

Background: The physiological activity of gut microbial metabolites has recently attracted much attention.
Results: A gut microbial metabolite of linoleic acid, 10-hydroxy-cis-12-octadecenoic acid (HYA), ameliorates intestinal epithelial barrier impairments by regulating TNFR2 expression via the GPR40-MEK-ERK pathway.
Conclusion: HYA-induced GPR40 signaling contributes to the intestinal homeostasis.
Significance: Our findings indicate a novel function of GPR40 in the inflamed intestine.
Keywords: Colitis, Epithelial Cell, Microbiome, Tight Junction, Tumor Necrosis Factor (TNF), Caco-2, G Protein-coupled Receptor, Linoleic Acid, Metabolite
Abstract
Gut microbial metabolites of polyunsaturated fatty acids have attracted much attention because of their various physiological properties. Dysfunction of tight junction (TJ) in the intestine contributes to the pathogenesis of many disorders such as inflammatory bowel disease. We evaluated the effects of five novel gut microbial metabolites on tumor necrosis factor (TNF)-α-induced barrier impairment in Caco-2 cells and dextran sulfate sodium-induced colitis in mice. 10-Hydroxy-cis-12-octadecenoic acid (HYA), a gut microbial metabolite of linoleic acid, suppressed TNF-α and dextran sulfate sodium-induced changes in the expression of TJ-related molecules, occludin, zonula occludens-1, and myosin light chain kinase. HYA also suppressed the expression of TNF receptor 2 (TNFR2) mRNA and protein expression in Caco-2 cells and colonic tissue. In addition, HYA suppressed the protein expression of TNFR2 in murine intestinal epithelial cells. Furthermore, HYA significantly up-regulated G protein-coupled receptor (GPR) 40 expression in Caco-2 cells. It also induced [Ca2+]i responses in HEK293 cells expressing human GPR40 with higher sensitivity than linoleic acid, its metabolic precursor. The barrier-recovering effects of HYA were abrogated by a GPR40 antagonist and MEK inhibitor in Caco-2 cells. Conversely, 10-hydroxyoctadacanoic acid, which is a gut microbial metabolite of oleic acid and lacks a carbon-carbon double bond at Δ12 position, did not show these TJ-restoring activities and down-regulated GPR40 expression. Therefore, HYA modulates TNFR2 expression, at least partially, via the GPR40-MEK-ERK pathway and may be useful in the treatment of TJ-related disorders such as inflammatory bowel disease.
Introduction
Increasing evidence suggests that polyunsaturated fatty acids, which contain more than one double bond, have various beneficial physiological effects (1–3). However, the details of the mechanisms of action are not fully understood. Recently, we reported that gut microorganisms generate hydroxy fatty acids, oxo fatty acids, conjugated fatty acid, and partially saturated (nonmethylene-interrupted) fatty acids from food-derived polyunsaturated fatty acids via the biohydrogenation pathway (4). We also revealed the existence of these unique fatty acids in mammalian tissues (4). Analyzing the physiological function of these fatty acids will help us understand the mechanisms of action of polyunsaturated fatty acids.
Lactobacillus plantarum has the ability to convert linoleic acid to oleic acid via 10-hydroxy-cis-12-octadecenoic acid (HYA),3 10-oxo-cis-12-octadecenoic acid (KetoA), 10-oxo-trans-11-octadecenoic acids (KetoC), 10-oxo-octadecanoic acid (KetoB), and 10-hydroxyoctadecanoic acid (HYB). Conjugated linoleic acids of 9c,11t- and 9t,11t-linoleic acid were generated via the branched pathway from KetoC. HYB and KetoB are also produced from oleic acid by initial reactions in biohydrogenation metabolism (5). In this paper, we evaluated the function of five representative fatty acids, HYA, HYB, KetoA, KetoB, and KetoC, in regard to their anti-inflammatory activity in the intestine.
Inflammatory bowel disease (IBD), including Crohn disease and ulcerative colitis, is characterized by an abnormal mucosal immune reaction (6). Although the pathogenesis of IBD is not yet fully understood, recent genome-wide association studies as well as animal models have suggested that dysfunction of tight junctions (TJ) in the intestine greatly contributes to the pathogenesis of IBD (7, 8). Increased intestinal permeability and impaired TJ integrity have really been observed in IBD (9, 10). In addition, excessive production of pro-inflammatory molecules, such as reactive oxidative species (11) and cytokines (12–14), is characteristic of IBD patients. Notably, tumor necrosis factor (TNF)-α is a pivotal pro-inflammatory cytokine in the pathogenesis of IBD (15, 16) and also causes disruption of the intestinal epithelial TJ barrier (17–19). Indeed, the blockade of TNFR1 and/or TNFR2 has recently been shown effective for suppression of inflammation and apoptosis in murine colitis (20).
The TJ is a multiprotein complex composed of transmembrane and intracellular proteins. To date, four transmembrane proteins, occludin, claudins, junctional adhesion molecule, and tricellulin, have been identified (21–24). The intracellular proteins, including zonula occludens (ZO) and cingulin, interact with these transmembrane proteins (25, 26). IBD-induced disruptions of the epithelial barrier have been associated with changes in expression of TJ proteins, including ZO-1 and occludin (27, 28). Dysregulation of ZO-1 and occludin corresponds to an increase in intestinal permeability and a decrease in transepithelial resistance (TER). In addition, using a colitis model induced by dextran sulfate sodium (DSS), a previous study has shown a decrease in ZO-1 (29). Therefore, the re-enforcement of TJ function is regarded as an effective therapy for intestinal disorders such as IBD.
G protein-coupled receptors are physiologically important membrane proteins that sense signaling molecules, such as hormones and neurotransmitters, and are the targets of several drug prescriptions (30). The G protein-coupled receptor 40 (GPR40), also known as free fatty acid receptor 1 (FFAR1), has been suggested to be an important component in the augmentation of insulin secretion and is activated by a range of medium- to long-chain saturated and unsaturated fatty acids of chain lengths of >6 carbons (31, 32). GPR40 is highly expressed in pancreatic islets and has been extensively studied for its role in insulin secretion by mouse pancreatic β cells in response to unsaturated long-chain fatty acids, oleic, linoleic and linolenic acids (33, 34). GPR40 has also been reported to be expressed in the brain and intestine. However, it is unclear whether GPR40 contributes to inflammation and immune responses in the intestine.
Therefore, we have evaluated the protective effects of five novel fatty acids generated by gut microbes from food-derived fatty acids on IFN-γ + TNF-α-induced intestinal barrier impairment in a human epithelial cell line, Caco-2, and in DSS-induced colitis mice. We demonstrate that HYA modulates TNFR2 expression partially via GPR40 signaling to ameliorate epithelial barrier impairment.
EXPERIMENTAL PROCEDURES
Reagents
The structures of gut microbial metabolites of linoleic and oleic acids used in this study are shown in Fig. 1. These were synthesized according to methods published in previous studies (4). The following were used for immunoblotting and immunohistochemistry: mouse anti-inhibitor κBα (IκBα) and anti-phospho-IκBα (Cell Signaling Technology, Danvers, MA); anti-MLCK (Sigma); anti-β-actin (Wako Pure Chemical Industries, Osaka, Japan); rabbit anti-nuclear factor (NF)-κB p65 (Cell Signaling Technology); anti-GPR40 (Santa Cruz Biotechnology, Paso Robles, CA); anti-TNFR2, ERK, phospho-ERK, p38, phospho-p38, JNK, phospho-JNK (Cell Signaling Technology); anti-occludin (Invitrogen); goat horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse immunoglobulin antibodies (Dako, Glostrup, Denmark); goat fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG (Invitrogen); and rat phycoerythrin-conjugated anti-mouse CD326 (eBioscience, San Diego). All other reagents were of reagent grade.
FIGURE 1.
