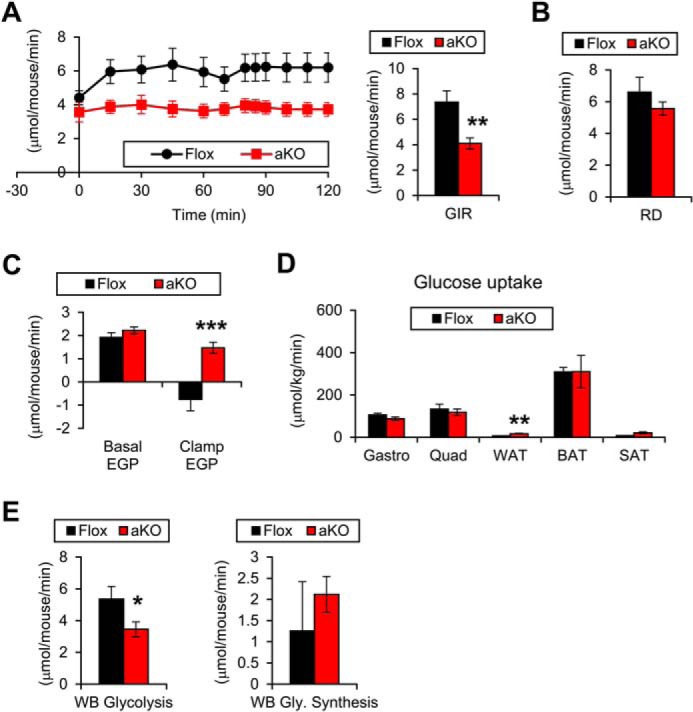
Hepatic insulin resistance in HFD-treated Fsp27ΔAd mice. HFD-treated female Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice presented in Fig. 10, D and E, were used for the hyperinsulinemic-euglycemic clamp assay. A, glucose infusion rate (GIR). B, rate of glucose disappearance (RD). C, endogenous glucose production (EGP). D, glucose uptake rate in muscle (gastrocnemius (Gastro) and quadriceps (Quad)) and adipose tissue. E, whole-body (WB) glycolysis activity and glycogen synthesis rate. Data were analyzed using the two-tailed Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with Flox mice. All values represent the mean ± S.E. n = 5–8/group.
Severe Hepatic Steatosis and Dyslipidemia in HFD-treated Fsp27ΔAd Mice
The livers of HFD-fed Fsp27ΔAd mice were enlarged and yellowish as compared with Fsp27F/F mice (Fig. 12A), and hepatic TG contents were significantly increased in Fsp27ΔAd mice (Fig. 12B). Microscopically, the livers of Fsp27ΔAd mice showed moderate-to-severe macrovesicular/microvesicular steatosis around central veins (Fig. 12C). Hepatocyte ballooning and inflammatory cell infiltration, key pathological features of NASH, were not detected. Reflecting severe hepatic steatosis, serum alanine aminotransferase levels were elevated in the Fsp27ΔAd mice (Fig. 12D). Additionally, serum concentrations of TG and NEFA were increased in these mice (Fig. 12D).
FIGURE 12.
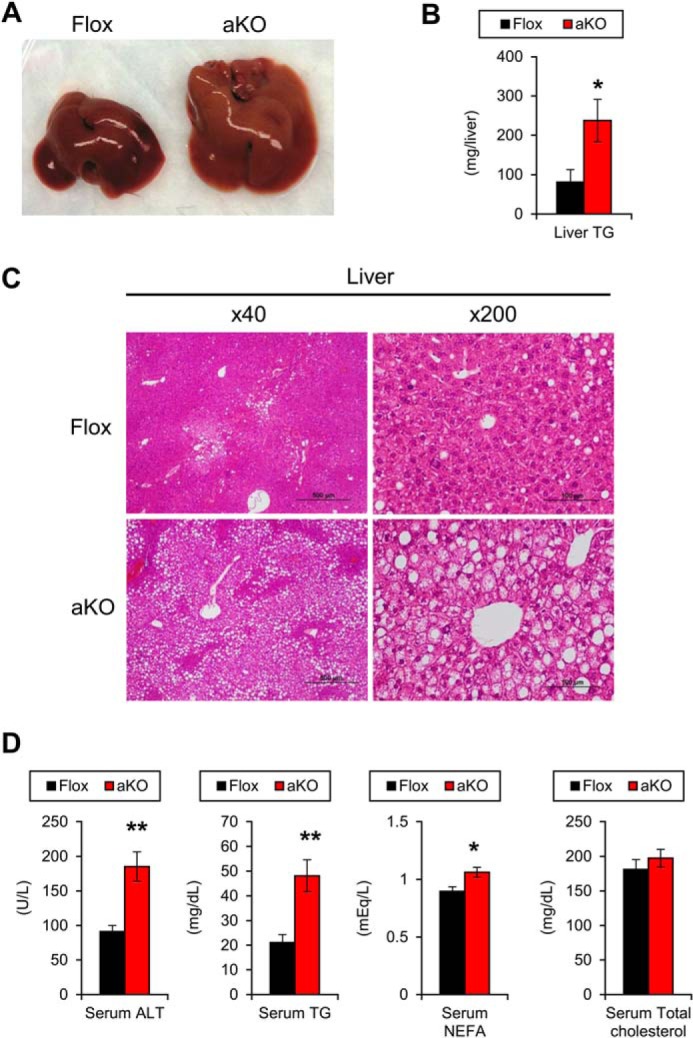
Severe hepatic steatosis and dyslipidemia in HFD-treated Fsp27ΔAd mice. A, gross appearance of liver of Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mouse treated with HFD for 17 weeks. B, hepatic TG contents. C, histological appearance of liver (H&E staining). Scale bars: 500 μm (×40 magnification) and 100 μm (×200 magnification), respectively. D, serum levels of alanine aminotransferase and lipids. Data were analyzed using the two-tailed Student's t test. *, p < 0.05; **, p < 0.01 compared with Flox mice. All values represent the mean ± S.E. n = 6–8/group.
