

Background: BMI1 regulates cancer stem cell phenotype and is overexpressed in cancer cells.
Results: PLK1 regulates BMI1 expression and its inhibitors suppress BMI1 expression, and induce premature senescence.
Conclusion: BMI1 acts downstream of PLK1, and its expression is strongly inhibited by PLK1 inhibitor BI 2536.
Significance: PLK1 inhibitors can be used to suppress growth of breast cancer cells overexpressing BMI1.
Keywords: Breast Cancer, Cancer Biology, Cell Growth, Cellular Regulation, Cellular Senescence, Gene Regulation, microRNA (miRNA), Polycomb
Abstract
The polycomb group protein BMI1 is an important regulator of cancer stem cell (CSC) phenotype and is often overexpressed in cancer cells. Its overexpression leads to increase in CSC fraction and therapy resistance in tumors. BMI1 functions via polycomb repressive complex 1 (PRC1)-mediated gene silencing and also via PRC1-independent transcriptional activities. At present, very little is known about the therapy reagents that can efficiently inhibit BMI1 expression, and the CSC phenotype. Here, we report that the polo-like kinase 1 (PLK1) regulates BMI1 expression, and that its inhibition can efficiently down-regulate BMI1 expression and PRC1 activity, and induce premature senescence in breast cancer cells. We also show that the exogenous BMI1 overexpression mitigates anti-oncogenic effects of PLK1 inhibition and overcomes senescence induction by PLK1 inhibitors. We further show that PLK1 inhibition down-regulates BMI1 by upregulating the miRNA-200c/141 cluster, which encodes miR-200c and miR-141, both of which are known to post-transcriptionally downregulate BMI1 expression. Thus, our data suggest that PLK1 inhibitors can be successfully used to inhibit growth of tumors in which PcG protein BMI1 is overexpressed or the PRC1 activity is deregulated.
Introduction
Polycomb group (PcG)3 proteins are evolutionarily conserved gene silencers, which determine cell fate decisions and body pattern during development (1). These proteins are often deregulated in cancer and other pathological conditions (2, 3). BMI1 and EZH2, the two most well characterized PcG proteins, are overexpressed in a number of human malignancies, including breast and prostate cancers (4–6). PcG proteins form polycomb repressive complexes (PRCs), which possess histone post-translational modification (PTM) activities that epigenetically silence gene expression (7, 8). BMI1, the main constituent of PRC1, promotes monoubiquitination of H2A, which plays a role in chromatin compaction during PRC-mediated gene silencing (7, 8). Overexpression of BMI1 in cancer cells has been shown to promote cancer stem cell phenotype (9, 10). It was found that BMI1 is required for proliferation and self-renewal of breast and prostate CSCs (9, 10). BMI1 is also required for self-renewal of normal stem cells, such as neural, hematopoietic, intestinal, and mammary stem cells (11–15). It has been shown that the overexpression of BMI1 results in repression of tumor suppressor p16INK4a (16, 17), which is epigenetically silenced in many cancers. BMI1 also plays an important role in senescence and aging (18). The exogenous overexpression of BMI1 results in extension of replicative life span of cells, and immortalization of certain epithelial cell types (16, 19). BMI1 can also regulate the expression of p16INK4a-independent targets that are relevant to aging and cancer (20–22). Recently, it was shown that the BMI1 targets include genes involved in transforming growth factor-β/bone morphogenetic protein (TGF-beta/BMP) and endoplasmic reticulum (ER) stress (23), and WNT pathways (24).
BMI1 is transcriptionally regulated by c-Myc and E2F1 transcription factors (25, 26). BMI1 is also regulated post-transcriptionally by several miRNAs. These miRNAs include miR-200c (10), and miR-141 (27), both of which are co-transcribed from the miR-200c/miR-141 loci on chromosome 12p13.31 (28, 29). The miR-200c has been shown to regulate breast cancer stem cell phenotype by targeting PcG protein BMI1 (10). Other miRNAs such as miR-15a, miR-16, miR-218, and miR-128 also have been shown to regulate expression of BMI1 and cancer stem cell phenotype to a varying degree in different cancer cell types (30–32). Very recently, we reported that by targeting BMI1, both miR-200c and miR-141 directly induce cellular senescence via pRb-p16 pathway in human diploid fibroblasts (HDFs) (27).
Polo-like kinase 1 (PLK1), the best characterized member of the PLK family plays an active role in carcinogenesis and contribute to transformed properties of cancer cells (33). Similar to BMI1, PLK1 is overexpressed in a broad spectrum of cancers and often correlates with a poor prognosis (34, 35) and is considered a strong therapy target for cancer treatment. Many small molecule inhibitors of PLK1 such as BI 2536 have been shown to inhibit oncogenic phenotypes and tumor growth in vivo (34, 36). Recently, it was shown that PLK1 inhibition can induce cellular senescence in human diploid fibroblasts (37). Since, BMI1 is a major regulator of senescence (16, 17), and its knockdown also induces senescent phenotype (25), it is possible that PLK1 inhibition induces senescence via regulation of BMI1. Here, we studied the effect of PLK1 inhibition on BMI1 expression and determined whether the PcG protein BMI1 is a target of PLK1 inhibitor BI 2536. Our data suggest that PLK1 regulates BMI1 expression and that its inhibition results in down-regulation of BMI1 and PRC1 activity. Furthermore, BMI1 overexpression can rescue inhibition of oncogenicity induced by PLK1 inhibition. Mechanistically, PLK1 inhibition appears to regulate BMI1 expression via up-regulation of miR-200c and miR-141.
EXPERIMENTAL PROCEDURES
Cells, Cell Culture Methods, and Reagents
The breast cancer cell lines MCF7, MDA-MB-231, MDA-MB-453 and MDA-MB-468, and 293T cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). The cells were cultured as described previously (24, 27). The PLK1 inhibitor BI 2536 was obtained from Selleck Chemicals (Houston, TX), and dissolved in DMSO prior to use. For EdU (5′ ethynyl-2′-deoxyuridine, a thymidine analog) staining, CF594-azide (red fluorescence), and CF488A- azide (green fluorescence), were obtained from Biotium (Hayward, CA).
Expression Vectors, Promoter-Reporters, 3′-UTR Reporters, Transient Transfections, Retrovirus, and Lentivirus production, and Luciferase Assays
Lentiviral vector pEZX-AM03 expressing miR-141, and miR-200c inhibitors, and a miRNA-scrambled control clone were obtained from the Genecopoeia (Rockville, MD). The PLK1 shRNAs and control shRNAs were from OriGene (Rockville, MD). The retroviral vectors overexpressing wild type BMI1 (pBabe-BMI1 (puro) or pMSCV-BMI1 (hygro)) and method for producing retroviruses, and transient transfection using calcium phosphate or FuGene 6 (Promega, Madison, WI), have been described previously (24, 38). The PLK1 overexpressing retroviral vector pWZL Neo Myr Flag PLK1 (originally from Dr. Jean Zhao), was obtained from Addgene (Cambridge, MA). The 3′-UTR reporter vectors pLS-miR-141WT, pLS-miR-141Mut, pLS-miR-200cWT and pLS-miR-200cMut contain wild type or mutant miR seed sequences of respective miRs and have been described previously (27). To generate the miR-200C/141 promoter reporter plasmid, the upstream sequence information of miR-200C/141 cluster was retrieved from NCBI database (NW_003871083.2), and the promoter region from −683 to −44 bp was amplified and cloned into pGL4.18 vector (Promega). The 3′-UTR reporter assays were performed as recommended by the manufacturer using LightSwitch Dual Assay System (SwitchGear Genomics, Menlo Park, CA) and described previously (27). Similarly, the promoter-reporter assays were performed using Dual-Luciferase® Reporter Assay System (Promega) as described previously (24).
Antibodies and Western Blot Analyses
Various antibodies were obtained from the commercial sources. The p53 mouse monoclonal antibody (mAb) and a p21 mAb were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The BMI1 mAb was from Invitrogen (Carlsbad, CA). The polyclonal antibody (pAb) against PLK1 and Phospho-PLK1 were obtained from Cell Signaling Technology (Danvers, MA). The β-actin mAb was from Sigma-Aldrich. The pAbs against H2AK119Ub and total H2A were obtained from Millipore (Billerica, MA) and Cell Signaling Technology (Danvers, MA) respectively. Western blot analyses were done using specific antibodies as described previously (20, 39).
Quantitative RT-PCR Analysis
The total RNA was extracted using the miRNeasy Mini Kit (Qiagen), and reverse transcribed using a miRNA cDNA synthesis kit from Quanta Biosciences, (Gaithersburg, MD). The quantitative real-time PCR (qRT-PCR) was performed using miR-141-, miR-200c- or RNU6B- specific primers from Quanta Biosciences, (Gaithersburg, MD), as per the manufacturer's protocol in a Step one plus RT-PCR machine from ABI (Foster, CA) as described (27) The comparative CT (ΔΔCT) method was used to calculate RQ of miRNA expression using RNU6B as the endogenous control.
Proliferation, Senescence-associated β-Galactosidase Assay, EdU Staining, Soft-agar Colony Formation, Mammosphere, and Aldefluor Assays
The proliferation assays were performed as described (25, 40). Senescence was determined using SA-β-Gal marker (25, 40). The EdU and SA-β-Gal co-staining was performed as described (41). Images were taken with a Nikon Eclipse Ti microscope camera under ×10 magnification and stained cells were counted as described (42). The soft-agar colony formation assay to determine oncogenic potential was done as described (21). For the study of Cancer Stem Cell (CSC) phenotype, Mammosphere formation, Aldefluor and flow cytometry assays were performed as described (24).
Statistical Analysis
All experiments were performed at least three times in triplicates for each group. The results are presented as the mean ± S.D. Statistical significance was determined using Student's t test, and p < 0.05 was considered significant.
RESULTS
PLK1 Regulates Expression of the PcG Protein BMI1 and Its Activity
To determine whether the PLK1 can regulate activity and function of PcG proteins, we analyzed expression of BMI1 in control and PLK1 knockdown cells by Western blot analysis. We also determined whether PLK1 knockdown results in down-regulation of H2AK119 ubiquitination activity of PRC1 by analyzing total H2A and H2AK119Ub levels by Western blot analysis. Our data indicated that PLK1 knockdown using two different shRNAs (PLKi46 and PLKi47) but not a control shRNA resulted in down-regulation of BMI1 as well as decreased levels of H2AK119Ub in MCF7, MDA-MB-231, and MDA-MB-468 cells (Fig. 1A). Next, we determined whether stable overexpression of PLK1 in MCF10A results in up-regulation of expression of BMI1 and H2AK119Ub levels indicative of PRC1 activity. Our results indicated that indeed PLK1 overexpression leads to up-regulation of BMI1 and PRC1 activity (Fig. 1B). Next, we determined whether BI 2536, a well-known and highly effective inhibitor of PLK1 (36), can down-regulate the expression of BMI1 and PRC1 activity. Our data showed that BI 2536 down-regulated BMI1 and H2AK119Ub levels in a dose-dependent manner in all three breast cancer cell lines (Fig. 1C). Similar results were obtained in MDA-MB-453 cell line (data not shown). Collectively, our results indicate that PLK1 regulates expression of BMI1 and PRC1 activity, and that PLK1 inhibitor BI 2536 is also a potent inhibitor of BMI1.
FIGURE 1.

PLK1 regulates expression of the PcG protein BMI1 and PRC1 activity. A, MCF7, MDA-MB-231, and MDA-MB-468 cells stably expressing a control and two PLK1-specific shRNAs (PLKi46 and PLKi47) were generated by infection with respective retroviral vectors and selection in puromycin (1 μg/ml). The expression of PLK1, phospho-PLK1 (P-PLK1), BMI1, total H2A, H2AK119Ub, and β-actin was determined by Western blot analysis. B, MCF10A cells overexpressing PLK1 were generated by infecting cells with PLK1 expressing retrovirus, and selecting cells in G418 for 7 days. After selection, control (vector) and PLK1 WT-expressing cells were analyzed for PLK1, P-PLK1, BMI1, total H2A, H2AK119Ub, and β-actin by Western blot analysis. C, indicated sets of cells were treated with 0 (DMSO), 50, and 100 nm of PLK1 inhibitor BI 2536 for 24 h. The total cell lysates were prepared and expression of PLK1, P-PLK1, BMI1, total H2A, H2AK119Ub, and β-actin was detected by Western blot analysis. The proteins were quantified by densitometry using ImageJ software and the fold induction of each protein normalized to β-actin was determined. The fold induction of relevant protein is shown below the immunoblot (IB) of each protein.
Exogenous BMI1 Expression Overcomes Inhibition of Oncogenic Phenotypes Induced by PLK1 Inhibition
The PLK1 inhibitor BI 2536 is a potent inhibitor of cell proliferation and oncogenic phenotypes (36). If BMI1 is a physiologically relevant downstream target of PLK1, the exogenous overexpression of BMI1 should overcome the growth inhibitory effect of BI 2536 or PLK1 knockdown. To test this hypothesis, we stably overexpressed BMI1 in MCF7 cells (Fig. 2). MCF7 control (MCF7-B0) and BMI1 overexpressing MCF7 cells (MCF7-BMI1) were treated with BI 2536, and their rate of proliferation was determined by counting and plotting cell numbers for 3, 5, 7, and 9 days. Our data indicated that BI 2536 strongly inhibited proliferation of MCF7 control cells, and that BMI1 overexpression rescued proliferation inhibition induced by BI 2536 (Fig. 2A). We also determined whether exogenous expression of BMI1 can rescue inhibition of other oncogenic phenotypes such as colony formation in soft agar induced by PLK1 inhibitor BI 2536. MCF7-B0 and MCF7-BMI1 cells were mock- or BI 2536-treated, and soft agar colony formation potential of treated cells was determined. The results indicated that exogenous BMI1 expression (in MCF7-BMI1) can restore colony formation in BI 2536-treated cells (Fig. 2B).
FIGURE 2.
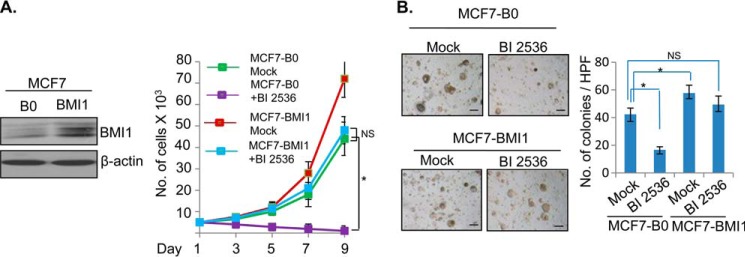
BMI1 overcomes inhibition of oncogenic phenotypes induced by PLK1 inhibitor BI 2536. A, left panel, MCF7 cells stably expressing BMI1 were generated by infecting cells with pBabe-BMI1 and selecting cells in puromycin (1 μg/ml for 5 days). MCF7 vector control (MCF7-B0) and MCF7-BMI1 cells were treated with 50 nm BI 2536 and allowed to grow for 3–9 days. Cells were counted at 3, 5, 7, and 9 days, and total number of cells plotted against number of days. B, MCF7-B0 control and MCF7-BMI1 cells were seeded in soft agar and treated with DMSO (mock) or 50 nm BI 2536 in DMSO added to the medium/soft agar, and allowed to form colonies for 10 days. The colonies were photographed under 10× magnification (mag), counted and plotted as a bar graph. The experiments were done in triplicates. Error bars represent ± S.D. *, p < 0.05 (significant); NS, not significant. Scale bars, 20 μm.
Next, we determined whether exogenous BMI1 can overcome growth inhibitory effect of PLK1 knockdown. For this experiment MCF7 cells stably expressing BMI1 were generated by infecting cells with pMSCV-BMI1 (hygro) vector. Control MCF7 (MCF7-pMSCV) and MCF7 cells expressing BMI1 (MCF7-pMSCV-BMI), were further infected with a control or 2 different PLK1 shRNA-expressing retroviral vectors. The cells were then selected in puromycin The resulting set of cell lines with combinations of vector or BMI1 expressing cells with a control shRNA or PLK1 shRNAs (PLKi46 and PLKi47) were further studied for cell proliferation and colony formation in soft agar (Fig. 3). The data indicated that the PLK1 knockdown resulted in strong inhibition in proliferation, and that BMI1 overexpression rescued proliferation inhibition caused by PLK1 knockdown (Fig. 3A). Similarly, soft agar colony formation assay indicated that PLK1 knockdown strongly inhibits colony formation in soft agar, whereas exogenous BMI1 overexpression rescues inhibition of colony formation potential of PLK1 knockdown cells (Fig. 3B). Western blot assays were done to confirm knockdown or inhibition of PLK1 and phospho-PLK1 by PLK1 shRNAs and BI 2536 (Fig. 3A, right panel). Western blot analysis also showed that the exogenous BMI1 overexpression can restore PLK1 and phospho-PLK1 expression (Fig. 3A, right panel).
FIGURE 3.

BMI1 overcomes inhibition of proliferation and oncogenic phenotype by PLK1 knockdown. A, MCF7-pMSCV (hygro) control and MCF7-pMSCV-BMI1 cells were generated (left panel), and subsequently infected with a control shRNA (Ctrl sh) or two different PLK1 shRNAs (PLKi46, PLKi47) to generate MCF7-Ctrl sh/pMSCV, MCF7-PLKi46/pMSCV, MCF7-PLKi47/pMSCV, MCF7-Ctrl sh/pMSCV-BMI1, MCF7-PLKi46/pMSCV-BMI1, and MCF7-PLKi47/pMSCV-BMI1 cells. The resulting cell lines were analyzed for cell proliferation over 9 days of growth by plotting cell number against number of days. The cell lines were also analyzed for the expression of PLK1, P-PLK1, BMI1, total H2A, H2AK119Ub, and β-actin. B, above described cell lines (as indicated) were plated for colony formation in soft agar. After 10 days, colonies were photographed and counted. The experiments were done in triplicates. Error bars represent ± S.D. *, p < 0.05 (significant); NS, not significant. Scale bars, 20 μm.