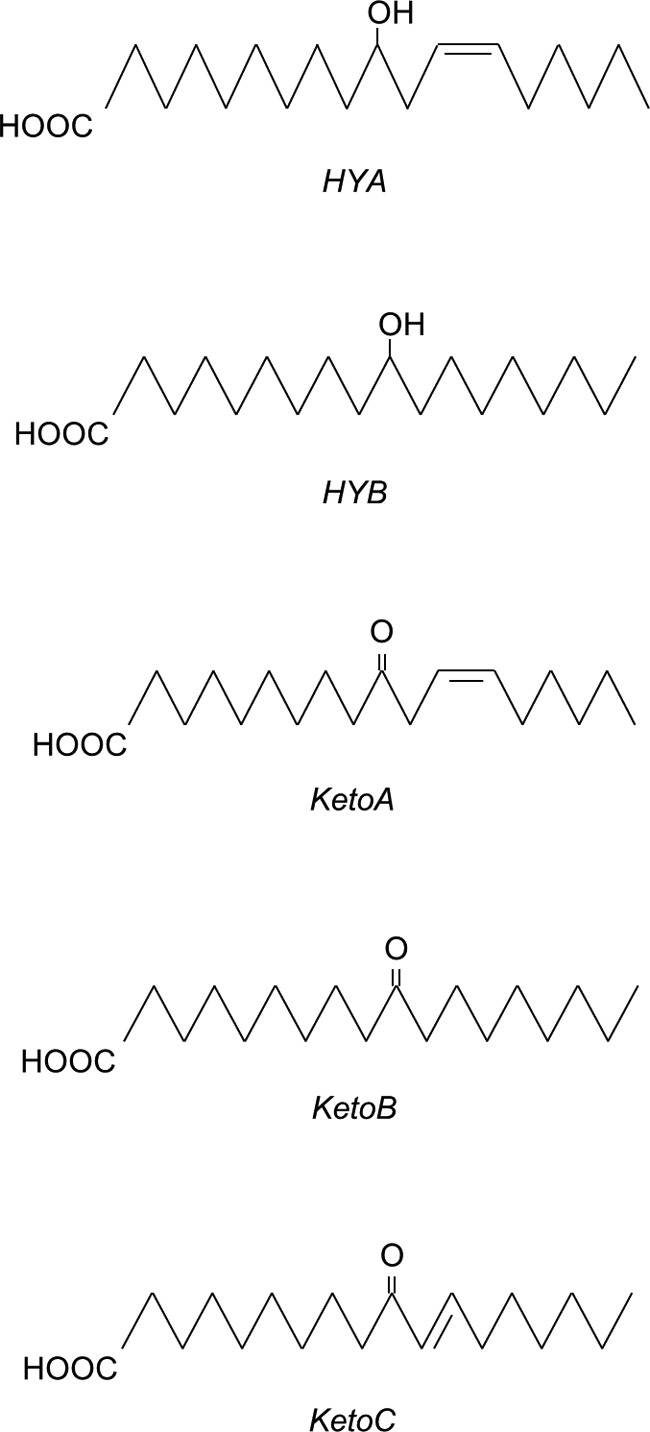
Structures of fatty acids used in this study. HYA, 10-hydroxy-cis-12-octadecenoic acid; HYB, 10-hydroxyoctadecanoic acid; KetoA, 10-oxo-cis-12-octadecenoic acid; KetoB, 10-oxo-octadecanoic acid; KetoC, 10-oxo-trans-11-octadecenoic acid. These fatty acids were synthesized by our previously published method (4).
Cell Culture
Caco-2 cells were obtained from the American Type Culture Collection (Manassas, VA) and used between passages 35 and 55. The culture medium consisted of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% nonessential amino acids, and antibiotics (100 units/ml penicillin, 100 mg/ml streptomycin, and 50 mg/ml gentamycin). DMEM, penicillin, streptomycin, and gentamycin were obtained from Invitrogen. Nonessential amino acids were obtained from Wako Pure Chemical Industries. Fetal bovine serum was obtained from ICN Biomedicals (Osaka, Japan).
Caco-2 cells were cultured at 37 °C under a humidified atmosphere of 5% CO2. They were grown in 75-cm2 tissue culture flasks to ∼80% confluence and then seeded into a 12-well Transwell cell culture chamber (0.4 μm in pore size and 12 mm in diameter) (Corning Costar, Cambridge, MA) at a density of 2 × 105 cells/cm2. Each well was placed in a cluster plate with an outside medium (basal side, 1.5 ml) and an inside medium (apical side, 0.5 ml). The Caco-2 cells were fed fresh medium every 48 h.
Cell Treatment
After 14 days of culture, the Caco-2 cells were treated with gut microbial metabolites (HYA, HYB, KetoA, KetoB or KetoC; 50 μm) in the apical side for 24 h. Next, IFN-γ (50 ng/ml) was added to the basal side, and the Caco-2 cells were incubated for 24 h. After that, the basal media were removed, and TNF-α (50 ng/ml) was added to the basal side, and Caco-2 cells were stimulated for 6 h. In some experiments, the Caco-2 cells were stimulated with IFN-γ for 24 h and were pretreated with the GPR40 antagonist GW1100 (1 μm, Cayman Chemical, Ann Arbor, MI) or the MEK inhibitor U0126 (10 μm, Wako Pure Chemical Industries) for 30 min prior to the addition of gut microbial metabolites.
Measurement of Intestinal TJ Barrier Function and IL-8 Concentration
Intestinal TJ barrier function was evaluated by measurement of TER and the unidirectional flux of FITC-conjugated dextran (FD-4; average molecular weight 4,000, Sigma) in Caco-2 cells in Transwell inserts. TER value was measured before and after the addition of TNF-α every 1 h by Millicell-ERS system (Millipore, Billerica, MA). FD-4 (100 μm) was injected into the apical side at the same time that TNF-α was added, and its flux into the basal wells was assessed for 6 h. The concentration of FD-4 in the basal solution was fluorometrically determined at 492 nm for excitation and 535 nm for emission (ARVO X4; PerkinElmer Life Sciences). After stimulation with TNF-α for 6 h, the basal culture supernatants were assayed for interleukin (IL)-8 concentration by sandwich ELISA (DuoSet, R&D Systems, Abingdon, UK) according to the manufacturer's instructions.
Real Time RT-PCR
Total RNA was extracted from the Caco-2 cells using TRIzol (Invitrogen) and from murine colon tissues using an RNeasy mini kit (Qiagen, Gaithersburg, MD). The reverse transcription (RT) reaction was performed with the high capacity cDNA reverse transcription kit (Invitrogen) at 25 °C for 10 min and at 37 °C for 120 min. The reaction was terminated by heating to 85 °C for 5 min followed by cooling at 4 °C.
Real time PCR was performed using the KAPA SYBR FAST ABI PRISM qPCR kit (Kapa Biosystems, Woburn, MA). Primers were designed using the Primer3Plus Program. Primer sequences are shown in Tables 1 and 2. The reaction was performed at 95 °C for 2 min, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. The dissociation stage was analyzed at 95 °C for 15 s, followed by 1 cycle of 60 °C for 15 s and 95 °C for 15 s. The fluorescence of the SYBR Green dye was determined as a function of the PCR cycle number, giving the threshold cycle number at which amplification reached a significant threshold. Data were analyzed by the ΔΔCt method and presented as fold changes in gene expression from the unstimulated level, after normalization to the β-actin gene for Caco-2 cells and GAPDH gene for colonic tissues.
TABLE 1.
Primer sequences used for in vitro experiments
| Gene | Sequence |
|---|---|
| β-Actin | Forward 5′-TTTTAGGATGGCAAGGGACTT-3′ |
| Reverse 5′-GATGAGTTGGCATGGCTTTA-3′ | |
| Occludin | Forward 5′-AAGAGTTGACAGTCCCATGGCATAC-3′ |
| Reverse 5′-ATCCACAGGCGAAGTTAATGGAAG-3′ | |
| ZO1 | Forward 5′-TCCGTGTTGTGGATACCTTG-3′ |
| Reverse 5′-GGATGATGCCTCGTTCTACC-3′ | |
| ZO2 | Forward 5′-AAAGCAGAGCGAACGAAGAG-3′ |
| Reverse 5′-TTTAGTTGCCAGACCCGTTC-3′ | |
| Claudin1 | Forward 5′-CCCTATGACCCCAGTCAATG-3′ |
| Reverse 5′-AAGGCAGAGAGAAGCAGCAG-3′ | |
| Claudin3 | Forward 5′-AAGGTGTACGACTCGCTGCT-3′ |
| Reverse 5′-GAAGTCCCGGATAATGGTGTT-3′ | |
| MLCK | Forward 5′-AACGAGATCAACATCATGAACCA-3′ |
| Reverse 5′-CAGCTGTGCTTGCTCTCGAA-3′ | |
| TNFR1 | Forward 5′-CTGCCAGGAGAAACAGAACAC-3′ |
| Reverse 5′-AGGGATAAAAGGCAAAGACCA-3′ | |
| TNFR2 | Forward 5′-TTCCAGAAAACCCCAGCA-3′ |
| Reverse 5′-TGGCCTGAGGTGATGCTT-3′ | |
| GPR40 | Forward 5′-GCCCACTTCTTCCCACTCT-3′ |
| Reverse 5′-ACCAGACCCAGGTGACACA-3′ | |
| GPR120 | Forward 5′-TCTCGTGGGATGTCTCTTTTG-3′ |
| Reverse 5′-CTTACCGTGAGCCTCTTCCTT-3′ | |
| PPARγ | Forward 5′-ATGGAGCCCAAGTTTGAGTTT-3′ |
| Reverse 5′-TGTCTGAGGTCCGTCATTTTC-3′ | |
| GPR41 | Forward 5′-CTTCATCCTCTGCCCACTCTC-3′ |
| Reverse 5′-CGCAGATATAGCCCACGACAT-3′ | |
| GPR43 | Forward 5′-ACGCAGAGGCAAAGACACA-3′ |
| Reverse 5′-TCTGTCGCTAGGCTGGAGT-3′ |
TABLE 2.