To investigate the mechanism of steatogenesis in HFD-treated Fsp27ΔAd mice, the mRNA levels associated with FA/TG metabolism were determined. Expression levels of some genes associated with FA uptake (FA translocase (Cd36)), FA desaturation/elongation (stearoyl-CoA desaturase 1 (Scd1) and elongation of long chain fatty acids family member 6 (Elovl6)), and TG synthesis (Dgat2) were increased in the Fsp27ΔAd mice (Fig. 13, A and B). Interestingly, the mRNA levels of Cidea, Fsp27, and Fsp27b, an alternative isoform of Fsp27 mainly present in the liver (20), were also elevated in these mice (Fig. 13C). The expression of genes associated with TG secretion/hydrolysis and inflammation did not change between the groups (Fig. 14, A and B). To further examine the function of hepatic TG hydrolysis, phosphorylation of HSL and ATGL was assessed using immunoblot analysis. The bands of hepatic HSL were very weak compared with those of WAT (Fig. 14C). Similar band intensities of total and phosphorylated ATGL between the two mouse groups (Fig. 14C) suggested that hepatosteatosis observed in the Fsp27ΔAd mice was unlikely due to suppressed hepatic lipolysis. Although the expression levels of SREBP-1c (Srebf1), Ppargc1b, and liver X receptor α (Lxra) mRNAs were not increased, the expression of Pparg2 was induced in the livers of HFD-treated Fsp27ΔAd mice (Fig. 14D). Recently, it was documented that cyclic AMP-responsive element-binding protein H (CREBH) can up-regulate Fsp27b in the liver (20). Hepatic mRNAs of genes encoding CREBH (Creb3l3) and its target apolipoprotein A4 (Apoa4) were also increased in the Fsp27ΔAd mice (Figs. 13C and 14D).
FIGURE 13.
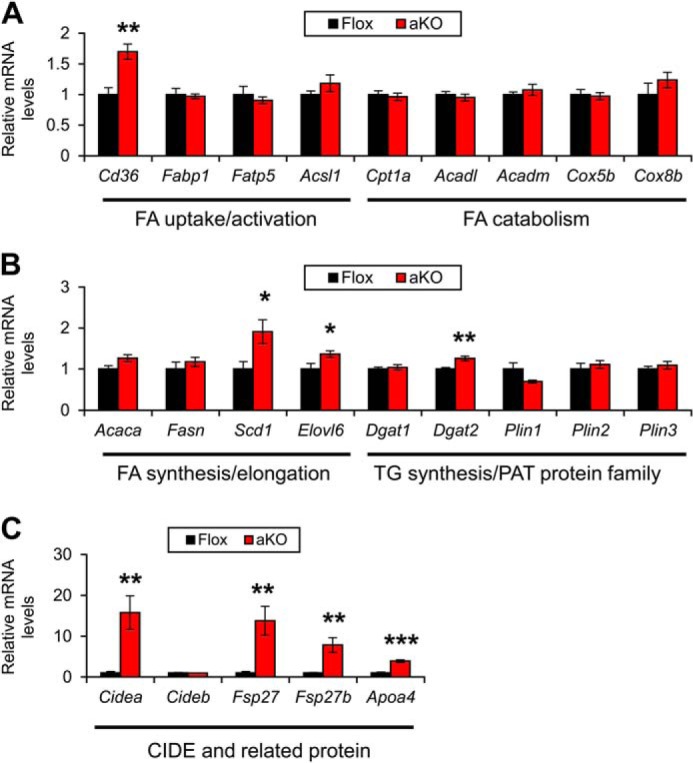
Hepatic expression of genes associated with FA/TG metabolism in HFD-treated Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice. Expression levels of genes associated with FA uptake/activation and catabolism (A), FA/TG synthesis and lipid coating (B), and CIDE (cell death-inducing DNA fragmentation factor-α-like effector) and related proteins (C). Data were analyzed using the two-tailed Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with Flox mice. All values represent the mean ± S.E. n = 6–8/group.
FIGURE 14.
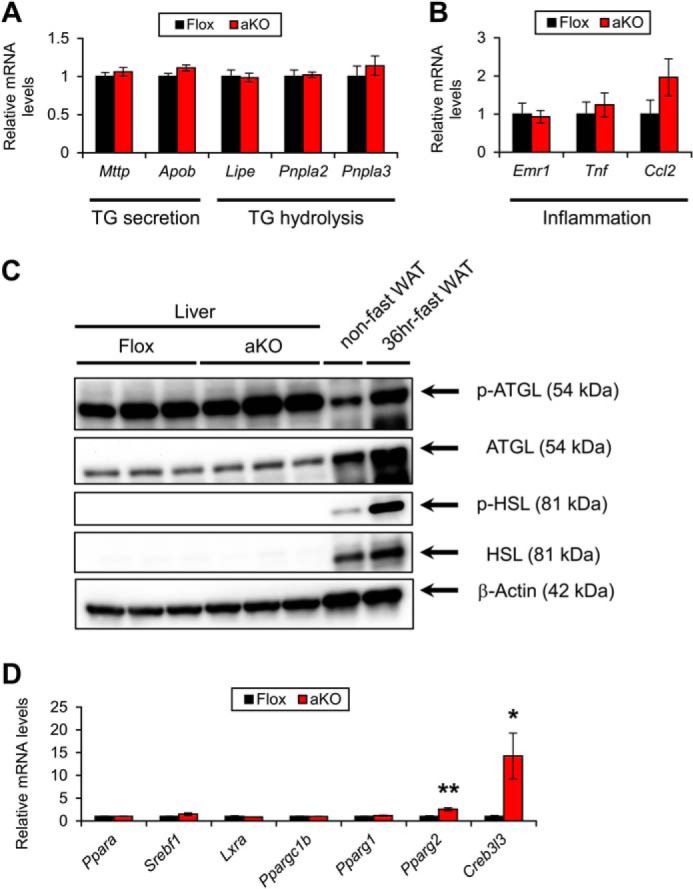
Hepatic expression of genes associated with TG secretion/hydrolysis and inflammation in HFD-treated Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice. A and B, Expression levels of genes associated with TG secretion/hydrolysis (A) and inflammation (B). C, immunoblot analysis of phosphorylated and total hormone-sensitive lipase (p-HSL and HSL, respectively) and adipose TG lipase (p-ATGL and ATGL, respectively). Liver homogenates (20 μg of protein) were loaded in each well. The band of β-actin was used as a loading control. Epididymal WAT isolated from a non-fasted and 36 h-fasted mouse was used for detecting the correct position of the bands. D, expression levels of transcription factors regulating FA/TG metabolism. Data were analyzed using two-tailed Student's t test. *, p < 0.05; **, p < 0.01 compared with Flox mice. All values represent the mean ± S.E. n = 6–8/group.
Contribution of WAT Lipolysis to the Development of Hepatic Steatosis and IR in HFD-treated Fsp27ΔAd Mice
Increased serum NEFA levels and hepatic Cd36 expression suggested that increased FA flux from WAT into the circulation might be associated with the steatogenesis of HFD-fed Fsp27ΔAd mice. Indeed, the expression of mRNAs encoding HSL (Lipe), ATGL (Pnpla2), and carboxylesterase 3 (Ces3), which are key enzymes for white adipose lipolysis, were significantly up-regulated in the WAT of HFD-treated Fsp27ΔAd mice (Fig. 15A). Up-regulated HSL and ATGL were corroborated by immunoblot analysis (Fig. 15B). Furthermore, actual lipolytic activities were determined using white adipocytes isolated from HFD-treated Fsp27F/F and Fsp27ΔAd mice. Basal lipolytic activities were markedly increased in the adipocytes of Fsp27ΔAd mice (340 ± 51% and 210 ± 7% compared with the Fsp27F/F adipocytes at 30 min and 2 h after isolation, respectively) (Fig. 15C). Similar increases in lipolytic activities were observed in Fsp27ΔAd mouse adipocytes incubated with CL316243 (180 ± 28% and 190 ± 7% of the Fsp27F/F adipocytes at 2 h of incubation with 10 and 100 nm CL316243, respectively) (Fig. 15C). However, the degree of increased lipolytic activities after β3-adrenoreceptor stimulation was similar between Fsp27ΔAd adipocytes and Fsp27F/F adipocytes (150 ± 23% versus 170 ± 23% by 10 nm CL316234; 180 ± 7% versus 190 ± 11% by 100 nm CL316234), suggesting that increased lipolytic activities in the Fsp27ΔAd adipocytes occur due to the lack of FSP27. Collectively, these results demonstrate marked acceleration of lipolysis and significant impairment of fat-storing function in the WAT of Fsp27ΔAd mice. Altered WAT function by adipocyte-specific Fsp27 disruption may induce severe fat deposition in the liver and the ensuing IR, especially under the over-nutritional state.
FIGURE 15.
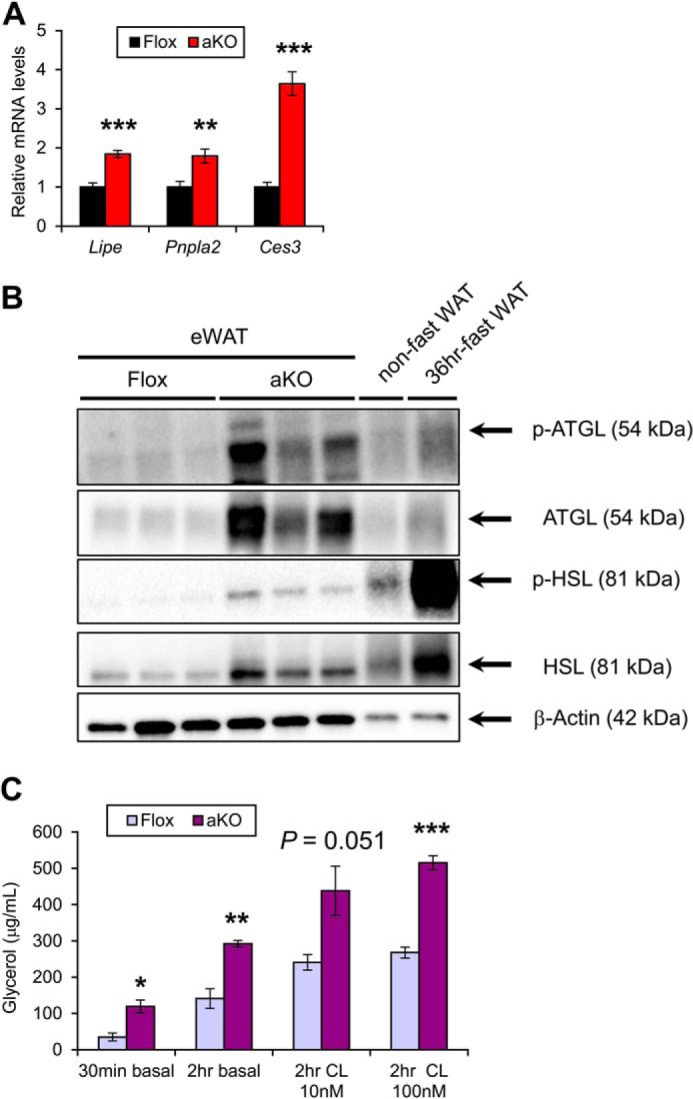
Enhanced lipolysis in WAT of HFD-treated Fsp27ΔAd mice. A, expression levels of genes encoding key enzymes for white adipose lipolysis in Fsp27ΔAd (aKO) and Fsp27F/F (Flox) mice (n = 6–8/group). B, immunoblot analysis of phosphorylated and total hormone-sensitive lipase (p-HSL and HSL, respectively) and adipose TG lipase (p-ATGL and ATGL, respectively). Cytosolic extracts of epididymal WAT (eWAT, 20 μg of protein) were loaded in each well. The band of β-actin was used as a loading control. The eWAT isolated from a non-fasted and 36 h-fasted mouse was used to identify the band position. C, lipolytic activities of adipocytes isolated from aKO and Flox mice treated with HFD for 20 weeks (n = 3/group). Comparisons were done between aKO adipocytes and Flox adipocytes in the same condition. CL indicates incubation with CL316243. Data were analyzed using the two-tailed Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with Flox mice. All values represent the mean ± S.E.
DISCUSSION
WAT is an important storehouse of surplus energy in the living body, and its dysfunction may disrupt energy homeostasis. This study confirmed the previous observations that FSP27 plays an important role in the maintenance of WAT functions and properties. Similar to the total body Fsp27-null mice, adipocyte-specific Fsp27 disruption protected against HFD-induced obesity. This anti-obesity effect was likely due to enhanced lipolytic activities and impaired capacity to store excess fat in WAT. However, different from Fsp27-null mice, excess fat overflowed from WAT accumulated in hepatocytes, causing systemic IR. The livers of adipocyte-specific Fsp27-disrupted mice demonstrated up-regulation of Fsp27 and severe steatosis likely due to activation of PPARγ- and CREBH-mediated pathways. Therefore, this study indicates a crucial role for adipose and extra-adipose tissue FSP27 in the control of whole-body energy/nutrient homeostasis (Fig. 16).
FIGURE 16.
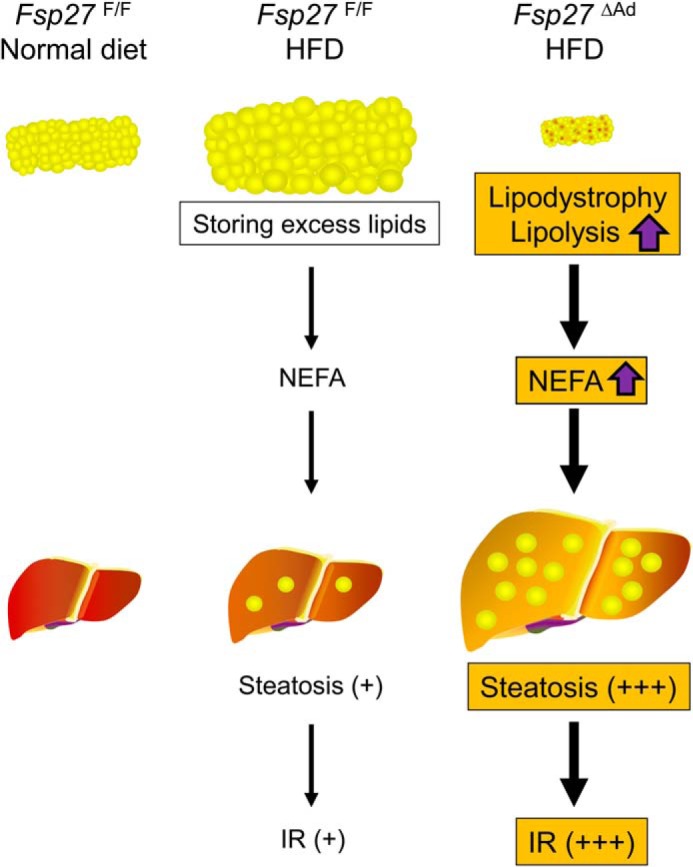
Proposed mechanism of the development of severe hepatic steatosis and insulin resistance in HFD-treated Fsp27ΔAd mice.
The WAT of Fsp27ΔAd mice had numerous small adipocytes containing multilocular lipid droplets. This finding is consistent with the previous observations that Fsp27 is required for the formation of large unilocular lipid droplets in cultured differentiated adipocytes (8, 9, 11). Fsp27-ablated adipocytes seemed to be unable to enlarge intracellular lipid droplets in response to dietary fat overload. Indeed, multiple proteins/enzymes regulate the lipolytic activities in WAT. Among several TG hydrolases, HSL and ATGL are the major contributors to adipose lipolysis. The expression levels of these enzymes were significantly augmented in the WAT of HFD-fed Fsp27ΔAd mice. A recent study uncovered that FSP27 directly interacts with ATGL to inhibit its activation (21). Additionally, acute disruption of FSP27 expression by siRNA in differentiated white adipose cell lines resulted in up-regulation of mRNA levels of Lipe and Pnpla2 (11). These findings support the results in the present study that Fsp27 disruption enhanced constitutive lipolytic activities independently of stimulation of β3-adrenergic receptor in isolated adipocytes. Thus, FSP27 is indispensable for stabilizing lipid droplets in mature white adipocytes.
The expression of mitochondrion- and BAT-related genes was increased in the WAT of HFD-fed Fsp27ΔAd mice, indicating the possible association between Fsp27 disruption and transformation of white adipocytes into brown adipocyte-like adipocytes. This notion is supported by results in the previous study that Fsp27-null mouse-derived embryonic fibroblasts differentiated in vitro displayed many characteristics of brown adipocytes in the presence of triiodothyronine (12). No increases in the mRNA levels of Tmem26 and Cd137 (data not shown) suggested the absence of increases in beige cells. Additionally, transcription factors determining BAT identity, such as EBF2 and FOXC2, were up-regulated in the HFD-treated Fsp27ΔAd mice. EBF2-overexpressing C2C12 myoblasts and WAT of mice overexpressing FOXC2 were reported to acquire BAT-like properties (22, 23). Therefore, FSP27 might play an important role in causing adipocytes to maintain the properties as mature white adipocytes. It may be of interest to examine the relationship between Fsp27 disruption and master regulators determining BAT identity.
The most intriguing finding in the present study was that HFD-treated Fsp27ΔAd mice demonstrated worsening of systemic IR despite protection from HFD-induced obesity. This was mainly due to hepatic steatosis and ensuing IR, as evidenced by the hyperinsulinemic-euglycemic clamp study. Fsp27-null mice were reported to show anti-obesity and attenuation of hepatic steatosis and IR upon HFD feeding (11, 12), suggesting that the attenuation of IR in these mice stemmed from accelerated FA β-oxidation in the WAT and the resultant increases in energy expenditure (7, 11). Although similar WAT phenotypes, i.e. small adipocytes with multiclocular droplets, increased lipolytic activity and overexpression of mitochondrial β-oxidation enzymes and increased energy expenditure were also detected in the Fsp27ΔAd mice, the conditional knock-out mice also revealed aggravation of hepatic steatosis and IR. This critical difference suggests the possible contribution of Fsp27 disruption in extra-adipose tissue to the anti-diabetic effect in total body Fsp27-null mice. The fact that forced expression of FSP27 in mouse primary hepatocytes suppressed mitochondrial FA β-oxidation activity (13) supports this possibility. In HFD-fed Fsp27-null mice, FA spilled from WAT may be consumed in Fsp27-disrupted extra-adipose tissues in which mitochondrial FA β-oxidation is presumably elevated to some degree leading to attenuated hepatic steatosis and ensuing improvement of insulin sensitivity. HFD-fed Fsp27ΔAd mice demonstrated a similar enhancement of FA spillover from WAT, but the surplus FA cannot be sufficiently accommodated in liver and other tissues probably because of the lack of Fsp27 disruption, resulting in increased ectopic fat accumulation and elevated systemic IR. The results obtained from Fsp27ΔAd mice in this study offer a new mechanism on how whole-body Fsp27 disruption in mice improves insulin sensitivity upon HFD feeding.
It is reasonable to consider that impaired ability to store excess fat in Fsp27-disrupted WAT and overflowing the lipids from WAT leads to hepatic steatosis. This “overflow hypothesis” has been well accepted in other lipodystrophic mouse models showing hepatosteatosis (4, 5). Up-regulation of hepatic Cd36 may reflect the enhancement of excess FA uptake into the hepatocytes. Hepatic Cidea expression was reported to be induced in response to increased FA in the blood promoting hepatic steatosis (24). Increases in Cd36/Cidea/Fsp27 and their upstream regulator Pparg2 suggest that the activation of PPARγ is one of the main mechanisms of steatogenesis in the Fsp27ΔAd mice. The direct and critical involvement of hepatic PPARγ in the pathogenesis of this type of fatty liver has been proved in AZIP mice demonstrating generalized lipodystrophy, severe hepatic steatosis, and systemic IR (25). Additionally, enhanced mRNA levels of Creb3l3 and its target genes Apoa4 and Fsp27b suggest that CREBH-FSP27 axis may contribute to the steatogenesis. It is of note that no induction of the mRNAs encoded by genes involved in de novo FA synthesis and the protein levels of ATGL were seen in the livers of HFD-fed Fsp27ΔAd mice. This may partially support the abovementioned hypothesis.
Major pathological changes detected in HFD-treated Fsp27ΔAd mice, i.e. small WAT, severe hepatic steatosis, increased circulating NEFA/TG levels, and IR, closely resembled the clinical features of humans having lipodystrophy. However, severe fat deposition was not found in the livers of Fsp27ΔAd mice fed a normal diet indicating that the lipodystrophic phenotypes caused by adipocyte-specific Fsp27 disruption appeared to be mild compared with other mouse models exhibiting generalized lipodystophy, such as Agpat2-null mice, BSCL/Seipin-null mice, and AZIP mice (5, 26, 27). Recently, a patient with a homozygous nonsense mutation in CIDEC was reported (28). This patient demonstrated partial lipodystrophy mainly affecting limb and femorogluteal fat pads and subcutaneous abdominal fat, insulin-resistant diabetes, severe hepatomegaly and hepatic steatosis, increased plasma NEFA/TG levels, increased urine glycerol levels suggesting increased white adipose lipolysis, augmented RER, and the presence of many adipocytes with multiple small lipid droplets in WAT. These clinical features are similar to the findings of HFD-treated Fsp27ΔAd mice obtained in the present study. Lipodystrophy caused by the CIDEC gene mutation is designated as familial partial lipodystrophy 5 (OMIM #615238). This novel mouse line might be useful to explore the pathogenesis of partial lipodystrophy and develop the therapeutic strategies against lipodystrophy-induced diabetes and NAFLD/NASH.
This study aimed to determine the role of adipose FSP27 in vivo on HFD-induced metabolic alterations. A previous in vitro experimentation unveiled the possible association between FSP27 and apoptosis (29). Assessment of how FSP27 regulates adipocyte fate in vivo deserves future investigation.
Lastly, reduced expression of CIDEC in WAT was reported to be associated with systemic IR in body mass index-matched humans (30), which is in agreement with the results in the present study. Additionally, clinicians sometimes encounter non-obese humans having NAFLD. Such disagreement might be caused by disproportional expression of fat-storing proteins, including CIDEC. Judging from the major contribution of FA originated from lipolysis in WAT to the development of NAFLD in humans (31), investigating the expression and function of adipose CIDEC in the patients having NAFLD and accompanying IR and diabetes might provide new information regarding the pathogenesis of these metabolic diseases.
This work was supported, in whole or in part, by National Institutes of Health Intramural Research Program (NCI) and Grant U54 ES16015 (to F. J. G.).
- WAT
- white adipose tissue
- eWAT
- epididymal WAT
- ATGL
- adipose triglyceride lipase
- BAT
- brown adipose tissue
- BW
- body weight
- CREBH
- cyclic AMP-responsive element-binding protein H
- FA
- fatty acid
- FSP27
- fat-specific protein 27
- HFD
- high-fat diet
- HSL
- hormone-sensitive lipase
- IR
- insulin resistance
- NAFLD
- non-alcoholic fatty liver disease
- NASH
- non-alcoholic steatohepatitis
- NEFA
- non-esterified FA
- PPAR
- peroxisome proliferator-activated receptor
- qPCR
- quantitative PCR
- RER
- respiratory exchange ratio
- TG
- triglyceride
- Neo
- neomycin
- CIDEC
- cell death-inducing DNA fragmentation factor-α-like effector c.
REFERENCES
- 1. Greenberg A. S., Coleman R. A., Kraemer F. B., McManaman J. L., Obin M. S., Puri V., Yan Q. W., Miyoshi H., Mashek D. G. (2011) The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Invest. 121, 2102–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lumeng C. N., Saltiel A. R. (2011) Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 121, 2111–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jiang C., Qu A., Matsubara T., Chanturiya T., Jou W., Gavrilova O., Shah Y. M., Gonzalez F. J. (2011) Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60, 2484–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garg A. (2011) Lipodystrophies: genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 96, 3313–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Savage DB. (2009) Mouse models of inherited lipodystrophy. Dis. Model Mech. 2, 554–562 [DOI] [PubMed] [Google Scholar]
- 6. Safar Zadeh E., Lungu A. O., Cochran E. K., Brown R. J., Ghany M. G., Heller T., Kleiner D. E., Gorden P. (2013) The liver diseases of lipodystrophy: the long-term effect of leptin treatment. J. Hepatol. 59, 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puri V., Czech M. P. (2008) Lipid droplets: FSP27 knockout enhances their sizzle. J. Clin. Invest. 118, 2693–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puri V., Konda S., Ranjit S., Aouadi M., Chawla A., Chouinard M., Chakladar A., Czech M. P. (2007) Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J. Biol. Chem. 282, 34213–34218 [DOI] [PubMed] [Google Scholar]
- 9. Keller P., Petrie J. T., De Rose P., Gerin I., Wright W. S., Chiang S. H., Nielsen A. R., Fischer C. P., Pedersen B. K., MacDougald O. A. (2008) Fat-specific protein 27 regulates storage of triacylglycerol. J. Biol. Chem. 283, 14355–14365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun Z., Gong J., Wu H., Xu W., Wu L., Xu D., Gao J., Wu J. W., Yang H., Yang M., Li P. (2013) Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. Commun. 4, 1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishino N., Tamori Y., Tateya S., Kawaguchi T., Shibakusa T., Mizunoya W., Inoue K., Kitazawa R., Kitazawa S., Matsuki Y., Hiramatsu R., Masubuchi S., Omachi A., Kimura K., Saito M., Amo T., Ohta S., Yamaguchi T., Osumi T., Cheng J., Fujimoto T., Nakao H., Nakao K., Aiba A., Okamura H., Fushiki T., Kasuga M. (2008) FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J. Clin. Invest. 118, 2808–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toh S. Y., Gong J., Du G., Li J. Z., Yang S., Ye J., Yao H., Zhang Y., Xue B., Li Q., Yang H., Wen Z., Li P. (2008) Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of Fsp27 deficient mice. PLoS ONE 3, e2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsusue K., Kusakabe T., Noguchi T., Takiguchi S., Suzuki T., Yamano S., Gonzalez F. J. (2008) Hepatic steatosis in leptin-deficient mice is promoted by the PPARγ target gene Fsp27. Cell Metab. 7, 302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. You Y., Bersgtram R., Klemm M., Nelson H., Jaenisch R., Schimenti J. (1998) Utility of C57BL/6J x 129/SvJae embryonic stem cells for generating chromosomal deletions: tolerance to gamma radiation and microsatellite polymorphism. Mamm. Genome 9, 232–234 [DOI] [PubMed] [Google Scholar]
- 15. Abel E. D., Peroni O., Kim J. K., Kim Y. B., Boss O., Hadro E., Minnemann T., Shulman G. I., Kahn B. B. (2001) Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409, 729–733 [DOI] [PubMed] [Google Scholar]
- 16. Tanaka N., Matsubara T., Krausz K. W., Patterson A. D., Gonzalez F. J. (2012) Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 56, 118–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanaka N., Moriya K., Kiyosawa K., Koike K., Gonzalez F. J., Aoyama T. (2008) PPARα activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 118, 683–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanaka N., Takahashi S., Fang Z. Z., Matsubara T., Krausz K. W., Qu A., Gonzalez F. J. (2014) Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochim. Biophys. Acta 1841, 1596–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ravussin Y., Gutman R., LeDuc C. A., Leibel R. L. (2013) Estimating energy expenditure in mice using an energy balance technique. Int. J. Obes. (Lond) 37, 399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu X., Park J. G., So J. S., Lee A. H. (2014) Transcriptional activation of Fsp27 by the liver-enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology 10.1002/hep.27371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grahn T. H., Kaur R., Yin J., Schweiger M., Sharma V. M., Lee M. J., Ido Y., Smas C. M., Zechner R., Lass A., Puri V. (2014) Fat-specific protein 27 (FSP27) interacts with adipose triglyceride lipase (ATGL) to regulate lipolysis and insulin sensitivity in human adipocytes. J. Biol. Chem. 289, 12029–12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rajakumari S., Wu J., Ishibashi J., Lim H. W., Giang A. H., Won K. J., Reed R. R., Seale P. (2013) EBF2 determines and maintains brown adipocyte identity. Cell Metab. 17, 562–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cederberg A., Grønning L. M., Ahrén B., Taskén K., Carlsson P., Enerbäck S. (2001) FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 106, 563–573 [DOI] [PubMed] [Google Scholar]
- 24. Zhou L., Xu L., Ye J., Li D., Wang W., Li X., Wu L., Wang H., Guan F., Li P. (2012) Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology 56, 95–107 [DOI] [PubMed] [Google Scholar]
- 25. Gavrilova O., Haluzik M., Matsusue K., Cutson J. J., Johnson L., Dietz K. R., Nicol C. J., Vinson C., Gonzalez F. J., Reitman M. L. (2003) Liver peroxisome proliferator-activated receptor 0.1002/hep.27371 contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 278, 34268–34276 [DOI] [PubMed] [Google Scholar]
- 26. Cortés V. A., Curtis D. E., Sukumaran S., Shao X., Parameswara V., Rashid S., Smith A. R., Ren J., Esser V., Hammer R. E., Agarwal A. K., Horton J. D., Garg A. (2009) Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 9, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cui X., Wang Y., Tang Y., Liu Y., Zhao L., Deng J., Xu G., Peng X., Ju S., Liu G., Yang H. (2011) Seipin ablation in mice results in severe generalized lipodystrophy. Hum. Mol. Genet. 20, 3022–3030 [DOI] [PubMed] [Google Scholar]
- 28. Rubio-Cabezas O., Puri V., Murano I., Saudek V., Semple R. K., Dash S., Hyden C. S., Bottomley W., Vigouroux C., Magré J., Raymond-Barker P., Murgatroyd P. R., Chawla A., Skepper J. N., Chatterjee V. K., Suliman S., Patch A. M., Agarwal A. K., Garg A., Barroso I., Cinti S., Czech M. P., Argente J., O'Rahilly S., Savage D. B., and LD Screening Consortium (2009) Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol. Med. 1, 280–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim J. Y., Liu K., Zhou S., Tillison K., Wu Y., Smas C. M. (2008) Assessment of fat-specific protein 27 in the adipocyte lineage suggests a dual role for FSP27 in adipocyte metabolism and cell death. Am. J. Physiol. Endocrinol. Metab. 294, E654–E667 [DOI] [PubMed] [Google Scholar]
- 30. Puri V., Ranjit S., Konda S., Nicoloro S. M., Straubhaar J., Chawla A., Chouinard M., Lin C., Burkart A., Corvera S., Perugini R. A., Czech M. P. (2008) Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc. Natl. Acad. Sci. U.S.A. 105, 7833–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., Parks E. J. (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]