PLK1 Inhibition Suppresses Cancer Stem Cell (CSC) Phenotype and Exogenous BMI1 Overcomes Inhibition of CSC Phenotype by BI 2536 in Breast Cancer Cells
PcG protein BMI1 is known to regulate breast CSC phenotype (10, 24). Hence, we determined whether PLK1 inhibition results in down-regulation of CSC phenotype in breast cancer cells and whether BMI1 overexpression can rescue inhibition of CSC phenotype by PLK1 inhibition. First, control (B0) or BMI1-overexpressing MCF7 and MDA-MB-231 cells were mock- or BI 2536-treated and plated for mammosphere formation. After 7 days, the number of mammospheres were counted and plotted. The results indicated that BI 2536 strongly inhibited number and size of mammospheres in control MCF7 (MCF7-B0) and MDA-MB-231 (MDA-MB-231-B0) but not in BMI1-overexpressing- MCF7 and MDA-MB-231 cells (Fig. 4A). Next, we studied the effect of PLK1 inhibitor on CSC phenotype by determining the ALDH1 activity using an Aldefluor assay kit. The data indicated that the treatment with BI 2536 led to decrease in fraction of ALDH-positive cells from 2.53% to 0.95% (62.45% decrease), while BMI1 overexpression partially restored fraction of ALDH-positive cells (Fig. 4B). Similar results were obtained in MCF7 cells (not shown). We also studied the effect of BI 2536 on CD44 expression in MDA-MB-231 cell line, which have high proportion of CD44-expressing cells. The results indicated that BI 2536 decreased the fraction of CD44 positive cells to 61%, while exogenous BMI1 restored the fraction of CD44-positive cells (Fig. 4C). We further confirmed inhibition of CSC phenotype by PLK1 inhibition using PLK1 knockdown. MCF7 and MDA-MB-231 cells expressing exogenous BMI1 (pMSCV-BMI1) or vector control (pMSCV), and PLKi46 and PLKi47 or control shRNA were studied for mammosphere formation. The results indicated that PLK1 knockdown strongly inhibited mammosphere formation, and that BMI1 overexpression rescued inhibition of mammosphere formation by PLK1 knockdown in both MCF7 and MDA-MB-231 cells (Fig. 5).
FIGURE 4.

BI 2536 inhibits breast cancer stem cell (CSC) phenotype, and BMI1 overexpression overcomes inhibition of breast CSC phenotype by BI 2536. A, MCF7-B0 and MCF7-BMI1 (top panel), and MDA-MB-231-B0 and MDA-MB-231-BMI1 (bottom panel) cells were plated in serum-free mammosphere culture medium supplemented with MammoCult Proliferation Supplements (StemCell Technologies, Vancouver, Canada). After 7 days of cell plating, number of mammospheres (>50 μm) were counted, photographed under phase contrast (10× mag) and plotted. Error bars represent ± S.D. *, p < 0.05 (significant); NS, not significant. B, MDA-MB-231-B0 and MDA-MB-231-BMI1 cells were mock- or BI 2536-treated and studied for ALDH1 activity using an Aldefluor assay kit (StemCell Technologies). DEAB (ALDH inhibitor)-treated cells were used to set the background. BI 2536 treatment led to 62.45% decrease in the fraction of ALDH-positive cells (from 2.53% to 0.95%) in control MDA-MB-231-B0 cells. BMI1 overexpression partially overcomes the inhibitory effect of BI 2536 on ALDH activity as MDA-MB-231-BMI1 cells showed only 32.8% decrease in the fraction of ALDH-positive cells (from 4.42% to 2.97%). C, MDA-MB-231-B0 and MDA-MB-231-BMI1 cells were mock- or BI 2536-treated and analyzed for the expression of CD44 marker using FACS analysis.
FIGURE 5.

BMI1 overexpression overcomes inhibition of mammosphere formation by PLK1 knockdown. Control (pMSCV) and BMI1 (pMSCV-BMI1) overexpressing MCF7 and MDA-MB-231 cells expressing a control sh RNA or PLK1 sh RNAs (PLKi46 and PLKi47) (as indicated) were studied for mammosphere formation as described in Fig. 4A. In this case, the shRNA vector expresses GFP, hence mammospheres were photographed using a FITC filter (10× mag). Error bars represent ± S.D. *, p < 0.05 (significant); NS, not significant.
PLK1 Inhibition Induces Premature Senescence and Exogenous BMI1 Overcome Premature Senescence Induction by BI 2536 in Breast Cancer Cells
PLK1 inhibition results in mitotic arrest and cell death in most cancer cells (36). It can also result in premature senescence in human diploid fibroblasts (HDFs) via induction of p53 (37). To determine whether BI 2536 can induce premature senescence in MCF7 cells and that exogenous BMI1 expression can overcome premature senescence induced by BI 2536, we treated MCF7-B0 (vector control) and MCF7-BMI1 cells with the 50 nm BI 2536 for 24 h and stained cells for senescence-associated β-galactosidase (SA-β-Gal), which is a well-known marker of cellular senescence (40). We also stained cells with EdU, a proliferation marker, which can be used together with SA-β-gal marker to identify senescent cells (41). Our data indicated that BI 2536 strongly induced premature senescence in control MCF7 cells (MCF7-B0), but not in BMI1 overexpressing MCF7 cells (Fig. 6A). We also determined the expression of BMI1, PLK1, p53, and p21 in BI 2536 and mock-treated cells by Western blot analysis. As expected, results indicated that BI 2536 did not downregulate exogenous BMI1, and H2AK119Ub activity mediated by exogenously overexpressed BMI1 (Fig. 6A, right panel). There was a robust induction of p53 and p21 by BI 2536 in control B0 cells, but in BMI1-overexpressing cells p53 and p21 induction was attenuated (Fig. 6A, right panel). The total PLK1 and phospho-PLK1 levels were also down-regulated by BI 2536 in B0 control cells indicating that BI 2536 not only inhibits the activity of PLK1 but also its expression. Alternatively, lower levels of phospho-PLK1 could simply reflect lower expression of total PLK1 in these cells. MCF7 cells do not express p16, which is also a major regulator of senescence (43), and a target of BMI1 (16, 17). Hence, a similar experiment was carried out using another breast cancer cell line MDA-MB-468, which expresses p16. Our data indicated that PLK1 inhibition also resulted in senescence induction (Fig. 6B, left panel) and p16 up-regulation in addition to p21 up-regulation in MDA-MB-468 cells, and exogenous BMI1 strongly attenuated p16 induction by BI 2536 (Fig. 6B, right panel). No p53 induction was observed in control MDA-MB-468 cells, which express a mutant p53 indicating p21 induction by BI 2536 is independent of p53 in MDA-MB-468 cell line. In summary, our results indicate that PLK1 inhibition results in induction of premature senescence, which can be bypassed by exogenous BMI1 overexpression indicating that PLK1 regulates senescence via PcG protein BMI1.
FIGURE 6.

PLK1 inhibition induces premature senescence and exogenous BMI1 overcomes premature senescence induction by BI 2536. A, MCF7-B0 and MCF7-BMI1 cells were generated as described in Fig. 2, plated and grown for 2 days, and treated with 50 nm BI 2536 in 6-well plates and co-stained for SA-β-gal and EdU. The nuclei were visualized by DAPI staining as described in “Experimental Procedures.” The cells were photographed under phase contrast (SA-β-gal), red-filter (EdU), and UV-filter (DAPI). The percentage of SA-β-gal- and EdU-positive cells were determined by counting 200 cells in randomly chosen field and plotted. In parallel, cells were also analyzed for the expression of PLK1, phospho-PLK1 (P-PLK1), BMI1, total H2A, H2AK119Ub, p53, p21, and β-actin by Western blot analysis as described in Fig. 1. B, MDA-MB-468-B0 and MDA-MB-468-BMI1 cells were either mock treated with DMSO or treated with 50 nm BI 2536 for 24 h, and studied for the induction of senescence by SA-β-gal/EdU co-staining and Western blot analysis of indicated markers (including p16) as described above for MCF7-derived cells. Error bars represent ± S.D. *, p < 0.05 (significant); NS, not significant.
PLK1 Inhibition Up-regulates miR-200c/141 Cluster
Next, we determined the mechanism of BMI1 down-regulation by PLK1 inhibition. The BMI1 expression is post-transcriptionally regulated by miR-200c/141 cluster (27). Both miR-200c and miR-141 are potential tumor suppressive miRs and can induce senescence when overexpressed in HDFs (27). These miRNAs are often but not always co-overexpressed (29). Hence, we hypothesized that PLK1 inhibition may result in up-regulation of the miR-200c/141 cluster, which could then down-regulate endogenous BMI1 expression, but not exogenously overexpressed BMI1, which lack miR-200c and miR-141 targeting sequences in the 3′ UTR (untranslated region) of the BMI1. To probe this hypothesis, we first examined the expression of miR-200c and miR-141 by qRT-PCR in MCF7 and MDA-MB-231 cells that were either mock- or BI 2536-treated. The data indicated that expression of both miR-200c and miR-141 was increased in a dose-dependent manner (Fig. 7A). To further confirm the potential regulation of miR-200c and miR-141 by PLK1, we determined whether PLK1 knockdown in MCF7 results in up-regulation of miR-200c and miR-141 (Fig. 7B). We also examined whether PLK1 overexpression can down-regulate miR-200c and miR-141 expression in MCF10A cells (Fig. 7C). Our data indicated that indeed PLK1 knockdown results in up-regulation of miR-200c and miR-141, while PLK1 overexpression resulted in down-regulation of miR-200c and miR-141 (Fig. 7, B and C).
FIGURE 7.

PLK1 inhibition up-regulates miR-200c/141 cluster. A, MCF7 and MDA-MB-231 cells were treated with 0 (mock), 50, and 100 nm of PLK1 inhibitor BI 2536 for 24 h, and total RNA was prepared and analyzed for the expression of BMI1, miR-141, and miR-200c by qRT-PCR analysis using primers specific for each gene as described in “Experimental Procedures.” B and C, MCF7 and MDA-MB-231 cells expressing a control and PLK shRNAs (B), and MCF10A control and PLK1-overexpressing cells (MCF10A-PLK1 WT) cells (C), were analyzed for the expression of BMI1, miR-141, and miR-200c by qRT-PCR analysis. D, promoter region of miR-200c/141 cluster was cloned in pGL4.18, and 293T, MCF7, and MDA-MB-231 cells (as indicated) were transiently transfected, and treated with 0, 50, and 100 nm of BI 2536. After 24 h, the cell lysates were prepared, and luciferase assays were performed as described in “Experimental Procedures.” E, 3′-UTR assays using indicated pLS-luciferase reporter vectors were performed by transient transfection of vectors and a pTK-Cluc control plasmid in 293T, MCF7, and MDA-MB-231 cells as indicated. After transfection, cells were treated with BI 2536 as described above and relative luciferase activity each reporter was determined and plotted as described in “Experimental Procedures.” Error bars represent ± S.D. *, p < 0.05.
As both miR-200c and miR-141 can be co-transcribed through a common promoter (29), and both appear to be co-regulated by PLK1, we surmised whether PLK1 transcriptionally regulates miR-200c/141 cluster. To determine if this is the case, we cloned 639 bp upstream region of miR-200c/141 cluster in pGL4.18 reporter vector and examined whether BI 2536 treatment leads to up-regulation of miR-200c/141 promoter in 293T, MCF7, and MDA-MB-231 cells. The cells were transiently transfected with the miR-200c/141 promoter reporter, treated with BI 2536, and the luciferase activity was measured as described in the “Experimental Procedures.” The results indicated that BI 2536 up-regulated miR-200c/141 promoter activity in all three cell types (Fig. 7D).
Next, using 3′-UTR reporters that contain either wild type or mutant seed sequences of miR-200c and miR-141 present in BMI1 3′-UTR, we further confirmed that BI 2536-treated cells compared with untreated cells have a higher expression of miR-200c and miR-141. The results show that activity of wild type 3′-UTR reporter (pLS-miR-200c WT and pLS-miR-141 WT) but not mutant 3′-UTR reporters (pLS-miR-200cMut and pLS-miR-141Mut) activity is down-regulated by BI 2536 treatment indicative of increased expression of miR-200c and miR-141 in BI 2536-treated cells (Fig. 7E). In summary, these data strongly suggest that BI 2536 down-regulates BMI1 via up-regulation of the miR-200c/141 cluster.
Inhibition of miR-200c and miR-141 Overcomes Senescence Induction by PLK1 Inhibitor
To further confirm the role of miR-200c/141 cluster in PLK1 regulation of BMI1, we generated MCF7 and MDA-MB-231 cells stably expressing either miR-200c or miR-141 inhibitors, and determined whether in these cells BI 2536 can down-regulate BMI1 and induce premature senescence. The control, miR-200c, and miR-141 inhibitors expressing cells were treated with BI 2536 and the expression of BMI1, H2AK119Ub, p53, and p21 was determined by Western blot analysis (Figs. 8 and 9). The data indicated that BI 2536 cannot down-regulate endogenous BMI1 and H2AK119Ub in cells that are stably expressing inhibitors of either miR-141 (Fig. 8A) or miR-200c (Fig. 8B). Furthermore, there was no induction of p53 and p21 in miR-141 inhibitor expressing cells by BI 2536 (Fig. 8A), while slight induction of p53 and p21 was noticed in miR-200c inhibitor expressing cells (Fig. 8B), which nevertheless was much less compared with their levels in control BI 2536 treated cells (Fig. 8, A and B). Next, the miR-200c and miR-141 inhibitors expressing cells were studied for the induction of the senescent phenotype by BI 2536. The mock (DMSO) and BI 2536 treated control, and miR-200c and miR-141 inhibitors expressing cells were stained for SA-β-gal and EdU. The stained cells were photographed, counted and plotted. The results indicated that BI 2536 induced premature senescence in control, but not in cells that are stably expressing inhibitors of either miR-200c or miR-141 (Fig. 8, A and B, right panels), indicating that both miR-200c and miR-141 mediates the senescence inducing phenotype of the PLK1 inhibitor BI 2536. Similar results were obtained in MDA-MB-231 cells indicating that miR-141/200c inhibitors can overcome growth inhibitory and senescence inducing effects of BI 2536 (Fig. 9, A and B). Collectively, our data suggest that PLK1 inhibition leads to up-regulation of miR-200c and miR-141, which then down-regulate BMI1 leading to up-regulation of molecular markers of senescence and induction of premature senescence.
FIGURE 8.

Inhibition of miR-200c/141 overcomes senescence induction by BI 2536 in MCF7 cells. A, MCF7 cells overexpressing either a control inhibitor (CtrlI) or miR-141 Inhibitor (miR-141I) were treated with BI 2536 or DMSO (mock) and analyzed for the expression of BMI1, phospho-PLK1, PLK1, H2AK119Ub, total H2A, p53, p21, and β-actin by the Western blot analysis as described in Fig. 1 (left panel). MCF7-CtrlI and MCF7-miR-141I cells were also analyzed for the expression of miR-141 by qRT PCR analysis. The SA-β-gal/EdU co-staining was carried out in BI 2536-treated MCF7 cells expressing (CtrlI), miR-141Inhibitor (miR-141I) and quantification of senescent (SA-β-gal-positive) and proliferating (EdU-positive) cells was done as described in Fig. 6. In this experiment EdU staining was detected using FITC-labeled secondary antibody (right panel). B, MCF7 cells expressing a control inhibitor (CtrlI) or miR-200c inhibitor (miR-200cI) were mock- and BI 2536-treated and studied by Western blot analysis and markers of senescence (SA-β-gal/Edu) co-staining as described above for miR-141 inhibitor experiment. Each experiment was done in triplicates. The error bars represent ± S.D. *, p < 0.05 (significant); NS, not significant.
FIGURE 9.

Inhibitors of miR-141 and miR-200c overcome senescence induction of BI 2536 in MDA-MB-231 cells. A, MDA-MB-231 cells expressing control inhibitor (CtrlI) or miR-141 inhibitor (miR-141I) were mock- or BI 2536- treated, and studied for markers of senescence by Western blot analysis and SA-β-gal/EdU co-staining as described in Fig. 8. B, mock or BI 2536-treated MDA-MB-231 cells with CtrlI or miR-200cI were analyzed for the markers of senescence by Western blot analysis (left panel) and co-staining for SA-β-gal/EdU. The error bars represent ± S.D. *, p < 0.05 (significant); NS, not significant.
DISCUSSION
PcG proteins are aberrantly expressed in most cancer cells and are required for the self-renewal and proliferation of CSCs. The CSCs differentiate into non-stem cancer cells that form the bulk of the tumor, and are resistant to most chemotherapeutic agents (44). Because CSCs are resistant to most therapeutics, they play a key role in tumor dormancy and disease recurrence. Hence, CSCs are attractive targets for cancer therapy as well as cancer prevention. BMI1, the most well characterized PcG protein is a critical regulator of CSCs (45). The self-renewal of breast, prostate and colorectal CSCs have been shown to be regulated by BMI1 (10, 46, 47). Hence, the therapeutics that target BMI1 and inhibit its PRC activity can potentially help in the treatment and prevention of breast, prostate, colorectal, and possibly other cancers.
Here, we report that BI 2536, a well-known inhibitor of PLK1, is a potent inhibitor of BMI1 and PRC1 activity. It also suppressed breast CSC phenotype, and induced cellular senescence, the two phenotypes strongly regulated by BMI1. The inhibition of BMI1 expression by BI 2536 suggests that this inhibitor has a broad anti-proliferative activity and that it can suppress the expression of oncogenic proteins such as BMI1, which are not structurally related to PLKs. It is very likely that such an inhibition by BI 2536 results indirectly via transcriptional, post-transcriptional, or posttranslational mechanisms. Indeed, our data suggest that BI 2536 up-regulates a tumor suppressive miRNA cluster, that encodes both miR-200c and miR-141, which inhibit epithelial to mesenchymal transition (EMT) and metastasis via posttranscriptional down-regulation of multiple targets, most notably ZEB1, and ZEB2 (48–50). The ZEB/miR-200c/141 feedback loop is thought to control cellular plasticity, which may be relevant to development and cancer (48, 49, 51). Another important target of miR-200c/141 cluster is PcG protein BMI1 (10, 27). It has been shown that miR-200c inhibits breast CSC phenotype by targeting BMI1 (10), and both miR-200c and miR-141 upregulate senescence, a strong tumor suppressive mechanism in HDFs (27).
Induction of senescence by miR-200c and miR-141 depends on BMI1 (27); hence, BI 2536 may exhibit anti-proliferative activity via induction of premature senescence in tumor cells. Our data suggests that indeed BI 2536 induces senescence in breast cancer cells, which can be abrogated either by inhibiting miR-200c and miR-141 or by overexpression of an exogenous BMI1, which lacks targeting sequence for miR-200c/141. These data are consistent with a molecular mechanism whereby BI 2536 induces expression of miR-200c and miR-141, and these miRNAs act post-transcriptionally to down-regulate BMI1, resulting in induction of senescence. The induction of senescence in MCF7 cells by BI 2536 appears to involve p53/p21 pathway as BI 2536 strongly induced p53 and p21 in MCF7 cells. Because BMI1 overexpression attenuated the induction of p53 and p21 in these cells by BI 2536, it also reversed senescence induction by BI 2536. The senescence induction by BI 2536 is mediated by p53/p21 and p16INK4a tumor suppressors, depending on the genetic makeup of the cancer cells. In MCF7 cells, which do not express p16INK4a, induction of p53/p21 is sufficient to induce senescence. On the other hand, in MDA-MB-468 cells that express mutant p53 and a functional p16INK4a, BI 2536 induces p16INK4a, and p21 but not p53, suggesting that p53-independent induction of p21, and p16INK4a are sufficient to induce senescence by PLK1 inhibition. Both miR-200c and miR-141 can target additional cancer relevant and growth regulatory factors such as ZEB1 and ZEB2; it is possible that in addition to BMI1, these factors are also induced by BI 2536 and that they may also play a role in up-regulation of p53/p21 and p16INK4a, and induction of senescence. The PcG protein BMI1 itself can regulate many growth regulatory pathways including WNT pathway and other tumor suppressors such as p57. Hence, it is likely that PLK1 inhibition acts via deregulation of multiple growth pathways that act downstream of miR-200c/141 and BMI1. Regulation of additional factors by BI 2536, and targets other than p53/p21 in senescence induction is currently under investigation in our laboratory.
Although, BI 2536 is thought to be a specific inhibitor of PLK1, it is possible that it inhibits BMI1 via non-canonical mechanism, which involves a hitherto unknown mechanism by which BI 2536 induces miR-200c and miR-141 leading to down-regulation of BMI1. However, our data do not favor this possibility, as the knockdown of PLK1 also downregulated BMI1 and induced both miR-200c and miR-141. Similarly, overexpression of PLK1 up-regulated BMI1, and suppressed the expression of miR-200c/141 cluster. Collectively, our data support a model whereby PLK1 inhibition leads to transcriptional up-regulation of the miR-200c/141 cluster, which results in down-regulation of BMI1 and suppression of the oncogenic phenotypes, including induction of premature senescence. Down-regulation of BMI1 and PRC1 activity very likely leads to the derepression of PRC targets, which include tumor suppressors and differentiation-specific growth inhibitors. There are many other PLK1 inhibitors in the clinical development (34, 35), which may exhibit similar or stronger inhibitory activity toward PcG proteins, in particular BMI1. Studies presented here also suggest that PLK1 inhibitors could be used to suppress other BMI1-regulated oncogenic phenotypes, in particular CSC phenotype. As CSCs are thought to promote therapy resistance and play a role in activation of dormant cancer cells, PLK1 inhibitors could be used to eliminate CSC population and overcome therapy resistance. In summary, our data support therapeutic use of PLK1 inhibitors in various cancers where PcG proteins, in particular BMI1 are overexpressed and their targets are epigenetically silenced.
This work was supported in part by R01 CA094150 from the NCI, National Institutes of Health (to G. P. D.).
- PcG
- polycomb group
- CSC
- cancer stem cell
- PRC
- polycomb repressive complex
- PTM
- post-translational modification
- PLK
- Polo-like kinase.
REFERENCES
- 1. Sparmann A., van Lohuizen M. (2006) Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 6, 846–856 [DOI] [PubMed] [Google Scholar]
- 2. Richly H., Aloia L., Di Croce L. (2011) Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Disease 2, e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Valk-Lingbeek M. E., Bruggeman S. W., van Lohuizen M. (2004) Stem cells and cancer; the polycomb connection. Cell 118, 409–418 [DOI] [PubMed] [Google Scholar]
- 4. Glinsky G. V., Berezovska O., Glinskii A. B. (2005) Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 115, 1503–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo W. J., Zeng M. S., Yadav A., Song L. B., Guo B. H., Band V., Dimri G. P. (2007) Mel-18 acts as a tumor suppressor by repressing Bmi-1 expression and down-regulating Akt activity in breast cancer cells. Cancer Res. 67, 5083–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim J. H., Yoon S. Y., Jeong S. H., Kim S. Y., Moon S. K., Joo J. H., Lee Y., Choe I. S., Kim J. W. (2004) Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast 13, 383–388 [DOI] [PubMed] [Google Scholar]
- 7. Di Croce L., Helin K. (2013) Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155 [DOI] [PubMed] [Google Scholar]
- 8. Simon J. A., Kingston R. E. (2013) Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49, 808–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lobo N. A., Shimono Y., Qian D., Clarke M. F. (2007) The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 23, 675–699 [DOI] [PubMed] [Google Scholar]
- 10. Shimono Y., Zabala M., Cho R. W., Lobo N., Dalerba P., Qian D., Diehn M., Liu H., Panula S. P., Chiao E., Dirbas F. M., Somlo G., Pera R. A., Lao K., Clarke M. F. (2009) Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138, 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lessard J., Sauvageau G. (2003) Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423, 255–260 [DOI] [PubMed] [Google Scholar]
- 12. Leung C., Lingbeek M., Shakhova O., Liu J., Tanger E., Saremaslani P., Van Lohuizen M., Marino S. (2004) Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 428, 337–341 [DOI] [PubMed] [Google Scholar]
- 13. Liu S., Ginestier C., Ou S. J., Clouthier S. G., Patel S. H., Monville F., Korkaya H., Heath A., Dutcher J., Kleer C. G., Jung Y., Dontu G., Taichman R., Wicha M. S. (2011) Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 71, 614–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Molofsky A. V., He S., Bydon M., Morrison S. J., Pardal R. (2005) Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 19, 1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sangiorgi E., Capecchi M. R. (2008) Bmi1 is expressed in vivo in intestinal stem cells. Nature Genetics 40, 915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Itahana K., Zou Y., Itahana Y., Martinez J. L., Beausejour C., Jacobs J. J., Van Lohuizen M., Band V., Campisi J., Dimri G. P. (2003) Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol. Cell. Biol. 23, 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacobs J. J., Kieboom K., Marino S., DePinho R. A., van Lohuizen M. (1999) The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168 [DOI] [PubMed] [Google Scholar]
- 18. Dimri G. P. (2004) The search for biomarkers of aging: next stop INK4a/ARF locus. Sci. Aging Knowledge Environ 2004, pe40. [DOI] [PubMed] [Google Scholar]
- 19. Dimri G. P., Martinez J. L., Jacobs J. J., Keblusek P., Itahana K., Van Lohuizen M., Campisi J., Wazer D. E., Band V. (2002) The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 62, 4736–4745 [PubMed] [Google Scholar]
- 20. Bommi P. V., Dimri M., Sahasrabuddhe A. A., Khandekar J., Dimri G. P. (2010) The polycomb group protein BMI1 is a transcriptional target of HDAC inhibitors. Cell Cycle 9, 2663–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Datta S., Hoenerhoff M. J., Bommi P., Sainger R., Guo W. J., Dimri M., Band H., Band V., Green J. E., Dimri G. P. (2007) Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 67, 10286–10295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim R. H., Lieberman M. B., Lee R., Shin K. H., Mehrazarin S., Oh J. E., Park N. H., Kang M. K. (2010) Bmi-1 extends the life span of normal human oral keratinocytes by inhibiting the TGF-β signaling. Exp. Cell Res. 316, 2600–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gargiulo G., Cesaroni M., Serresi M., de Vries N., Hulsman D., Bruggeman S. W., Lancini C., van Lohuizen M. (2013) In Vivo RNAi Screen for BMI1 Targets Identifies TGF-β/BMP-ER Stress Pathways as Key Regulators of Neural- and Malignant Glioma-Stem Cell Homeostasis. Cancer cell 23, 660–676 [DOI] [PubMed] [Google Scholar]
- 24. Cho J. H., Dimri M., Dimri G. P. (2013) A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J. Biol. Chem. 288, 3406–3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo W. J., Datta S., Band V., Dimri G. P. (2007) Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol. Biol. Cell 18, 536–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nowak K., Kerl K., Fehr D., Kramps C., Gessner C., Killmer K., Samans B., Berwanger B., Christiansen H., Lutz W. (2006) BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 34, 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dimri M., Carroll J. D., Cho J. H., Dimri G. P. (2013) microRNA-141 regulates BMI1 expression and induces senescence in human diploid fibroblasts. Cell Cycle 12, 3537–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burk U., Schubert J., Wellner U., Schmalhofer O., Vincan E., Spaderna S., Brabletz T. (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Reports 9, 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neves R., Scheel C., Weinhold S., Honisch E., Iwaniuk K. M., Trompeter H. I., Niederacher D., Wernet P., Santourlidis S., Uhrberg M. (2010) Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Research Notes 3, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhattacharya R., Nicoloso M., Arvizo R., Wang E., Cortez A., Rossi S., Calin G. A., Mukherjee P. (2009) MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 69, 9090–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He X., Dong Y., Wu C. W., Zhao Z., Ng S. S., Chan F. K., Sung J. J., Yu J. (2012) MicroRNA-218 inhibits cell cycle progression and promotes apoptosis in colon cancer by downregulating BMI1 polycomb ring finger oncogene. Mol. Med. 18, 1491–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jin M., Zhang T., Liu C., Badeaux M. A., Liu B., Liu R., Jeter C., Chen X., Vlassov A. V., Tang D. G. (2014) miRNA-128 suppresses prostate cancer by inhibiting BMI-1 to inhibit tumor-initiating cells. Cancer Res. 74, 4183–4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cholewa B. D., Liu X., Ahmad N. (2013) The role of polo-like kinase 1 in carcinogenesis: cause or consequence? Cancer Res. 73, 6848–6855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Strebhardt K. (2010) Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nature Reviews. Drug Discovery 9, 643–660 [DOI] [PubMed] [Google Scholar]
- 35. Strebhardt K., Ullrich A. (2006) Targeting polo-like kinase 1 for cancer therapy. Nature Reviews. Cancer 6, 321–330 [DOI] [PubMed] [Google Scholar]
- 36. Steegmaier M., Hoffmann M., Baum A., Lénárt P., Petronczki M., Krssák M., Gürtler U., Garin-Chesa P., Lieb S., Quant J., Grauert M., Adolf G. R., Kraut N., Peters J. M., Rettig W. J. (2007) BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 17, 316–322 [DOI] [PubMed] [Google Scholar]
- 37. Kim H. J., Cho J. H., Kim J. R. (2013) Downregulation of Polo-like kinase 1 induces cellular senescence in human primary cells through a p53-dependent pathway. J. Gerontol. 68, 1145–1156 [DOI] [PubMed] [Google Scholar]
- 38. Dimri G. P., Itahana K., Acosta M., Campisi J. (2000) Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol. Cell. Biol. 20, 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sahasrabuddhe A. A., Dimri M., Bommi P. V., Dimri G. P. (2011) βTrCP regulates BMI1 protein turnover via ubiquitination and degradation. Cell Cycle 10, 1322–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U. S. A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Itahana K., Itahana Y., Dimri G. P. (2013) Colorimetric detection of senescence-associated β galactosidase. Methods Mol. Biol. 965, 143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dimri M., Naramura M., Duan L., Chen J., Ortega-Cava C., Chen G., Goswami R., Fernandes N., Gao Q., Dimri G. P., Band V., Band H. (2007) Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 67, 4164–4172 [DOI] [PubMed] [Google Scholar]
- 43. Dimri G. P. (2005) What has senescence got to do with cancer? Cancer Cell 7, 505–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Malik B., Nie D. (2012) Cancer stem cells and resistance to chemo and radio therapy. Front. Biosci. 4, 2142–2149 [DOI] [PubMed] [Google Scholar]
- 45. Siddique H. R., Saleem M. (2012) Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells 30, 372–378 [DOI] [PubMed] [Google Scholar]
- 46. Kreso A., van Galen P., Pedley N. M., Lima-Fernandes E., Frelin C., Davis T., Cao L., Baiazitov R., Du W., Sydorenko N., Moon Y. C., Gibson L., Wang Y., Leung C., Iscove N. N., Arrowsmith C. H., Szentgyorgyi E., Gallinger S., Dick J. E., O'Brien C. A. (2014) Self-renewal as a therapeutic target in human colorectal cancer. Nature Medicine 20, 29–36 [DOI] [PubMed] [Google Scholar]
- 47. Lukacs R. U., Memarzadeh S., Wu H., Witte O. N. (2010) Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 7, 682–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brabletz S., Brabletz T. (2010) The ZEB/miR-200 feedback loop–a motor of cellular plasticity in development and cancer? EMBO Rep. 11, 670–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gregory P. A., Bert A. G., Paterson E. L., Barry S. C., Tsykin A., Farshid G., Vadas M. A., Khew-Goodall Y., Goodall G. J. (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biology 10, 593–601 [DOI] [PubMed] [Google Scholar]
- 50. Park S. M., Gaur A. B., Lengyel E., Peter M. E. (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 22, 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brabletz S., Bajdak K., Meidhof S., Burk U., Niedermann G., Firat E., Wellner U., Dimmler A., Faller G., Schubert J., Brabletz T. (2011) The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 30, 770–782 [DOI] [PMC free article] [PubMed] [Google Scholar]