Primer sequences used for in vivo experiments
| Gene | Sequence |
|---|---|
| GAPDH | Forward 5′-TCAAGAAGGTGGTGAAGCAG-3′ |
| Reverse 5′-AAGGTGGAAGAGTGGGAGTTG-3′ | |
| Occludin | Forward 5′-CACACTTGCTTGGGACAGAG-3′ |
| Reverse 5′-TAGCCATAGCCTCCATAGCC-3′ | |
| ZO1 | Forward 5′-CTTCTCTTGCTGGCCCTAAAC-3′ |
| Reverse 5′-TGGCTTCACTTGAGGTTTCTG-3′ | |
| ZO2 | Forward 5′-AACGGATGCTGGAAGTTAAT-3′ |
| Reverse 5′-TCTGCTTGCTGTCTCTCAACA-3′ | |
| Claudin1 | Forward 5′-GATGTGGATGGCTGTCATTG-3′ |
| Reverse 5′-CCTGGCCAAATTCATACCTG-3′ | |
| Claudin3 | Forward 5′-AACTGCGTACAAGACGAGACG-3′ |
| Reverse 5′-ATCCCTGATGATGGTGTTGG-3′ | |
| MLCK | Forward 5′-GCGTGATCAGCCTGTTCTTTCTAA-3′ |
| Reverse 5′-GCCCCATCTGCCCTTCTTTGACC-3′ | |
| TNFR1 | Forward 5′-AAAGGTCTCTAAGGGGGAAGG-3′ |
| Reverse 5′-CAAGTGGGCACCAGATACATT-3′ | |
| TNFR2 | Forward 5′-GCCATCCCAAGGACACTCTA-3′ |
| Reverse 5′-AGGGCTTCTTTTTCCTCTGC-3′ |
Immunoblotting
After the appropriate incubation, the Caco-2 cells were washed with ice-cold PBS three times and lysed in RIPA lysis buffer (Atto, Tokyo, Japan). The cell suspensions were centrifuged at 14,000 × g for 30 min at 4 °C to yield a clear lysate. Cell extracts were mixed with a one-half volume of Laemmli sample buffer (3× concentrated; 4% SDS, 10% glycerol, 10% β-mercaptoethanol, and 0.04% bromphenol blue in 125 mm Tris-HCl, pH 6.8) and heated at 95 °C for 10 min. The proteins were separated using a 10% polyacrylamide gel and were then transferred onto blotting membranes (Immobilon-P PVDF, Millipore). Membranes were blotted for IκBα, phospho-IκBα, NF-κB p65, TNFR2, GPR40, ERK, phospho-ERK, p38, phospho-p38, JNK, phospho-JNK and β-actin using specific antibodies in combination with HRP-conjugated anti-mouse or anti-rabbit immunoglobulin antibodies. Blots were developed using the ECL chemiluminescence method (PerkinElmer Life Sciences). Quantification was performed by densitometric analysis of specific bands on the immunoblots using ImageJ software (National Institutes of Health, Bethesda).
Immunohistochemistry
Caco-2 cells were washed with ice-cold PBS after the indicated treatment, fixed with 4% (w/v) paraformaldehyde for 10 min, and permeabilized with 0.2% Triton X-100 in PBS for 5 min. The cells were then blocked in 4% nonfat milk and incubated with rabbit polyclonal anti-GPR40 (35, 36) at 4 °C overnight, followed by incubation for 1 h with FITC-conjugated anti-rabbit IgG. Fluorescence was visualized using a confocal microscope (LSM700, Carl Zeiss Microscopy, Jena, Germany).
GPR40 Functional Activity Assay
For generation of HEK293 cells expressing human GPR40, Flp-In T-REx HEK293 cells were transfected with a mixture of mouse FLAG-GPR40 cDNA in pcDNA5/FRT/TO and pOG44, respectively, using the Lipofectamine reagent (Invitrogen). After 48 h, the medium was exchanged to medium supplemented with 200 μg/ml hygromycin B to initiate selection of stably transfected cells. Following isolation of resistant cells, expression of GPR40 from the Flp-In locus was induced by treatment with 1 μg/ml doxycycline for 48 h. Expression of GPR40 was confirmed by RT-PCR and FACSCalibur (BD Biosciences) using the FLAG tag. For [Ca2+]i measurement, cells were seeded at a density of 2 × 105 cells/well on collagen-coated 96-well plates, incubated at 37 °C for 21 h, and then incubated in Hanks' balanced salt solution, pH 7.4, containing calcium assay kit component A (Molecular Devices, Sunnyvale, CA) for 1 h at room temperature. Fatty acids used in the Functional Drug Screening System (Hamamatsu Photonics, Shizuoka, Japan) assay were dissolved in Hanks' balanced salt solution (with 1% EtOH) and prepared in another set of 96-well plates. These plates were set on the Functional Drug Screening System, and mobilization of [Ca2+]i evoked by agonists was monitored. The same assay was done also on Caco-2 cells. When using the GPR40 antagonist, cells were pretreated with GW1100 for 15 min prior to the addition of the fatty acids. HEK293 cells not expressing GPR40 were used as negative controls. Data analysis was performed using Igor Pro (WaveMetrics, Lake Oswego, OR).
Animal Studies
BALB/c mice (female, 6 weeks old) were purchased from Charles River Japan (Kanagawa, Japan). All experimental protocols involving animals were approved by the Animal Care Committee, Graduate School of Biosphere Science, Hiroshima University (No. C10-17). Acute colitis was induced in mice by adding 3.5% (w/v) DSS (molecular weight, 36,000–50,000; MP Biomedicals, Aurora, OH) to their drinking water for 5 days. To assess the effects of HYA and HYB, mice (n = 6 for each group) were intragastrically administered the suspension of HYA or HYB (30 μg each in 100 μl of water containing 2% DMSO) by gavage. This administration was started 5 days before the DSS treatment and continued for a total 10 days.
The level of colonic inflammation was evaluated daily according to the weight loss, stool consistency, and gross rectal bleeding (37). Colon length was measured as a marker of inflammation, and then colon tissue mRNA and protein expression were analyzed as described above. For histological analysis, frozen and paraffin sections (7 μm thick) of colonic tissue were stained by hematoxylin and eosin. Histological examination was performed in a blinded manner by assigning a score for epithelial damage and leukocyte infiltration on microscopic cross-sections of the colon. The score used was as follows: (i) epithelial damage (0 = intact epithelium, 1 = disruption of architectural structure, 2 = erosion, and 3 = ulceration) and (ii) leukocyte infiltration (0 = normal, 1 = focal, 2 = multifocal, and 3 = diffuse). The final score was expressed as the sum of scores for epithelial damage and leukocyte infiltration.
Mouse colon tissue was embedded in OCT compound (Sakura Finetek, Tokyo, Japan), and frozen sections (7 μm thick) were prepared on glass slides. The sections were fixed with 3% (w/v) paraformaldehyde for 10 min and washed with PBS. The sections were blocked in 5% normal goat serum (Wako Pure Chemical Industries) and incubated with rabbit polyclonal anti-occludin, anti-TNFR2, anti-NF-κB p65, or mouse polyclonal anti-MLCK at 4 °C overnight, followed by incubation for 1 h with goat FITC-conjugated anti-rabbit IgG (Invitrogen) for the detection of occludin, TNFR2, and NF-κB p65 and goat Alexa Fluor 546-conjugated anti-mouse IgG (Invitrogen) for MLCK detection. In some experiments, after incubation with secondary antibody, the sections were washed with PBS and incubated with phycoerythrin-conjugated anti-CD326 antibody for 1 h. The specimens were preserved in a mounting medium, and fluorescence was visualized using a fluorescence microscope (FW4000, Leica Microsystems). Colon cryosections were also immunolabeled for NF-κB p65 and stained with DAPI, and the number of NF-κB p65-positive cells were counted.
Isolation of Murine Intestinal Epithelial Cells
The distal colon segment was opened longitudinally, washed with ice-cold PBS, and incubated in 1 mm DTT/PBS (Wako Pure Chemical Industries) at room temperature for 10 min under gentle agitation to remove the mucin layer. The intestinal epithelial cells (IECs) were isolated by incubation with 30 mm EDTA/PBS (Wako Pure Chemical Industries) at 37 °C for 10 min followed by vigorous vortexing for 1 min. Harvested IECs were treated with 2 mg/ml dispase (Invitrogen) and 50 mg/ml DNase I (Roche Applied Science) at 37 °C for 60 min with vortexing every 5 min. The resultant single IECs were purified by centrifugation through a 20/40% Percoll gradient (GE Healthcare).
Flow Cytometric Analysis
IECs (n = 4 for each group) were stained with phycoerythrin-conjugated anti-TNFR2 (BioLegend, San Diego) antibody at 4 °C for 30 min. For cytokeratin staining, the cells were fixed/permeabilized with Cytofix/Cytoperm solution (BD Biosciences) and then stained with FITC-conjugated anti-pan-cytokeratin (Sigma) antibody at 4 °C for 30 min. Species- and isotype-matched antibodies of irrelevant specificity were used as controls. Fluorescence intensity was measured by a Guava EasyCyte flow cytometry system (Millipore). IEC viability (>70%) was assessed by Guava ViaCount (Millipore), and purity (> 80%) was assessed by flow cytometry using anti-pan-cytokeratin and anti-CD326 antibodies.
Statistical Analysis
Results are expressed as the mean ± S.E. Statistical analysis was by using one-way analysis of variance followed by Tukey's post hoc test by the Statcel 2 program (OMS Publishing, Saitama, Japan). A difference of p < 0.05 was considered statistically significant.
RESULTS
HYA Restores TJ Permeability and TJ-related Molecule Expression in Intestinal Epithelial Caco-2 Cells Impaired by IFN-γ + TNF-α
Caco-2 cells were treated with IFN-γ for 24 h followed by TNF-α for 6 h. This treatment significantly decreased the TER value. We then investigated the effect of five kinds of fatty acids on IFN-γ + TNF-α-induced changes in TER. The pretreatment of HYA showed a protective effect on TER in a dose-dependent manner (0.5–50 μm) (Fig. 2, A and B). In contrast, the other fatty acids (50 μm each) did not recover TER; the pretreatments of KetoA, KetoB, and KetoC even caused a greater decrease in TER. Caco-2 cells had almost the same response to linoleic acid as they did to HYA (data not shown). For further analysis, we compared the effects of HYA and HYB.
FIGURE 2.

Effects of fatty acids on IFN-γ + TNF-α-induced barrier impairment in Caco-2 cells. A, Caco-2 cells were treated with the fatty acids (50 μm each) for 24 h and then stimulated with IFN-γ + TNF-α. Time course changes in TER were monitored (n = 3) (left). ●, untreated; ○, IFN-γ + TNF-α (−); ■, IFN-γ + TNF-α + HYA; □, IFN-γ + TNF-α + HYB; ♦, IFN-γ + TNF-α + KetoA; ♢, IFN-γ + TNF-α + KetoB; ▴, IFN-γ + TNF-α + KetoC. The data on TER at 6 h is shown (right). p < 0.05 and **, p < 0.01, compared between untreated (●) and IFN-γ + TNF-α (−); #, p < 0.05 and ##, p < 0.01, compared between IFN-γ + TNF-α + HYA (■) and IFN-γ + TNF-α (−) (Tukey-Kramer). B, recovery of TER at 6 h was examined with varying concentrations of HYA (0, 0.5, 5, and 50 μm) (n = 3). *, p < 0.05, and **, p < 0.01, compared with untreated; #, p < 0.05, and ##, p < 0.01, compared with IFN-γ + TNF-α (without HYA) (Tukey-Kramer). C, FD-4 permeability into the basal wells was assessed for 6 h (n = 3). LA, linoleic acid. D, after TER measurement at 6 h, the basal medium was collected and the IL-8 concentration was determined (n = 3). LA, linoleic acid. E, total RNA was extracted from Caco-2 cells at 6 h, and mRNA expression was examined by real time RT-PCR. Data are presented as the fold change in gene expression from the control (untreated), after normalization to the β-actin gene (n = 3). *, p < 0.05, and **, p < 0.01, compared with untreated; ##, p < 0.01, compared with IFN-γ + TNF-α (−); $, p < 0.05, and $$, p < 0.01, compared with HYA (Tukey-Kramer). Results are expressed as means ± S.E. Each result (in A–E) is representative of three independent similar experiments.
IFN-γ + TNF-α induced an increase in FD-4 permeability; however, the pretreatment of HYA and linoleic acid significantly suppressed this increase (Fig. 2C). In contrast, the pretreatment of HYB did not alter IFN-γ + TNF-α-induced changes in TER and FD-4 permeability. As shown in Fig. 2D, IFN-γ + TNF-α induced a marked increase in the secretion of IL-8, which is a major pro-inflammatory cytokine. However, the pretreatment of HYA significantly suppressed IL-8 secretion.
Changes in mRNA expression of TJ-related molecules were examined to investigate the barrier-recovering effects of HYA (Fig. 2E). IFN-γ + TNF-α significantly down-regulated the mRNA expression of occludin, ZO-1, ZO-2, and claudin-3, and up-regulated the mRNA expression of claudin-1 and MLCK. Pretreatment of HYA, but not HYB, significantly restored IFN-γ + TNF-α-induced changes in the expression of occludin, ZO-1, claudin-1, and MLCK. In particular, a significant difference in the expression of occludin and ZO-1 between HYA and HYB pretreatments was observed. Similarly, IFN-γ + TNF-α significantly down-regulated the protein expression of occludin and ZO-1, and up-regulated the protein expression of MLCK. However, pretreatment of HYA, but not HYB, significantly restored these changes in expression (data not shown).
HYA Decreases TNFR2 Expression in Caco-2 Cells
To further investigate the molecular mechanisms of the protective effects of HYA on TJ-related molecules, we evaluated the mRNA expression of the TNFRs and the protein expression of IκBα and NF-κB p65. IFN-γ significantly increased mRNA expression of TNFR1 and TNFR2 (Fig. 3A), and IFN-γ + TNF-α significantly induced an increase in the ratio of phospho-IκBα/IκBα (Fig. 3B) and NF-κB p65 expression (Fig. 3C). The pretreatment of HYA suppressed IFN-γ-induced up-regulation of TNFR2 (but not TNFR1), and it subsequently decreased the ratio of phospho-IκBα/IκBα and NF-κB p65 expression induced by cytokines (Fig. 3, B and C). In contrast, pretreatment of HYB did not show the same inhibitory effects as HYA.
FIGURE 3.

Effects of HYA and HYB on the expression of TNFRs. A, after stimulation with IFN-γ for 24 h, total RNA was extracted from Caco-2 cells, and the mRNA expression of TNFRs was examined by real time RT-PCR (n = 3). B and C, protein was extracted from Caco-2 cells, and protein expression of phospho-IκBα/IκBα (B) and NF-κB p65 (C) was examined by immunoblotting (n = 3). Results are expressed as means ± S.E. *, p < 0.05, and **, p < 0.01, compared with untreated; #, p < 0.05, compared with IFN-γ (A), IFN-γ + TNF-α (B and C) (−); $$, p < 0.01, compared with HYA (Tukey-Kramer). Data (in A–C) are representative of three independent similar experiments.
HYA Augments the Expression of GPR40, a Free Fatty Acid Receptor
The mRNA expression of various fatty acid receptors was also examined. We focused on GPR40, GPR120, peroxisome proliferator-activated receptor γ (PPARγ), GPR41, and GPR43. It was noteworthy that the pretreatment of HYA augmented the mRNA expression of GPR40 and GPR43 but not GPR120, PPARγ, and GPR41 in Caco-2 cells (Fig. 4A). However, the pretreatment of HYB did not up-regulate GPR40 and GPR43 expression (Fig. 4A).
FIGURE 4.

Effects of HYA and HYB on expression of fatty acid receptors. A, after stimulation with TNF-α for 6 h, total RNA was extracted from Caco-2 cells, and the mRNA expression of fatty acid receptors (GPR40, GPR120, PPARγ, GPR41, and GPR43) was examined by real time RT-PCR (n = 3). B, protein was extracted from Caco-2 cells (n = 3), and expression of GPR40 was examined by immunoblotting. Results are expressed as means ± S.E. *, p < 0.05, and **, p < 0.01, compared with untreated; #, p < 0.05, and ##, p < 0.01, compared with IFN-γ + TNF-α (−); $, p < 0.05, and $$, p < 0.01, compared with HYA (Tukey-Kramer). Each result (A and B) is representative of three independent similar experiments. C and D, after stimulation with IFN-γ + TNF-α, Caco-2 cells were labeled for GPR40 (green) and DAPI (blue) (C) or GPR40 (green) and rhodamine-phalloidin (red) (D). The x-z as well as x-y fluorescence images of HYA-treated cells were obtained (C). Scale bars, 20 μm. Each image (C and D) is representative of two independent similar experiments.
The protein expression of GPR40 was then evaluated. Similar to Fig. 4A, the pretreatment of HYA significantly increased GPR40 expression in Caco-2 cells (Fig. 4, B and D). It was confirmed that GPR40 was localized on the apical side of Caco-2 cells where HYA would presumably bind to GPR40 (Fig. 4C).
HYA Induces Ca2+ Responses in HEK293 Cells Expressing GPR40
GPR40 reportedly couples to Gq as the α-subunit of the heterotrimeric G protein and has the ability to mobilize intracellular calcium [Ca2+]i (38). As shown in Fig. 5A, [Ca2+]i influx in HYA- and linoleic acid-treated Caco-2 cells was detected. To clarify whether HYA has a potent agonistic effect on GPR40 signaling, we performed a GPR40 functional activity assay using HEK293 cells expressing human GPR40. HYA and linoleic acid induced [Ca2+]i responses in the HEK293 cells in a dose-dependent manner, but the addition of the GPR40 antagonist GW1100 completely negated these responses (Fig. 5, B and C). The activity of HYA was higher than that of linoleic acid, the endogenous agonists (EC50: 6.0 ± 2.8 and 11.3 ± 0.6 μm in HYA and linoleic acid, respectively), whereas GPR40-expressing HEK293 and Caco-2 cells exhibited no responses to HYB (Fig. 5, A–C).
FIGURE 5.

Induction of [Ca2+]i rise by HYA in Caco-2 cells and HEK293 cells expressing human GPR40. A, mobilization of [Ca2+]i induced by HYA, HYB, and linoleic acid (100 μm each) was monitored in Caco-2 cells, and data are presented as relative Ca2+ intensity (n = 3). LA, linoleic acid. **, p < 0.01, and *, p < 0.05, compared with untreated; $$, p < 0.01, compared with HYA (Tukey-Kramer). B, mobilization of [Ca2+]i induced by HYA, HYB, and linoleic acid (10 μm each) was monitored in GPR40-expressing HEK293 cells. The cells were pretreated with or without the GPR40 antagonist GW1100 for 15 min prior to the addition of the fatty acids. HEK293 cells not expressing GPR40 were used as negative controls (doxycycline (Dox (−)) (n = 3).**, p < 0.01, compared with untreated; ##, p < 0.01, compared with GW1100 (+). NS, not significant (Tukey-Kramer). C, representative dose-response curve of HYA-induced [Ca2+]i rise in HEK293-hGPR40 cells (n = 3). ●, linoleic acid; ■, HYA; ▾, HYB. Inactive, no response at 1,000 μm. EC50 indicates the concentration of a sample that produces 50% of the maximal response and was calculated from dose-response curves. Results are expressed as means ± S.E. Each result (A–C) is representative of three similar experiments.
Blockade of GPR40 Signaling Cancels the Recovering Effects of HYA
Next, we confirmed the involvement of GPR40 signaling using the GPR40 antagonist. The effects of HYA and HYB on IFN-γ + TNF-α-induced changes in TJ barrier functions (TER and FD-4 permeability) and IL-8 secretion were evaluated with or without the addition of GW1100 (Fig. 6). GW1100 alone did not affect TER, FD-4 permeability or IL-8 secretion.
FIGURE 6.

Inhibitory effects of a GPR40 antagonist on the barrier-recovering and TNFR2-suppressive activity of HYA. Caco-2 cells were pretreated with the GPR40 antagonist GW1100 for 30 min, and then the barrier-recovering effects of HYA were evaluated as per Fig. 2. A, time course of changes in TER. Open symbols are values from cells without GW1100 treatment, and closed symbols are with GW1100 treatment (left). ○ and ●, untreated; □ and ■, IFN-γ + TNF-α; ▵ and ▴, IFN-γ + TNF-α + HYA. The data on TER at 6 h is shown (right). B and C, evaluation of FD-4 permeability and IL-8 concentration. D, effects of HYA on TNFR2 expression were evaluated as per Fig. 3 by immunoblotting. *, p < 0.05, and **, p < 0.01, compared with untreated; #, p < 0.05, and ##, p < 0.01, compared with IFN-γ + TNF-α (−) or IFN-γ (−);$, p < 0.05, and $$, p < 0.01, compared with HYA (Tukey-Kramer). ND, not detected. NS, not significant. Data represent the means ± S.E. Each result (A–D) is representative of three independent similar experiments.
Similar to Fig. 2, A–C, IFN-γ + TNF-α induced a decrease in TER, an increase in FD-4 permeability, and also a marked increase in the secretion of IL-8. Again, the pretreatment of HYA showed restorative effects on TER, FD-4 permeability, and IL-8 secretion. However, GW1100 abolished these effects (Fig. 6, A–C). GW1100 did not alter the inactivity of HYB on barrier impairment in Caco-2 cells (data not shown). We next evaluated the effect of GPR40 inhibition on TNFR2 expression (Fig. 6D). Similar to Fig. 3A, IFN-γ increased TNFR2 expression, regardless of the addition of GW1100. The pretreatment of HYA significantly decreased protein expression of TNFR2 in the absence of GW1100. Notably, however, this effect was abolished by the addition of GW1100. These results clearly suggested that the recovering effects of HYA were attributed to GPR40 signaling.
Blockade of the MEK-ERK Pathway Weakens the Effects of HYA
We explored the possible signaling pathway underlying the HYA-GPR40 axis. As shown in Fig. 7, HYA phosphorylated ERK (but not p38 and JNK), and the MEK inhibitor U0126 restored the phosphorylation level of ERK. The effects of HYA on IFN-γ and the TNF-α-induced changes in TJ barrier functions and IL-8 secretion were evaluated with or without the addition of U0126 (Fig. 8). U0126 alone did not affect TER, FD-4 permeability, and IL-8 secretion.
FIGURE 7.
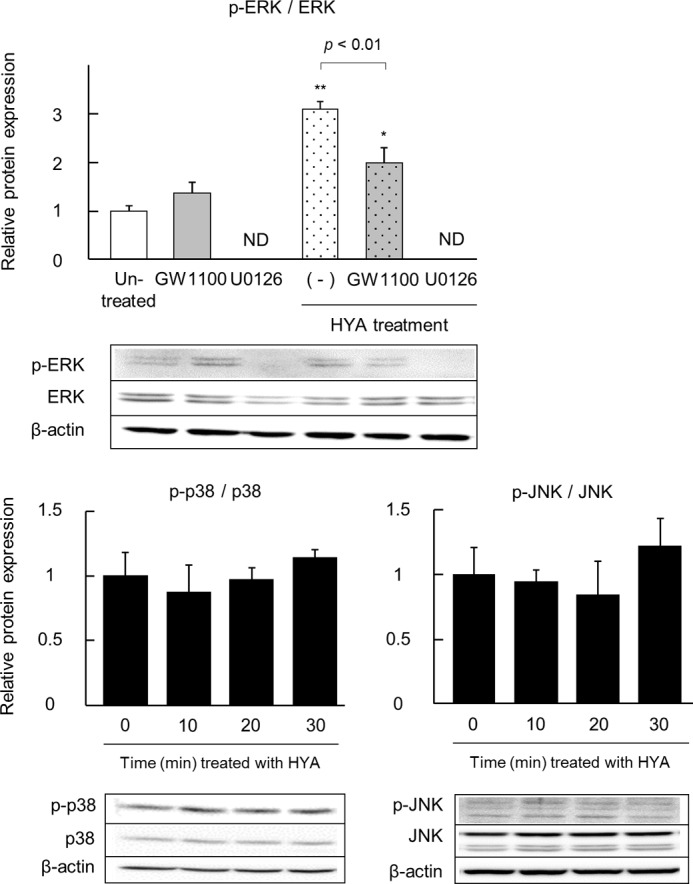
Effect of HYA on the phosphorylation of MAPK. Caco-2 cells were treated with HYA (50 μm) for 0-30 min (p38 and JNK) or 20 min (ERK). Also, some cells were pretreated with the GPR40 antagonist GW1100 or the MEK inhibitor U0126 for 30 min (ERK). Protein was extracted from Caco-2 cells, and phosphorylated and total protein expression of ERK, p38, and JNK was examined by immunoblotting (n = 3). *, p < 0.05, and **, p < 0.01, compared with untreated (Tukey-Kramer). Each result is representative of two similar experiments. ND, not detected.
FIGURE 8.

Inhibitory effects of a MEK inhibitor on the barrier-recovering activity of HYA. Caco-2 cells were pretreated with the MEK inhibitor U0126 for 30 min and then the barrier-recovering effects of HYA and TNFR2 expression were evaluated as per Figs. 2 and 3, respectively. A, time course of changes in TER. Open symbols are values from cells without U0126 treatment, and closed symbols are with U0126 treatment (left). ○ and ●, untreated; □ and ■, IFN-γ + TNF-α; ▵ and ▴, IFN-γ + TNF-α + HYA. The data on TER at 6 h is shown (right). B–D, evaluation of FD-4 permeability, IL-8 concentration, and TNFR2 expression (n = 3). **, p < 0.01, compared with untreated; #, p < 0.05, and ##, p < 0.01, compared with IFN-γ + TNF-α with or without U0126 (−); $, p < 0.05, and $$, p < 0.01, compared with HYA without U0126 (Tukey-Kramer). ND, not detected. NS, not significant. Results are expressed as means ± S.E. Each result (A–C) is representative of two similar experiments.
IFN-γ + TNF-α induced a decrease in TER, an increase in FD-4 permeability, and also a marked increase in the secretion of IL-8, and the pretreatment of HYA showed restorative effects on these parameters. However, U0126 weakened the positive effects of HYA on TER and FD-4, although U0126 did not alter the IL-8-suppressive effect of HYA. In addition, the pretreatment of HYA significantly decreased TNFR2 protein expression, but this effect was abolished by the addition of U0126.
Oral Administration of HYA Alleviates DSS-induced Colitis
We investigated whether oral administration of HYA improved DSS-induced colitis in mice. The oral administration of HYA prevented body weight loss, improved stool scores, and restored colon length (Fig. 9, A–D). Histological assessment confirmed that DSS treatment induced marked destruction of epithelial cells, with crypt loss and infiltration of leukocytes, and the degree of tissue damage was attenuated in HYA-treated mice (Fig. 9E).
FIGURE 9.

Anti-inflammatory effects of HYA in DSS-induced colitis in mice. A and C, mice were monitored daily for weight (A) and stool score (C), which ranges from 1 to 4 (total) representing the average of scores from 1 to 4 for rectal bleeding (1 = normal, 2 = loose stools, 3 = diarrhea, 4 = watery diarrhea) and stool consistency (1 = normal, 2 = slight bleeding, 3 = severe bleeding, 4 = gross bleeding). The data from the last 5 days of the experimental period are shown. ●, untreated; ○, DSS-treated mice; □, DSS-treated mice with HYA administration; and ▵, DSS-treated mice with HYB administration (n = 6). The data for HYA or HYB alone groups were similar to the untreated group. Weight loss was calculated (B) and the colon length was measured on day 10 (D). E, colonic tissue sections were stained with hematoxylin and eosin for histological examination. Scale bars, 100 μm. Histological examination was performed by assigning a score for epithelial damage and leukocyte infiltration on microscopic cross-sections of the colon in each mice. *, p < 0.05, and **, p < 0.01, compared with untreated; #, p < 0.05, and ##, p < 0.01, compared with DSS-treated mice without HYA and HYB administration (−); $, p < 0.05, and $$, p < 0.01, compared with DSS+HYA (Tukey-Kramer). Each result (A–E) is representative of two independent similar experiments.
Oral Administration of HYA Restores TJ-related Molecules
The changes in expression and localization of TJ-related molecules in DSS-induced colitis mice were examined to investigate the barrier-restoring effects of HYA. In the colitis mice, the mRNA expressions of occludin, ZO-1, ZO-2, claudin-1, and claudin-3 were altered (Fig. 10A). The oral administration of HYA significantly recovered the altered expression of occludin, claudin-1, and MLCK. However, oral administration of HYB did not change the expression of all molecules tested.
FIGURE 10.

Recovering effects of HYA on DSS-induced TJ impairment. A, total RNA was extracted from colonic tissue of DSS-induced colitis mice, and the mRNA expression of TJ-related molecules were examined by real time RT-PCR. Data are presented as the fold change in gene expression from the control (untreated), after normalization to the GAPDH gene (n = 6). B and C, cryosections of colonic tissue were immunolabeled for occludin (green) (B) or MLCK (yellow) (C), and DAPI (blue). Scale bars, 50 μm (B) or 100 μm (C). Results are expressed as means ± S.E. *, p < 0.05, and **, p < 0.01, compared with untreated; #, p < 0.05, and ##, p < 0.01, compared with DSS-treated mice without HYA and HYB administration (−) (Tukey-Kramer). Each result (A–C) is representative of two independent similar experiments.
DSS significantly down-regulated occludin protein expression and up-regulated MLCK protein expression. However, the oral administration of HYA restored the altered expression of occludin and MLCK. As shown in Fig. 10B, occludin was observed predominantly in the epithelial cells of normal mice, but the localization of occludin could not be determined in DSS-induced colitis mice. However, the oral administration of HYA improved the disturbed localization of occludin induced by DSS treatment. In contrast, markedly elevated MLCK expression was detected in DSS-induced colitis mice but was suppressed by the oral administration of HYA (Fig. 10C). In contrast to HYA, the oral administration of HYB did not change the expression of occludin and MLCK (Fig. 10, B and C).
HYA Decreases TNFR2 Expression in the Colitis Mice
In DSS-induced colitis mice, the mRNA expressions of TNFR1 and TNFR2 were significantly elevated, but the oral administration of HYA significantly attenuated their expression (Fig. 11A). In contrast, the oral administration of HYB only suppressed TNFR1 transcription. We next investigated TNFR2 expression by immunohistochemistry and flow cytometry. In DSS-induced colitis mice, the expression of TNFR2 was increased on murine IECs (Fig. 11, B and C). However, the oral administration of HYA suppressed the expression of TNFR2 on IECs, whereas the oral administration of HYB did not show a similar recovering effect as HYA.
FIGURE 11.

Effects of HYA on the TNFR expression in DSS-induced colitis mice. A, total RNA was extracted from colonic tissue of DSS-induced colitis mice, and the mRNA expression of TNFRs was examined by real time RT-PCR (n = 6). B, cryosections of colonic tissue were immunolabeled for TNFR2 (green), DAPI (blue), and CD326 (red). Scale bars, 100 μm. C, intestinal epithelial cells were stained for pan-cytokeratin and TNFR2 and analyzed by flow cytometry. The representative histogram analysis (left) and the percentage of TNFR2-positive IECs (right) are shown. D, cryosections of colonic tissue were immunolabeled for NF-κB p65 (green) and DAPI (blue) (left). Scale bars, 50 μm. The number of NF-κB p65-positive cells were counted (right). *, p < 0.05, and **, p < 0.01, compared with untreated; #, p < 0.05, and ##, p < 0.01, compared with DSS-treated mice without HYA and HYB administration (−) (Tukey-Kramer). Each result (A–D) is representative of two independent similar experiments.
The NF-κB p65 staining was faint in IECs in normal mice, but it was strongly induced in DSS-induced colitis mice. However, the oral administration of HYA attenuated the increased expression of NF-κB p65 induced by DSS treatment (Fig. 11D).
Collectively, we have demonstrated that HYA modulates TNFR2 expression via the GPR40-MEK-ERK pathway and suppresses IFN-γ + TNF-α-induced epithelial damage and DSS colitis (Fig. 12).
FIGURE 12.
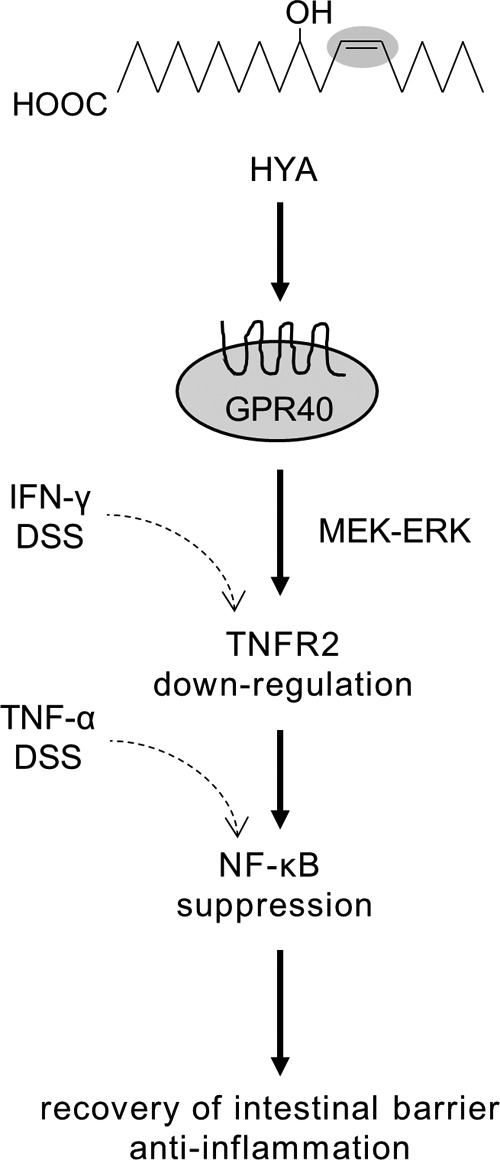
Schematic showing the association between the barrier recovering activity of HYA and GPR40 signaling.
DISCUSSION
It has previously been shown that conjugated linoleic acids exert beneficial effects on human health, including protection from intestinal injury and anti-inflammatory and anti-carcinogenesis effects (39, 40). However, only limited information is available on the physiological functions of the gut microbial metabolites of linoleic acid such as HYA. A notable finding in this study was that HYA ameliorated intestinal barrier impairment by suppressing TNFR2 expression via GPR40.
To induce barrier impairment by inflammatory cytokines in Caco-2 cells, we treated IFN-γ primed cells with TNF-α, which is reported to be overexpressed in patients with IBD (18, 41). TNF-α activates MAPK and NF-κB to induce inflammatory cytokines and chemokines such as IL-8, triggers inflammatory responses, and leads to changes in the expression of TJ-related molecules; for example, it increases MLCK expression and decreases ZO-1 expression through activation of p38 (42–45). In this study, the pretreatment of HYA normalized the expression of occludin, ZO-1, and MLCK in Caco-2 cells (Fig. 2, D and E) by suppressing TNF-α signaling; it also inhibited the phosphorylation of IκBα and the expression of NF-κB p65 (Fig. 4, B and C). It has been reported that GPR40 induced phosphorylation of cAMP-response element-binding protein and that ERK inhibited the activation of MLCK (46, 47). Thus, HYA might also inhibit MLCK expression by regulating the MEK-ERK pathway. As shown in Fig. 7, we confirmed that HYA induced ERK phosphorylation. Similar to our result, it has recently been shown that GW9508 (a GPR40 agonist) treatment leads to a significant increase in phospho-ERK levels in primary cultures of neurons from the mouse central nervous system (46).
To explore the mechanism underlying the protective effects of HYA on the intestinal epithelium, we focused on TNFR, whose expression is reported to be high in the inflamed intestine (48). Indeed, it was confirmed that TNFR2 was highly expressed in the epithelial cells in DSS-colitis mice (Fig. 4, F and G). It has been shown that TNFR2 contributes to exacerbation of inflammation using TNFR2−/− mice (48, 49). TNFR2 signaling mediates the activation of MLCK and the change in localization of occludin. TNFR2 signaling also induces infiltration of macrophages and neutrophils into the lamina propria and activates Th17 cells (50, 51). Su et al. (20) reported that the induced expression of TNFR2 in murine colitis promoted apoptosis of intestinal epithelial cells, chemokine overexpression, and exacerbation of inflammation. Therefore, regulation of TNFR2 is a promising candidate for the treatment of inflammatory disorders such as IBD. In this study, HYA suppressed the protein expression of TNFR2, which was increased by IFN-γ (Figs. 8C and 9). Furthermore, the oral administration of HYA suppressed the expression of TNFR2 in epithelial cells (Fig. 4, F and G), which was correlated with the recovery from colitis (Fig. 3, A–E).
PPARγ and GPR120 have previously been reported to be receptors for long-chain fatty acids and shown to mediate anti-inflammatory activities. Conjugated linoleic acid represses activation of the NF-κB pathway via PPARγ (52). GPR120 is involved in the suppression of inflammation by docosahexaenoic acid and eicosapentaenoic acid by inhibiting phosphorylation of TGF-β-activated kinase-1 (TAK-1) and activation of NF-κB (53). GPR43, also known as FFAR2, recognizes short-chain fatty acid (SCFA). SCFAs regulate the function and size of regulatory T cells (Tregs) in the intestine and protect against intestinal inflammation in a GPR43-dependent manner (54). We show here that HYA exerts its anti-inflammatory property, at least in part, via GPR40 signaling. GPR40, a receptor for long-chain fatty acids, is reported to enhance secretion of glucagon-like peptide 1 and insulin from pancreatic β cells (53, 55, 56), and it has a crucial role in the pathogenesis of obesity and type 2 diabetes. Although there are no reports on the involvement of GPR40 in intestinal disorders, it has recently been reported that topical application of GW9508, a GPR40 agonist, suppresses skin lesions (57). Thus, it is reasonable to hypothesize that GPR40 also mediates the suppression of inflammation in the intestine. Indeed, GW1100, a GPR40 antagonist, canceled the effect of HYA on the suppression of TJ permeability, IL-8 production, and TNFR2 expression in epithelial cells (Fig. 8). Therefore, although there are only a few other studies that have demonstrated GPR40 expression in the intestine (58), this study suggests a novel important role for GPR40 in regulating inflammation in the intestine.
Various fatty acids, such as long-chain fatty acids (for example, linoleic acid, linolenic acid, and oleic acid) and middle-chain fatty acids (capric acid and lauric acid), are reported to be endogenous ligands for GPR40. Among them, linoleic acid shows the highest affinity for GPR40 (32, 56). To clarify the interaction between HYA and GPR40, we performed a GPR40 functional assay using HEK293 cells expressing human GPR40. As the sensitivity of HYA was higher than linoleic acid (Fig. 6), HYA is a promising candidate to modulate GPR40 signaling. In addition, GPR40 exhibited no response to HYB (Fig. 6), which indicated that the carbon-carbon double bond at Δ12 position in HYA is indispensable for the binding between HYA and GPR40.
Notably, increasing evidence has suggested that the host gut microbiota is tightly associated with the gut immune system, and thus far the role of only some microbial metabolites has been revealed. For example, microbially derived butyrate regulates the differentiation of Tregs (59). GPR43, which recognizes SCFAs such as butyrate, was reported to be essential for neutrophil recruitment in DSS-induced colitis (54, 60). Examples of the role of SCFAs are not limited to immune modulation. Kimura et al. (61) reported that gut microbiota suppresses insulin-mediated fat accumulation via GPR43. GPR41 is also reported to be involved in energy regulation in response to SCFAs produced by gut microbiota (62). In addition, GPR41 functions as an energy sensor in the peripheral nervous system to maintain energy homeostasis (63). Further investigations are required to reveal the additional roles of gut microbial metabolites that exert their effects via G protein-coupled receptors.
In this study, we examined the activity of initial metabolites of linoleic and oleic acid contained in the diet. HYA, one of the initial metabolites of linoleic acid, suppresses intestinal inflammation via GPR40 (Figs. 5 and 6). In contrast, HYB, one of the initial metabolites of oleic acid, did not exert a colitis-suppressive activity. These data imply the possibility that the linoleic/oleic acid ratio in the dietary fat and/or the balance of fatty acid-metabolizing enzymes in gut microbes could regulate intestinal disorders.
In conclusion, we demonstrated that HYA attenuates epithelial barrier impairment in Caco-2 cells and ameliorates intestinal inflammation in DSS-induced colitis in mice by the suppression of TNFR2 up-regulation. Furthermore, it was clearly shown that GPR40 recognizes HYA with higher affinity than linoleic acid, an endogenous agonist, and that the blockade of GPR40 signaling cancels the suppressive effects of HYA on intestinal inflammation. This is the first study that indicates the crucial role of GPR40 in the intestinal barrier. Although further in vivo evaluation of the optimal dosage and administration route for HYA is necessary, HYA is indeed promising not only as a gut microbial metabolite of linoleic acid but also as a novel endogenous GPR40 agonist.
This work was supported in part by the Japanese Ministry of Education, Science, Sports and Culture Grants-in-aid KAKENHI 22380076 and 25292074 (to S. T.), the Bio-Oriented Technology Research Advancement Institution of Japan (to J. O.), NEDO Innovation Commercialization Venture Support Project (to collaboration of NITTO PHARMA and J. O.) and by Agreement of Scientific Cooperation Grant CNR-JSPS 2010-11 (to S. T. and M. R.).
- HYA
- 10-hydroxy-cis-12-octadecenoic acid
- DSS
- dextran sulfate sodium
- GPR
- G protein-coupled receptor
- HYB
- 10-hydroxyoctadecanoic acid
- IBD
- inflammatory bowel disease
- IκB
- inhibitor of κB
- MLCK
- myosin light chain kinase
- NF-κB
- nuclear factor-κB
- TER
- transepithelial resistance
- TJ
- tight junction
- TNFR
- tumor necrosis factor receptor
- ZO
- zonula occludens
- IEC
- intestinal epithelial cell
- PPARγ
- peroxisome proliferator-activated receptor γ
- SCFA
- short-chain fatty acid
- KetoA
- 10-oxo-cis-12-octadecenoic acid
- KetoB
- 10-oxo-octadecanoic acid
- KetoC
- 10-oxo-trans-11-octadecenoic acid.
REFERENCES
- 1. Gerasimenko J. V., Gryshchenko O., Ferdek P. E., Stapleton E., Hébert T. O., Bychkova S., Peng S., Begg M., Gerasimenko O. V., Petersen O. H. (2013) Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc. Natl. Acad. Sci. U.S.A. 110, 13186–13191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cao Z., Mulvihill M. M., Mukhopadhyay P., Xu H., Erdélyi K., Hao E., Holovac E., Haskó G., Cravatt B. F., Nomura D. K., Pacher P. (2013) Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology 144, 808–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. von Moltke J., Trinidad N. J., Moayeri M., Kintzer A. F., Wang S. B., van Rooijen N., Brown C. R., Krantz B. A., Leppla S. H., Gronert K., Vance R. E. (2012) Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 490, 107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kishino S., Takeuchi M., Park S. B., Hirata A., Kitamura N., Kunisawa J., Kiyono H., Iwamoto R., Isobe Y., Arita M., Arai H., Ueda K., Shima J., Takahashi S., Yokozeki K., Shimizu S., Ogawa J. (2013) Polyunsaturated fatty acid saturation by gut lactic acid bacteria affecting host lipid composition. Proc. Natl. Acad. Sci. U.S.A. 110, 17808–17813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogawa J., Kishino S., Ando A., Sugimoto S., Mihara K., Shimizu S. (2005) Production of conjugated fatty acids by lactic acid bacteria. J. Biosci. Bioeng. 100, 355–364 [DOI] [PubMed] [Google Scholar]
- 6. Strober W., Fuss I., Mannon P. (2007) The fundamental basis of inflammatory bowel disease. J. Clin. Invest. 117, 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khor B., Gardet A., Xavier R. J. (2011) Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poritz L. S., Garver K. I., Green C., Fitzpatrick L., Ruggiero F., Koltun W. A. (2007) Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J. Surg. Res. 140, 12–19 [DOI] [PubMed] [Google Scholar]
- 9. Meddings J. (2008) The significance of the gut barrier in disease. Gut 57, 438–440 [DOI] [PubMed] [Google Scholar]
- 10. Schulzke J. D., Ploeger S., Amasheh M., Fromm A., Zeissig S., Troeger H., Richter J., Bojarski C., Schumann M., Fromm M. (2009) Epithelial tight junctions in intestinal inflammation. Ann. N.Y. Acad. Sci. 1165, 294–300 [DOI] [PubMed] [Google Scholar]
- 11. Rezaie A., Parker R. D., Abdollahi M. (2007) Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig. Dis. Sci. 52, 2015–2021 [DOI] [PubMed] [Google Scholar]
- 12. Tanabe S. (2013) The effect of probiotics and gut microbiota on Th17 cells. Int. Rev. Immunol. 32, 511–525 [DOI] [PubMed] [Google Scholar]
- 13. Eken A., Singh A. K., Treuting P. M., Oukka M. (2014) IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 7, 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanai T., Mikami Y., Sujino T., Hisamatsu T., Hibi T. (2012) RORγt-dependent IL-17A-producing cells in the pathogenesis of intestinal inflammation. Mucosal Immunol. 5, 240–247 [DOI] [PubMed] [Google Scholar]
- 15. Vivinus-Nébot M., Frin-Mathy G., Bzioueche H., Dainese R., Bernard G., Anty R., Filippi J., Saint-Paul M. C., Tulic M. K., Verhasselt V., Hébuterne X., Piche T. (2014) Functional bowel symptoms in quiescent inflammatory bowel diseases: role of epithelial barrier disruption and low-grade inflammation. Gut 63, 744–752 [DOI] [PubMed] [Google Scholar]
- 16. Fischer A., Gluth M., Pape U. F., Wiedenmann B., Theuring F., Baumgart D. C. (2013) Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G970–G979 [DOI] [PubMed] [Google Scholar]
- 17. Baker O. J., Camden J. M., Redman R. S., Jones J. E., Seye C. I., Erb L., Weisman G. A. (2008) Proinflammatory cytokines tumor necrosis factor-α and interferon-γ alter tight junction structure and function in the rat parotid gland Par-C10 cell line. Am. J. Physiol. Cell Physiol. 295, C1191–C1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye D., Ma I., Ma T. Y. (2006) Molecular mechanism of tumor necrosis factor-α modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G496–G504 [DOI] [PubMed] [Google Scholar]
- 19. Weber C. R., Nalle S. C., Tretiakova M., Rubin D. T., Turner J. R. (2008) Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Invest. 88, 1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su L., Nalle S. C., Shen L., Turner E. S., Singh G., Breskin L. A., Khramtsova E. A., Khramtsova G., Tsai P. Y., Fu Y. X., Abraham C., Turner J. R. (2013) TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology 145, 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arrieta M. C., Bistritz L., Meddings J. B. (2006) Alterations in intestinal permeability. Gut 55, 1512–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Al-Sadi R., Khatib K., Guo S., Ye D., Youssef M., Ma T. (2011) Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G1054–G1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petit C. S., Barreau F., Besnier L., Gandille P., Riveau B., Chateau D., Roy M., Berrebi D., Svrcek M., Cardot P., Rousset M., Clair C., Thenet S. (2012) Requirement of cellular prion protein for intestinal barrier function and mislocalization in patients with inflammatory bowel disease. Gastroenterology 143, 122–132 [DOI] [PubMed] [Google Scholar]
- 24. Hossain Z., Hirata T. (2008) Molecular mechanism of intestinal permeability: interaction at tight junctions. Mol. Biosyst. 4, 1181–1185 [DOI] [PubMed] [Google Scholar]
- 25. Hamada K., Shitara Y., Sekine S., Horie T. (2010) Zonula occludens-1 alterations and enhanced intestinal permeability in methotrexate-treated rats. Cancer Chemother. Pharmacol. 66, 1031–1038 [DOI] [PubMed] [Google Scholar]
- 26. Fukui A., Naito Y., Handa O., Kugai M., Tsuji T., Yoriki H., Qin Y., Adachi S., Higashimura Y., Mizushima K., Kamada K., Katada K., Uchiyama K., Ishikawa T., Takagi T., Yagi N., Kokura S., Yoshikawa T. (2012) Acetyl salicylic acid induces damage to intestinal epithelial cells by oxidation-related modifications of ZO-1. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G927–G936 [DOI] [PubMed] [Google Scholar]
- 27. Salim S. Y., Söderholm J. D. (2011) Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 362–381 [DOI] [PubMed] [Google Scholar]
- 28. Piche T., Barbara G., Aubert P., Bruley des Varannes S., Dainese R., Nano J. L., Cremon C., Stanghellini V., De Giorgio R., Galmiche J. P., Neunlist M. (2009) Impaired intestinal barrier integrity in the colon of patients with irritable bowel syndrome: involvement of soluble mediators. Gut 58, 196–201 [DOI] [PubMed] [Google Scholar]
- 29. Mennigen R., Nolte K., Rijcken E., Utech M., Loeffler B., Senninger N., Bruewer M. (2009) Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1140–G1149 [DOI] [PubMed] [Google Scholar]
- 30. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 31. Burant C. F. (2013) Activation of GPR40 as a therapeutic target for the treatment of type 2 diabetes. