

Background: The redox activity of Ref-1 activates the binding of several transcription factors important in cancer.
Results: Repression of Ref-1 potently activates NRF2 resulting in up-regulation of target gene expression.
Conclusion: Activation of NRF2 is a potential mechanism of resistance to therapies based on Ref-1 inhibition.
Significance: Dual blockade of Ref-1 and NRF2 or the specific downstream target, HMOX-1, represents a strategy for overcoming resistance.
Keywords: Hypoxia, Nuclear Factor 2 (Erythroid-derived 2-like Factor) (NFE2L2) (Nrf2), Reactive Oxygen Species (ROS), Redox Signaling, Transcription Factor, APE1/Ref-1, HMOX1, Pancreatic Adenocarcinoma
Abstract
Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) (henceforth referred to as Ref-1) is a multifunctional protein that in addition to its base excision DNA repair activity exerts redox control of multiple transcription factors, including nuclear factor κ-light chain enhancer of activated B cells (NF-κB), STAT3, activator protein-1 (AP-1), hypoxia-inducible factor-1 (HIF-1), and tumor protein 53 (p53). In recent years, Ref-1 has emerged as a promising therapeutic target in cancer, particularly in pancreatic ductal carcinoma. Although a significant amount of research has centered on Ref-1, no wide-ranging approach had been performed on the effects of Ref-1 inhibition and transcription factor activity perturbation. Starting with a broader approach, we identified a previously unsuspected effect on the nuclear factor erythroid-related factor 2 (NRF2), a critical regulator of cellular defenses against oxidative stress. Based on genetic and small molecule inhibitor-based methodologies, we demonstrated that repression of Ref-1 potently activates NRF2 and its downstream targets in a dose-dependent fashion, and that the redox, rather than the DNA repair function of Ref-1 is critical for this effect. Intriguingly, our results also indicate that this pathway does not involve reactive oxygen species. The link between Ref-1 and NRF2 appears to be present in all cells tested in vitro, noncancerous and cancerous, including patient-derived tumor samples. In particular, we focused on understanding the implications of the novel interaction between these two pathways in primary pancreatic ductal adenocarcinoma tumor cells and provide the first evidence that this mechanism has implications for overcoming the resistance against experimental drugs targeting Ref-1 activity, with clear translational implications.
Introduction
Redox factor-1 (Ref-1)5 is a dual function protein that in addition to DNA repair function controls the activity of multiple transcription factors, including NF-κB (nuclear factor-κB), STAT3, AP-1 (activator protein-1), and HIF-1 (hypoxia inducible factor) (1–4). The redox activity of Ref-1 reduces specific cysteine residues in the DNA binding domain of these transcription factors, thus stimulating their DNA binding activity (5). As most transcription factors stimulated by Ref-1 are well recognized regulators of tumorigenesis, this protein has emerged as a viable therapeutic target in cancer (3, 5–7). Particular attention has been given to pancreatic ductal adenocarcinoma (PDAC) as Ref-1 levels are known to be elevated in a variety of human PDAC-derived cell lines as well as in neoplastic tissue and peri-pancreatic metastases. We and others demonstrated that silencing Ref-1 in pancreatic cancer cells resulted in apoptosis and decreased proliferative capacity (8, 9). Furthermore, blockade of Ref-1 redox activity delayed tumor progression in xenograft models of human PDAC, including patient-derived tumor cells (4).
However, much remains to be elucidated about the functions of Ref-1, particularly with respect to the biochemical consequences of its inhibition. Detailed knowledge at this level is anticipated to increase the effectiveness of Ref-1 inhibitors and help delay/overcome therapeutic resistance to such agents. To this end, we performed a comprehensive survey of the effects of Ref-1 inhibition on the activity of a broad spectrum of transcription factors, using a library of reporters (Attagene, Inc.) (10). Our approach led to a novel connection between Ref-1 and nuclear factor erythroid-related factor 2 (NRF2), a critical regulator of cellular defenses against oxidative stress (11).
MATERIALS AND METHODS
Cell Lines and Patient-derived PDAC Cells
MIA-PaCa-2 were purchased from and authenticated by ATCC (Manassas, VA). Pa03C, Panc10.05, Panc 198, and Pa02C were obtained from Dr. Anirban Maitra, The Johns Hopkins University (12). All cells were maintained at 37 °C in 5% CO2 and grown in DMEM (Invitrogen) with 10% FBS (Hyclone, Logan, UT) and routinely tested for mycoplasma.
Inhibitors
E3330 and RN7–58 were synthesized as previously described (13–15), and tin porphyrin (Sn-PP) was purchased from Sigma. As a negative control, the E3330 analog, RN7–58 was used. RN7–58, although structurally similar to E3330, does not inhibit the redox activity of Ref-1 (16).
Transfection of PDAC Cells with siRNA
All siRNA transfections (Ref-1 (referred to as siRef-1#1) or scrambled control) were performed as previously described (3, 13, 17–19). Samples for the Attagene screen and quantitative PCR (QPCR) were collected 72 h after transfection of cancer cells with Ref-1 siRNA. For additional Ref-1 siRNA experiments, we purchased prevalidated siRNAs from LifeTech (#s1446, siRef-1#2, and s1447, siRef-1#3).
Attagene cis-FACTORIAL Screen
Our initial screen for the effects of Ref-1 on the activity of a diverse panel of transcription factors was performed using cis-FACTORIAL technology from Attagene followed by validation of the NRF2 pathway using the relevant reporter from the library (10). PaCa-2 cells were transfected with siRef-1 or scrambled siRNA as above. Twenty-four hours later cells were washed with fresh medium and transiently transfected with cis-FACTORIALTM. Twenty-four hours after transfection cells were supplied with fresh medium (containing 10% FBS) and incubated for an additional 24 h. Profiles of the FACTORIALTM end point activities were determined as fold-induction values of siRef-1 or scrambled RNA-transfected cells divided by values from untransfected cells. The methodology is described in detail in Refs. 10, 20, and 21).
QPCR Reactions
This method was used to measure the mRNA expression levels of NRF2 and its downstream target genes, HMOX-1 (heme oxygenase-1), GCLC (glutamate-cysteine ligase, catalytic subunit), and GCLM (glutamate-cysteine ligase, modifier subunit). Total RNA was extracted from cells using the Qiagen RNeasy Mini kit (Valencia, CA) according to the manufacturer's instructions. The extracted RNA was quantified using a Qubit fluorometer (Invitrogen). First-strand cDNA was prepared from RNA using random hexamers and MultiScribe reverse transcriptase (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using TaqMan Gene Expression assays and Universal PCR master mix (Applied Biosystems) in a 7900HT Sequence detection system (Applied Biosystems). The relative quantitative mRNA level was determined using the comparative Ct method using Actin (PaCa-2) or large ribosomal protein, P0 (RPLP0, patient lines) as the reference gene (4). The primers for NRF2, HMOX-1, GCLC, GCLM, Actin, and RPLP0 are commercially available (Applied Biosystems). Experiments were performed in triplicate for each sample.
Western Blot Analysis
Whole cell lysates were prepared by lysing the cells in RIPA buffer (Santa Cruz Biotechnology, Santa Cruz, CA), followed by quantification of protein concentration (Lowry protein assay). Proteins were separated by SDS-PAGE, electroblotted onto nitrocellulose, and immunoblotting was performed using the following antibodies: Ref-1 and NRF2 (Abcam 62352), HMOX-1 (Abcam, Cambridge, MA), and Ku70, tubulin, or Actin (Sigma).
Reactive Oxygen Species (ROS) Measurement
The production of ROS was determined by detecting the fluorescent intensity of the oxidation-sensitive probe dihydrorhodamine 123 (DHR) (Molecular Probes, Invitrogen). PaCa-2 and Pa03C cells were treated with E3330 for 24 h. As a positive control for ROS production, tert-butyl hydroperoxide solution (1 mm, 30 min) was utilized. After washing with PBS, the cells were incubated with 1 μm DHR in fresh PBS for 30 min. Excessive probe was washed off using PBS. Cells were harvested with trypsin, and ROS fluorescence of labeled cells was measured by using a Coulter EPICS XL flow cytometer (Coulter). An average of 10,000 cells from each sample was counted, and each experiment was done in triplicate.
Co-immunoprecipitation (Co-IP)
Samples were co-immunoprecipitated using the Pierce Co-IP kit (Thermo Scientific) with the following modifications. Cells were washed twice with ice-cold PBS and the proteins were cross-linked using the water soluble, membrane permeable, imidoester cross-linker dimethyl 3,3′-dithiobispropionimidate (Thermo Scientific, 5 mm, for 30 min on ice). Dimethyl 3,3′-dithiobispropionimidate was quenched by sequential washing with cold inactivation buffer (100 mm Tris-HCl, pH 8, 150 mm NaCl) and PBS. Cells were lysed by the addition of IP lysis buffer supplemented with protease inhibitors (800 μl/10-cm dish; 4 °C, 20 min on rocking platform) and then scraped and transferred to a microcentrifuge tube. Cell debris was removed by centrifugation and the protein concentration of the cleared lysate was determined using the Pierce BCA Protein assay. To reduce nonspecific protein binding, 1.4 mg of cell lysate was pre-cleared using the control agarose resin, prior to adding to columns of either Ref-1 or NRF2 antibody or control rabbit IgG that had been covalently coupled onto an amine-reactive agarose resin. After extensive washing, the bound proteins were eluted and prepared for SDS-PAGE analysis by the addition of 5× sample buffer containing 100 mm DTT.
ChIP Assay
We performed ChIP assay according to the manufacturer's protocol (Millipore). PaCa-2 cells were incubated for 16 h in the presence of 67.5 μm E3330, or 0.17% DMSO in DMEM with 2% FBS. Proteins were cross-linked to DNA by the addition of 37% formaldehyde (to a final concentration of 1%, 10 min, room temperature) and then quenched by adding glycine (125 mm, 5 min, room temperature). Cell lysates were sonicated (Sonic Dismembrator model 100 (Fisher) setting number 4, 12 cycles, 10 s) until cross-linked DNA was sheared to achieve lengths of 200–600 bp. The antibodies used were Ref-1 (2 μg), NRF2 (3 μg), or control rabbit IgG (Cell Signaling). PCR primer sequences used are in Table 1.
TABLE 1.
Primers utilized in ChIP assay
| Primer name | Sequence 5′ to 3′ | HMOX1 promoter location | Reference |
|---|---|---|---|
| E1 forward | CAGTGCCTCCTCAGCTTCTC | − 3899 to − 3880 | 30 |
| E1 reverse | CTCGGTGGATTGCAACATTA | − 3716 to − 3697 | 30 |
| E2 forward | TAATCCTTTCCCGAGCCA | − 9083 to − 9065 | 30 |
| E2 reverse | GGAACTCTGAGGAAAACAAATC | − 8941 to − 8918 | 30 |
| MCM5 forward | AGACCATGCGTCAGGAAA | 30 | |
| MCM5 reverse | CTGGCTGGGAAGGAAGTG | 30 |
Transient Luciferase Reporter Assays
To generate an NRF2 activity reporter, we inserted into the pGL4 backbone vector the following sequence that corresponds to the NAD(P)H:quinone oxidoreductase antioxidant response element (underlined): CCG CTC GAG AAA TCG CAG TCA CAG TGA CTC AGC AGA ATC TGA GCC TGG GCT ATA AAA GGG GGT GGG GGC GCG TTC GTC CTC AAG CTT GGG. Bases in italics indicate the minimal TATA-like sequences (10). PaCa-2 and Pa02C cells were co-transfected with the reporter construct and a Renilla luciferase control, pRL-TK (Promega Corp., Madison, WI), in a 20:1 ratio by using Lipofectamine 2000 (Invitrogen). Sixteen hours after transfection, cells were treated with E3330 in 2–5% serum-containing media for 24 h, followed by assay for Firefly and Renilla luciferase activity using the Dual Luciferase Reporter Assay System (Promega Corp.) Each time, a medium-only control was included, and the final data are expressed as the fold-change of the relative luciferase units compared with the vehicle control. DMSO concentration was equivalent in all samples and the final concentration was less than 0.25%. All of the transfection experiments were performed in triplicate and repeated at least 3 times in independent experiments.
Proliferation Studies
The proliferative capacity of PDAC cells was assessed using the MTS assay as previously described (4). Cells were treated with varying doses of either E3330 or tin protoporphyrin, alone, or in combination with either hypoxia exposure (0.2% O2, Ruskinn InVivo200 hypoxia work station) or low glucose containing medium for 48 h.
Tumors for HMOX-1 Immunohistology
The tumors in this study had been maintained as a live PancXenoBank (4, 12). Female nu/nu athymic mice (Harlan) were treated with either vehicle control or E3330, administered twice daily, 8 h apart, at 25 mg/kg for a total of 20 doses (5 days on 2 days off, 5 days on and 2 days off schedule) as previously described (4). At the end of this regimen, we collected tumor samples for immunostaining.
Statistical Analysis
All QPCR data points for vehicle, E3330, and RN7–58 treatments were analyzed using the 2−ΔΔCT method and analysis of covariance (ANCOVA) models (22). Specifically, for each target gene (HMOX-1, GCLC, and GCLM) and each dose of Ref-1 inhibitor (25, 35, and 50 μm E3330) or negative control, RN7–58 (RN25, RN35, and RN50), an ANCOVA model using threshold cycle number, CT of both treatment and vehicle control (DMSO) for both the target gene, and the reference gene (Actin or RPLP0, for normalization) were used as the response variable in the ANCOVA model. Covariates including group (treatment or control), target (target or reference gene), and their interaction were adjusted. If multiple experiments were conducted on different days, days were adjusted. The coefficient of the interaction terms in the ANCOVA model is the estimate of the ΔΔCt, which is the difference of normalized target gene CT between treatment and control sample, e.g. (CT,HMOX-1(E3330) − CT,RPLP0(E3330)) − (CT,HMOX-1(DMSO) − CT,RPLP0–1(DMSO)). The 95% confidence interval of ΔΔCt and p value was also estimated based on the ANCOVA model. The ΔΔCt estimates and the bounds of 95% confidence interval were transformed to 2−ΔΔCt value as the fold-change estimate and its confidence interval. Differences between the treatment and control groups were considered significant if p < 0.05. All Statistical analyses were performed using SAS (Version 9.3, SAS Institute Inc., Cary, NC).
RESULTS
Knockdown of Ref-1 Increases NRF2 Activity and Expression of NRF2 Target, HMOX-1
Using the cis-FACTORIAL assay from Attagene (10) to screen for activity of >50 transcription factors, following Ref-1 knockdown we observed changes in previously reported Ref-1 targets such as AP-1 (data not shown). Unexpectedly, the most robust change in response to blockade of Ref-1 blockade was the induction of NRF2 (NFE2L2) activity. When Ref-1 protein levels were knocked down by more than 85%, NRF2 activity was increased 3.5-fold compared with Scrambled (SCR) control in PaCa-2 cells (Fig. 1A). In contrast, the impact of Ref-1 knockdown on NRF1 activity was minimal (1.3-fold, Fig. 1A). To ensure this was not a cell-specific observation, we expanded these findings in PaCa-2 cells to patient-derived pancreatic cancer cells as well as a cancer-associated fibroblast line. As shown in Fig. 1B, the knockdown of Ref-1 was effective in all cell lines tested. The effect of Ref-1 knockdown on the well recognized NRF2 target, HMOX-1, was assessed using QPCR and immunoblotting. In 4 of the 6 lines tested, decreased expression of Ref-1 corresponds to a statistically significant and generally robust increase in HMOX-1 as shown by QPCR (2–6-fold, Fig. 1C, 95% confidence interval of ΔΔCt and p value based on the ANCOVA model). To demonstrate that this effect is dependent upon Ref-1, patient-derived Pa03C cells were treated with increasing amounts of Ref-1 siRNA#1 (2–25 nm). On day 3 following transfection, we observed a dose-dependent increase in HMOX-1 protein as Ref-1 protein levels decrease (Fig. 1D). This was confirmed with two additional siRNAs against Ref-1, which also led to significant increases of HMOX-1 levels (Fig. 1E).
FIGURE 1.

Transcriptional activity of NRF2 and NRF1 after Ref-1 knockdown in PaCa-2 cells. A, PaCa-2 cells were transfected on sequential days with siRef-1#1 and then a cis-FACTORIAL system. After an additional 24 h, NRF1 and NRF2 activities were determined as fold-induction values of scrambled or siRef-1-transfected cells divided by values from mock transfected cells. Data are shown as fold-induction values. *, p < 0.05 comparing siRef-1 versus scrambled control; n = 3. B, immunoblotting for Ref-1 on day 3 following transfection of 100 nm siRNA; C, NRF2 target gene HMOX-1 RNA levels are elevated in patient-derived tumor cells and cancer-associated fibroblast line (CAF19) following knockdown of Ref-1 with si#1. HMOX-1 QPCR was performed on day 3 following transfection of siRNA. D, Western blot for HMOX-1 protein levels in Pa03C cells following Ref-1 knockdown (day 3) with increasing amounts of siRNA#1; E, Western blot showing Ref-1 protein levels are decreased following transfections with Ref-1 siRNAs. QPCR for HMOX-1 levels following transfection of Ref-1 siRNA#1, siRNA#2 (s1446), and siRNA#3 (s1447; 50 nm, PaCa-2 day 3 following transfection). *, p < 0.05: **, p < 0.01 comparing siRef-1 versus scrambled control; n = 3–6.
Inhibition of Ref-1 Using E3330 Activates NRF2 Transcriptional Activity
To more specifically address the role of the redox function of Ref-1 on NRF2 transcriptional activity, we generated a luciferase construct with an antioxidant response element sequence known to be activated by NRF2 (23).
As the siRNA-based approach removes both redox and DNA repair functions of Ref-1, as well as any protein-protein interactions involving this protein, we used the small molecule, E3330, that directly inhibits the redox function of Ref-1 without affecting its well documented endonuclease activity (14, 24–26). In both PaCa-2 and low passage patient-derived cell Pa02C, inhibition of Ref-1 with E3330 markedly stimulates NRF2 activity in a dose-dependent manner in transient luciferase assays (Fig. 2, gray bars, p < 0.05). Furthermore, treatment with the E3330 analog RN7–58 did not show significant stimulation of NRF2 activity in Pa02C cells (Fig. 2B, white bars). RN7–58, although structurally similar to E3330, does not inhibit the redox activity of Ref-1 (16) and serves as a negative control compound. To demonstrate that the Ref-1 protein is critical in activation of the NRF2 pathway and expression of HMOX-1, we knocked Ref-1 protein levels down using siRNA (Fig. 2D) and then treated with E3330. As expected when the Ref-1 protein is decreased in the absence of treatment, there is a 1.5-fold increase in HMOX-1 expression. When we treat with E3330 PDAC cells that have a reduction in Ref-1 protein, we observe a comparable stimulation (1.8-fold) of HMOX-1 expression when normalizing for the increase observed with siRNA treatment (Fig. 2C).
FIGURE 2.

NRF2 activity is increased in a dose-dependent manner when Ref-1 redox activity is inhibited via E3330. Pancreatic cancer cells, PaCa-2 (A) and Pa02C (B), were assayed for NRF2 activity via luciferase reporter assay. At 24 h post-transfection of luciferase and firefly constructs, PDAC cells were treated with vehicle control (DMSO), E3330, or negative control compound, RN7–58, for 24 h and then the luciferase assay was conducted. Doses were based on survival data from previously published data (4). C, quantitation of HMOX-1 expression by QPCR using PaCa-2 cells transfected with Ref-1 siRNA#1 or scrambled control and then treated with E3330 at 50 μm for 24 h; and D, immunoblot for Ref-1 for the samples in C with Actin as loading control. *, p < 0.05, comparing DMSO control to drug-treated samples; n = 3–4.
To further consolidate the evidence for induction of NRF2 activity following inhibition of Ref-1, we quantitated the expression of three classic NRF2 target genes GCLC, GCLM, and HMOX-1, in four patient-derived lines as well as PaCa-2 cells. Our results demonstrate a significant and dose-dependent increase in GCLC, GCLM, and HMOX-1 in all five cell lines (Fig. 3, black bars, 95% confidence interval of ΔΔCt and p value based on ANCOVA model). The increase in gene expression for these three genes is abrogated when the negative control compound RN7–58 is utilized (Fig. 3, gray bars), again supporting a negative impact of the redox activity of Ref-1 on NRF2.
FIGURE 3.

Inhibition of Ref-1 increases NRF2 activity in 4 patient-derived cells as well as established PDAC line, PaCa-2. Using three genes (HMOX-1, GCLC, and GCLM) as markers of NRF2 activity, we quantitatively determined the impact of Ref-1 inhibition (black bars) on gene expression by QPCR. PaCa-2 and patient-derived lines were treated with increasing amounts of E3330 for 24 h in medium containing 5% serum. An analog of E3330, RN7–58 (gray bars), was also used but did not induce NRF2 activity. *, p < 0.05; ** p < 0.01; #, p < 0.001, comparing drug-treated cells to DMSO vehicle control, n = 3–5.
Inhibition of Ref-1 Increases NRF2 at Protein Levels
We next determined whether the elevated NRF2 activity in response to Ref-1 inhibition is caused by increased NRF2 expression. To this end, we analyzed RNA and protein levels of NRF2 in PaCa-2 cells as well as two patient-derived cell lines treated with E3330. There is a dose-dependent increase of NRF2 in the levels of both RNA and protein by E3330 (Fig. 4A). RN7–58 failed to elicit such a response in all three cell lines tested (Fig. 4A). A similar dose-dependent response was also observed for the NRF2 target, HMOX-1 (Fig. 4B).
FIGURE 4.

Inhibition of Ref-1 induces expression of NRF2 and HMOX-1. A, patient-derived cells, Pa03C and Panc10.05, and PaCa-2 cells show dose-dependent, significant up-regulation of NRF2 protein levels following treatment with the Ref-1 inhibitor but not with RN7–58 (RN, 50 μm). Cells were treated with E3330 or RN7–58 for 24 h for PaCa-2 and 5 h for patient lines. *, p < 0.05 versus DMSO control; n = 4. B, similar increases are observed with the NRF2 target gene, HMOX-1, following inhibition of Ref-1. Cells were treated with E3330 or RN7–58 for 24 h and then collected for immunoblotting. *, p < 0.05 versus DMSO control; n = 4. Error bars, ± S.E.
Inhibition of Ref-1 Did Not Increase ROS Generation
Because NRF2 can be activated by various stressors including ROS (27–29), we next tested the effect of E3330 treatment on ROS production in pancreatic cancer cells (PaCa-2, Pa03C, Panc10.05, and Panc-1). ROS levels were quantified at various time points following increasing amounts of E3330, using the oxidant-sensitive probe DHR-123 for analysis. Although ROS levels dramatically increased with the positive control tert-butyl hydroperoxide in all cell lines, at 24–48 h we did not see increased ROS generation in the cell lines (Fig. 5). Based on the data in Fig. 5, we conclude that the activation of NRF2 by E3330 was independent of ROS generation. We have previously demonstrated that Ref-1 knockdown does not lead to detectable amounts of ROS, therefore suggesting that ROS generation is not a significant contributor to the impact of Ref-1 on NRF2 activity (8).
FIGURE 5.

The increase in NRF2 activity is not due to an increase in ROS. Quantitation of ROS by DHR-123 fluorescence following inhibition of Ref-1 demonstrates no increase in ROS in either patient-derived or PaCa-2 cells. Cells were treated with E3330 for 24 h and harvested for ROS levels. Positive control tert-butyl hydroperoxide (TBHP) shows a significant increase in ROS generation as expected. Error bars, ±S.E.
Ref-1 and NRF2 Co-immunoprecipitate
Based on existing knowledge that Ref-1 regulates the activity of several transcription factors by direct interaction, we also tested the possibility that Ref-1 and NRF2 can exist in the same protein complex. Indeed, we readily detected the presence of Ref-1 following immunoprecipitation of lysates with NRF2 antibody (Fig. 6A) and in reverse experiments in the presence of NRF2 in the Ref-1 immunoprecipitates (Fig. 6B).
FIGURE 6.
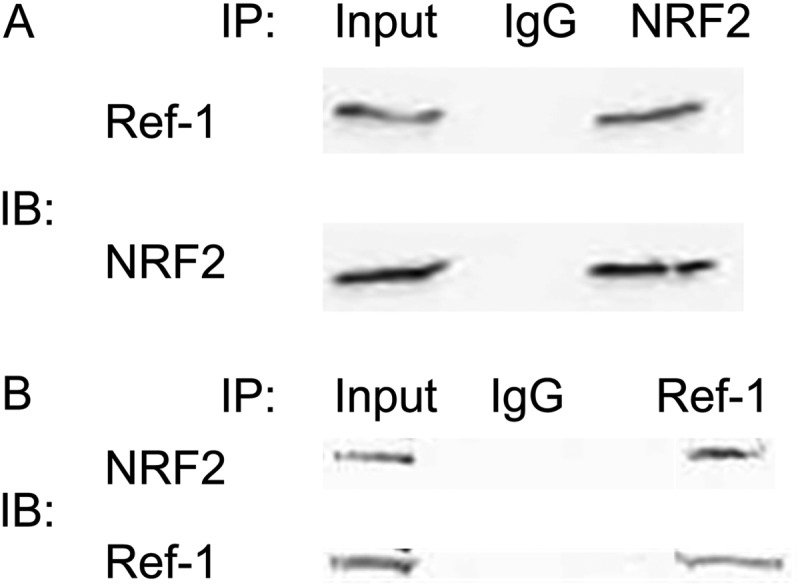
NRF2 interacts with Ref-1 in PDAC cells. Cell extracts were prepared from PaCa-2 cells that overexpress Ref-1 (A) and were treated with E3330 for 24 h (A and B). Extracts were immunoprecipitated with anti-NRF2 (A) or anti-Ref-1 (B) antibody or IgG. The immunoprecipitated complexes were then probed for NRF2 or Ref-1. IB, immunoblot.
To test whether the interaction also occurs at sites where NRF2 exerts its transcription factor activity, ChIP was performed at specific sequences on the HMOX-1 promoter. These NRF2 binding sites were chosen based on published data showing these sites were bound by NRF2 (E1 and E2) (30) (Fig. 7A). We also used the published MCM5 site downstream of HMOX-1 as a negative control for nonspecific chromatin binding. PaCa-2 were treated with E3330, RN7–58, vehicle control (Fig. 7, B and C), or Ref-1 siRNA#1, Ref-1 pooled siRNA #2 and #3, or scrambled control (Fig. 7, D and E); followed by cross-linkage, chromatin fragmentation, and immunoprecipitation using anti-Ref-1, anti-NRF2, or IgG control antibodies. Recovered DNA was subjected to PCR using primers specific for NRF2 binding sites on HMOX-1 promoter. As presented in Fig. 7B, NRF2 binding sequences on the HMOX-1 promoter were co-precipitated by Ref-1 under control conditions, whereas treatment with E3330 dramatically decreased the amount of DNA co-precipitated with Ref-1. When chromatin IP was performed using the anti-NRF2 antibody we reproducibly detected increased recruitment of NRF2 at the same promoter sites following treatment with E3330, but not with negative control compound RN7–58 (Fig. 7C). Finally, when Ref-1 was depleted using multiple siRNAs we observed a similarly increased NRF2 recruitment to the HMOX-1 promoter versus scrambled control (Fig. 7, D and E). In all cases, there is no binding to the negative control MCM5 site.
FIGURE 7.

Ref-1 associates with NRF2 binding sites on the HMOX-1 promoter and inhibition of Ref-1 diminishes this binding. A, schematic of the NRF2 binding sites on the HMOX-1 promoter (30). B, ChIP assay to show Ref-1 is part of a complex on the HMOX-1 promoter and treatment with the Ref-1 inhibitor can significantly diminish the amount of Ref-1 associated with the HMOX-1 promoter. C, ChIP assay with anti-NRF2 antibody demonstrating that there is more NRF2 bound to the chromatin following Ref-1 inhibition. However, RN7–58 does not increase the amount of NRF2 bound to the HMOX-1 promoter. D and E, Ref-1 siRNA#1 and pooled Ref-1 siRNA#3 and #4 decrease the amount of NRF2 bound to the HMOX1 promoter. MCM5 is included as a negative control to demonstrate that the NRF2-Ref-1 pulldown is not due to nonspecific chromatin binding. IgG and NTC (no template control) serve as controls for the ChIP assay.
To summarize, we provide the first evidence of a complex functional relationship between Ref-1 and NRF2 in PDAC cells. On the one hand, inactivation of Ref-1 leads to increased levels of NRF2 transcript and protein. On the other hand, Ref-1 and NRF2 coexist in protein complexes, at least in part at defined promoter sites where NRF2 exerts its transcriptional activity. The evidence suggests that Ref-1 exerts inhibitory effects on this activity, potentially by direct redox regulation and independently of readily detectable ROS levels.
Blockade of Ref-1 and HMOX-1 under Hypoxic Stress, but Not Glucose Deprivation, Results in Synergistic Effects on Human PDAC Cells
Targeting Ref-1 redox activity in PDAC exerts anti-tumor effects both in vitro and in vivo (4), and our data demonstrates that upon inhibition of Ref-1, NRF2 signaling is activated. Activation of the NRF2 pathway has been previously associated with resistance to anti-cancer drugs in PDAC and other cancers (31–33). Based on this information and the data in Fig. 3 demonstrating strong induction of HMOX-1 following Ref-1 inhibition, we hypothesized that targeting HMOX-1 in combination with Ref-1 would lead to sensitization of PDAC cells. Furthermore, combination treatment with the Ref-1 redox inhibitor and HMOX-1 inhibitor may potentiate common stressors such as hypoxia and glucose deprivation in tumor cells as they adapt to low oxygen levels as well as a reduction in local glucose levels. Based on our data demonstrating that inhibition of Ref-1 is activating NRF2 signaling, we evaluated whether the combined blockade of HMOX-1 using Sn-PP (34) and Ref-1 redox activity using E3330 synergizes to more effectively inhibit the growth of human PDAC cells. Cell survival was assessed using the MTS assay following 48 h of drug treatment. E3330 was used at doses that effectively inhibit Ref-1 redox activity, and HMOX-1 inhibitor at increasing doses. As single agents, dose-dependent decreases in proliferation of PDAC cells was observed in the presence of Sn-PP (Fig. 8A) or E3330 (Fig. 8B) (4). We observed that Ref-1 redox inhibition by E3330 synergizes with HMOX-1 blockade most dramatically under hypoxic stress conditions, but not glucose deprivation (Fig. 8A, bottom panel). The effects were seen both in PaCa-2 cells, and primary patient-derived Pa03C cells (Fig. 8B).
FIGURE 8.

Combination treatment of Ref-1 inhibitor and HMOX-1 inhibitor is synergistic under hypoxic conditions, but not low glucose. A, PaCa-2 cells were treated with increasing amounts of HMOX-1 inhibitor (Sn-PP) in combination with Ref-1 inhibitor E3330 (50 or 80 μm) simultaneously under several conditions. Top, normal oxygen; middle panel, 0.2% hypoxia; and bottom panel, low glucose. B, patient-derived cells, Pa03C were treated with increasing amounts of E3330 in combination with 25 μm Sn-PP and then placed under hypoxic conditions (0.2%). Cell survival was assessed via MTS assay 48 h after treatment for both cell lines. Error bars, ±S.E.
HMOX-1 Protein Is Expressed in Low Passage Patient-derived Xenografts Treated with Ref-1 Inhibitor
For the immunohistology studies, grafted patient tumors that had been maintained as a live PancXenoBank (12) were utilized as described in Ref. 4. Female nu/nu athymic mice (Harlan) treated with either vehicle control or E3330 at 25 mg/kg. At the end of this regimen, we collected tumor samples for immunostaining. This dose was effective at reducing tumor volume in previously published studies (4). A total of seven vehicle-treated tumors and 15 E3330-treated tumors were evaluated for HMOX-1 immunostaining. Moderate cytoplasmic immunostaining is seen in tumor cell epithelia in a majority of pancreatic adenocarcinoma cases examined. There is significant higher staining intensity of HMOX-1 in patient tumors that were treated with E3330 versus vehicle-treated (*, p < 0.05) (Fig. 9). Elevated levels of HMOX-1 after Ref-1 inhibition are consistent with our hypothesis that up-regulation of NRF2 signaling may contribute to the resistance of pancreatic cancer to Ref-1 inhibition.
FIGURE 9.
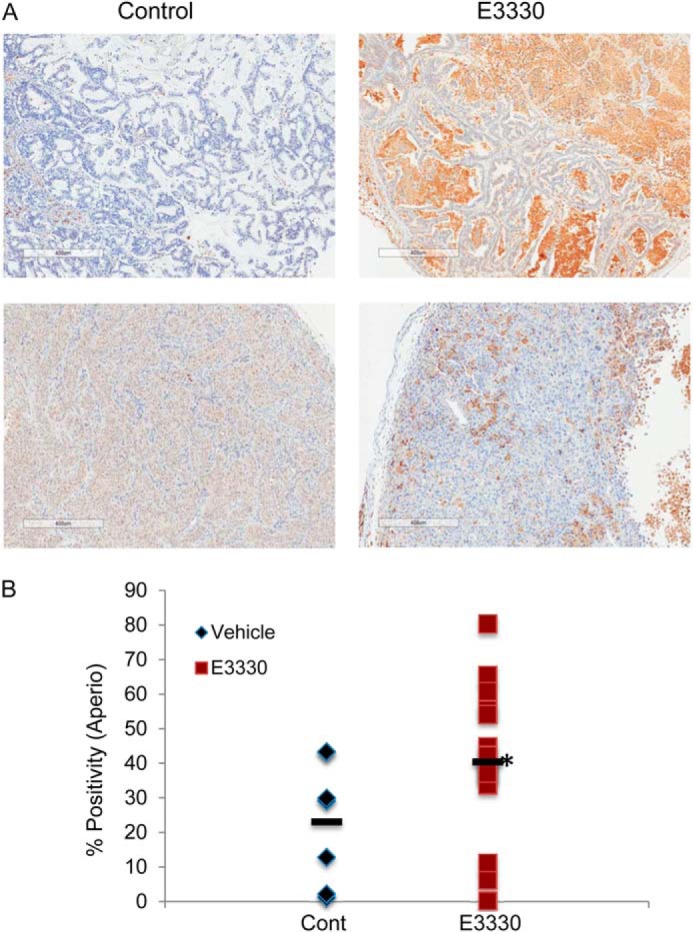
Immunohistochemical staining of HMOX-1 in PDAC patient-derived tissue from live PancXenoBank. Mice were treated with either vehicle control (n = 7, Cremophor:EtOH (4%) or E3330 (n = 15, 25 mg/kg)) for 2 weeks and tumors were harvested following the last dose (4) (p < 0.05 using unpaired t test with Welch's correction, one-tailed).
DISCUSSION
Our study characterizes for the first time a novel functional interaction between the dual function protein, Ref-1, and NRF2, a regulator of cellular redox responses. Although the pathways regulated by Ref-1 have been studied in detail for more than a decade, this is the first report of a negative interaction mediated by Ref-1. The available published information from initial studies suggesting a potential link, however, implied a positive effect on NRF2, analogous to the effect on HIF, p53, and AP-1 (35).
We determined that the repression of Ref-1 leads to activation of NRF2 and its function in all the cell types tested, primary and transformed. The induction at least in part occurs at the transcriptional level, as the blockade of transcription with actinomycin D abolished the effect (data not shown). The level of NRF2 protein is induced in a dose-dependent fashion, as is the level of multiple NRF2 targets, such as HMOX-1 or enzymes involved in glutathione synthesis.
Although activation of the NRF2 pathway frequently involves an increased level of ROS, as in the case of treatment with sulforaphane or tert-butylhydroquinone (36), we saw no evidence that ROS are generated in higher amounts following Ref-1 blockade.
Based on the general response in all the cells tested, activation of NRF2 as a consequence of Ref-1 inactivation may represent a major shift in the ability of cells to cope with oxidative stress. This may have implications for the resistance to drug therapy that directly or indirectly inhibit Ref-1.
To our knowledge the only study linking Ref-1 to NRF2 activity indicated a stimulatory effect on ferritin H gene transcription following Hemin treatment (35). The caveat is that previous work was performed in one cell line (K562) rather than patient-derived tumor cells and in vivo tumors as described here. Employing multiple siRNAs, and small molecules, we tested a panel of well established NRF2 targets, as well as NRF2 transcript, protein levels, and direct activity in reporter assays. We certainly cannot exclude the possibility that the interplay between Ref-1 and NFR2 may lead to differential or even opposite effects for some NRF targets, in specific cellular contexts. However, our results indicate that the portrayal of Ref-1 as a general “helper” of specific transcription factors may need to be revisited.
Although our work does not dissect the biochemistry of the Ref-1-NRF2 relationship, it indicates at least two major mechanisms of repression, one being control of the NRF2 mRNA level. In a timely fashion, Ref-1 was recently reported to repress expression of the p21 gene (37), in association with the AP4 factor at specific sites within the p21 promoter. Interestingly, the NRF2 promoter contains two AP4 sites (not shown), which may mediate a similar repressive effect of Ref-1.
Finally, our work anticipates novel strategies to improve the efficacy of experimental anti-cancer agents acting on Ref-1 function. Among NRF2 targets, HMOX-1 is a candidate protumorigenic gene product and preclinical development of targeted small molecule inhibitors is being actively pursued (38). More importantly, HMOX-1 is emerging as an important contributor to tumor cell resistance to chemotherapeutic agents, such as imatinib (39). In pancreatic cancer cells, gemcitabine or radiation strongly induced HMOX-1 expression and its knockdown was shown to inhibit growth while increasing radio- and chemosensitivity (40, 41).
Our work predicts a mechanism of resistance to therapies based on Ref-1 inhibition and opens the road for development of synergizing strategies, dependent on the dual blockade of Ref-1 and NRF2 or specific downstream targets, such as HMOX-1. The main justification for focusing on the latter in the present work is the availability of small molecule inhibitors that could be tested in conjunction with pharmacological Ref-1 blockade. Moreover, in pancreatic tumors treated with E3330, HMOX-1 is induced, conceivably as part of resistance.
In vitro, Ref-1 redox inhibition appears to be synthetically lethal with HMOX-1 blockade in particular under low oxygen conditions. Speculating future translational implications, such combinations may exhibit lower toxicity on (well oxygenated) normal tissues and target in particular tumor cells surviving in hypoxic compartments, which represent important sources of therapeutic failure. We are aware of the limitations of porphyrin-based compounds, however, our investigation was intended to serve as proof of principle, in anticipation of newer generations of HMOX-1 inhibitors such as imidazole-dioxolane derivatives, currently under evaluation (42).
Furthermore, it has been demonstrated that targeted inactivation of Ref-1 sensitizes cells in vitro to a variety of genotoxic agents, including antineoplastic agents: TMZ, BCNU, etoposide, cisplatin, and doxorubicin (6, 7, 19, 37). Our work suggests that these effects may be further enhanced by the concomitant blockade of the NRF2 pathway or select targets such as HMOX-1.
Acknowledgments
We thank Dr. George Sandusky, Department of Pathology and Laboratory Medicine, for help with interpretation and scoring of the HMOX-1 patient-derived xenografts, and Dr. Angelo Cardoso, Department of Pediatrics, Wells Center for Pediatric Research, for help with the statistical analysis of the HMOX-1 patient-derived xenografts.
This work was supported, in whole or in part, by National Institutes of Health Grants NCI CA167291 (to M. L. F.), NCI 1R01 CA155332-01 (to M. I.), CA121168, CA121168S1, and CA167291, and a grant from the Riley Children's Foundation (to M. R. K.), a grant from the Indiana University Simon Cancer Center Indiana Translational Research Acceleration Collaboration (IUSCC ITRAC) basic science pilot funding mechanism (to M. I. and M. L. F.), Stand Up to Cancer Dream Team Translational Research Grant SU2C-AACR and Program of the Entertainment Industry Foundation Grant SU2C-AACR-DT0509 (to N. V. R.).
This manuscript is dedicated to the fond memory of Victor Fung, Ph.D., a former Program Officer at NCI and former Scientific Review Officer of the Cancer Etiology study section of CSR, National Institutes of Health, for his wisdom, compassion, integrity, his love of sciences and the arts, his incredible culinary skills, and above all, his contributions to the career development of so many investigators during his own distinguished career.
- Ref-1
- redox factor-1
- PDAC
- pancreatic ductal adenocarcinoma
- NRF2
- nuclear factor erythroid-related factor 2
- QPCR
- quantitative PCR
- ROS
- reactive oxygen species
- DHR
- dihydrorhodamine
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- DMSO
- dimethyl sulfoxide
- ANCOVA
- analysis of covariance
- IP
- immunoprecipitation
- HMOX-1
- heme oxygenase-1
- GCLC
- glutamate-cysteine ligase, catalystic subunit
- GCLM
- glutamate-cystein ligase, modifier subunit.
REFERENCES
- 1. Fishel M. L., Kelley M. R. (2007) The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol. Aspects Med. 28, 375–395 [DOI] [PubMed] [Google Scholar]
- 2. Tell G., Quadrifoglio F., Tiribelli C., Kelley M. R. (2009) The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid. Redox Signal. 11, 601–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cardoso A. A., Jiang Y., Luo M., Reed A. M., Shahda S., He Y., Maitra A., Kelley M. R., Fishel M. L. (2012) APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 7, e47462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fishel M. L., Jiang Y., Rajeshkumar N. V., Scandura G., Sinn A. L., He Y., Shen C., Jones D. R., Pollok K. E., Ivan M., Maitra A., Kelley M. R. (2011) Impact of APE1/Ref-1 redox inhibition on pancreatic tumor growth. Mol. Cancer Ther. 10, 1698–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhakat K. K., Mantha A. K., Mitra S. (2009) Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid. Redox Signal. 11, 621–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kelley M. R., Fishel M. L. (eds). (2013) DNA Repair and Cancer Therapeutics, Lippincott Williams & Wilkins, Baltimore, MD [Google Scholar]
- 7. Fishel M. L., Vascotto C., Kelley M. R. (eds) (2013) DNA Base Excision Repair Therapeutics: Summary of BER Targets with a focus on APE1, Science Publishers, Edenbridge Ltd., British Channel Islands, Enfield, NH [Google Scholar]
- 8. Jiang Y., Zhou S., Sandusky G. E., Kelley M. R., Fishel M. L. (2010) Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Invest. 28, 885–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zou G. M., Maitra A. (2008) Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol. Cancer Ther. 7, 2012–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Romanov S., Medvedev A., Gambarian M., Poltoratskaya N., Moeser M., Medvedeva L., Gambarian M., Diatchenko L., Makarov S. (2008) Homogeneous reporter system enables quantitative functional assessment of multiple transcription factors. Nat. Methods 5, 253–260 [DOI] [PubMed] [Google Scholar]
- 11. Higgins L. G., Hayes J. D. (2011) The cap'n'collar transcription factor Nrf2 mediates both intrinsic resistance to environmental stressors and an adaptive response elicited by chemopreventive agents that determines susceptibility to electrophilic xenobiotics. Chem. Biol. Interact. 192, 37–45 [DOI] [PubMed] [Google Scholar]
- 12. Jones S., Zhang X., Parsons D. W., Lin J. C., Leary R. J., Angenendt P., Mankoo P., Carter H., Kamiyama H., Jimeno A., Hong S. M., Fu B., Lin M. T., Calhoun E. S., Kamiyama M., Walter K., Nikolskaya T., Nikolsky Y., Hartigan J., Smith D. R., Hidalgo M., Leach S. D., Klein A. P., Jaffee E. M., Goggins M., Maitra A., Iacobuzio-Donahue C., Eshleman J. R., Kern S. E., Hruban R. H., Karchin R., Papadopoulos N., Parmigiani G., Vogelstein B., Velculescu V. E., Kinzler K. W. (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fishel M. L., Colvin E. S., Luo M., Kelley M. R., Robertson K. A. (2010) Inhibition of the redox function of APE1/Ref-1 in myeloid leukemia cell lines results in a hypersensitive response to retinoic acid-induced differentiation and apoptosis. Exp. Hematol. 38, 1178–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Su D., Delaplane S., Luo M., Rempel D. L., Vu B., Kelley M. R., Gross M. L., Georgiadis M. M. (2011) Interactions of APE1 with a redox inhibitor: evidence for an alternate conformation of the enzyme. Biochemistry 50, 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nyland R. L., Luo M., Kelley M. R., Borch R. F. (2010) Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/Ref-1). J. Med. Chem. 53, 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelley M. R., Luo M., Reed A., Su D., Delaplane S., Borch R. F., Nyland R. L., 2nd, Gross M. L., Georgiadis M. M. (2011) Functional analysis of novel analogs of E3330 that block the redox signaling activity of the multifunctional AP endonuclease/redox signaling enzyme APE1/Ref-1. Antioxid. Redox Signal. 14, 1387–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang D., Luo M., Kelley M. R. (2004) Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol. Cancer Ther. 3, 679–686 [PubMed] [Google Scholar]
- 18. Fan Z., Beresford P. J., Zhang D., Xu Z., Novina C. D., Yoshida A., Pommier Y., Lieberman J. (2003) Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat. Immunol. 4, 145–153 [DOI] [PubMed] [Google Scholar]
- 19. Fishel M. L., He Y., Reed A. M., Chin-Sinex H., Hutchins G. D., Mendonca M. S., Kelley M. R. (2008) Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair 7, 177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burdelya L. G., Brackett C. M., Kojouharov B., Gitlin I. I., Leonova K. I., Gleiberman A. S., Aygun-Sunar S., Veith J., Johnson C., Haderski G. J., Stanhope-Baker P., Allamaneni S., Skitzki J., Zeng M., Martsen E., Medvedev A., Scheblyakov D., Artemicheva N. M., Logunov D. Y., Gintsburg A. L., Naroditsky B. S., Makarov S. S., Gudkov A. V. (2013) Central role of liver in anticancer and radioprotective activities of Toll-like receptor 5 agonist. Proc. Natl. Acad. Sci. U.S.A. 110, E1857–E1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiwaji M., Daly R., Gibriel A., Barkess G., McLean P., Yang J., Pansare K., Cumming S., McLauchlan A., Kamola P. J., Bhutta M. S., West A. G., West K. L., Kolch W., Girolami M. A., Pitt A. R. (2012) Unique reporter-based sensor platforms to monitor signalling in cells. PLoS ONE 7, e50521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan J. S., Reed A., Chen F., Stewart C. N., Jr. (2006) Statistical analysis of real-time PCR data. BMC Bioinformatics 7, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dhakshinamoorthy S., Jaiswal A. K. (2000) Small maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:quinone oxidoreductase1 gene. J. Biol. Chem. 275, 40134–40141 [DOI] [PubMed] [Google Scholar]
- 24. Zhang J., Luo M., Marasco D., Logsdon D., LaFavers K. A., Chen Q., Reed A., Kelley M. R., Gross M. L., Georgiadis M. M. (2013) Inhibition of apurinic/apyrimidinic endonuclease I's redox activity revisited. Biochemistry 52, 2955–2966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo M., Delaplane S., Jiang A., Reed A., He Y., Fishel M., Nyland R. L., 2nd, Borch R. F., Qiao X., Georgiadis M. M., Kelley M. R. (2008) Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid. Redox Signal. 10, 1853–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo M., Zhang J., He H., Su D., Chen Q., Gross M. L., Kelley M. R., Georgiadis M. M. (2012) Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry 51, 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Surh Y. J., Kundu J. K., Na H. K. (2008) Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 74, 1526–1539 [DOI] [PubMed] [Google Scholar]
- 28. Enomoto N., Koshikawa N., Gassmann M., Hayashi J., Takenaga K. (2002) Hypoxic induction of hypoxia-inducible factor-1α and oxygen-regulated gene expression in mitochondrial DNA-depleted HeLa cells. Biochem. Biophys. Res. Commun. 297, 346–352 [DOI] [PubMed] [Google Scholar]
- 29. Rensing H., Jaeschke H., Bauer I., Pätau C., Datene V., Pannen B. H., Bauer M. (2001) Differential activation pattern of redox-sensitive transcription factors and stress-inducible dilator systems heme oxygenase-1 and inducible nitric oxide synthase in hemorrhagic and endotoxic shock. Crit. Care Med. 29, 1962–1971 [DOI] [PubMed] [Google Scholar]
- 30. Meng D., Wang X., Chang Q., Hitron A., Zhang Z., Xu M., Chen G., Luo J., Jiang B., Fang J., Shi X. (2010) Arsenic promotes angiogenesis in vitro via a heme oxygenase-1-dependent mechanism. Toxicol. Appl. Pharmacol. 244, 291–299 [DOI] [PubMed] [Google Scholar]
- 31. Shibata T., Kokubu A., Gotoh M., Ojima H., Ohta T., Yamamoto M., Hirohashi S. (2008) Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135, 1358–1368, 1368.e1–4 [DOI] [PubMed] [Google Scholar]
- 32. Arlt A., Sebens S., Krebs S., Geismann C., Grossmann M., Kruse M. L., Schreiber S., Schäfer H. (2013) Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 32, 4825–4835 [DOI] [PubMed] [Google Scholar]
- 33. Hong Y. B., Kang H. J., Kwon S. Y., Kim H. J., Kwon K. Y., Cho C. H., Lee J. M., Kallakury B. V., Bae I. (2010) Nuclear factor (erythroid-derived 2)-like 2 regulates drug resistance in pancreatic cancer cells. Pancreas 39, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoshinaga T., Sassa S., Kappas A. (1982) Purification and properties of bovine spleen heme oxygenase: amino acid composition and sites of action of inhibitors of heme oxidation. J. Biol. Chem. 257, 7778–7785 [PubMed] [Google Scholar]
- 35. Iwasaki K., Mackenzie E. L., Hailemariam K., Sakamoto K., Tsuji Y. (2006) Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol. Cell Biol. 26, 2845–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tkachev V. O., Menshchikova E. B., Zenkov N. K. (2011) Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry 76, 407–422 [DOI] [PubMed] [Google Scholar]
- 37. Sengupta S., Mitra S., Bhakat K. K. (2013) Dual regulatory roles of human AP-endonuclease (APE1/Ref-1) in CDKN1A/p21 expression. PLoS ONE 8, e68467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salerno L., Pittalà V., Romeo G., Modica M. N., Siracusa M. A., Di Giacomo C., Acquaviva R., Barbagallo I., Tibullo D., Sorrenti V. (2013) Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorg Med. Chem. 21, 5145–5153 [DOI] [PubMed] [Google Scholar]
- 39. Tibullo D., Barbagallo I., Giallongo C., La Cava P., Parrinello N., Vanella L., Stagno F., Palumbo G. A., Li Volti G., Di Raimondo F. (2013) Nuclear translocation of heme oxygenase-1 confers resistance to imatinib in chronic myeloid leukemia cells. Curr. Pharm. Des. 19, 2765–2770 [DOI] [PubMed] [Google Scholar]
- 40. Nuhn P., Künzli B. M., Hennig R., Mitkus T., Ramanauskas T., Nobiling R., Meuer S. C., Friess H., Berberat P. O. (2009) Heme oxygenase-1 and its metabolites affect pancreatic tumor growth in vivo. Mol. Cancer 8, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Berberat P. O., Dambrauskas Z., Gulbinas A., Giese T., Giese N., Künzli B., Autschbach F., Meuer S., Büchler M. W., Friess H. (2005) Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 11, 3790–3798 [DOI] [PubMed] [Google Scholar]
- 42. Pittalà V., Salerno L., Romeo G., Modica M. N., Siracusa M. A. (2013) A focus on heme oxygenase-1 (HO-1) inhibitors. Curr. Med. Chem. 20, 3711–3732 [DOI] [PubMed] [Google Scholar]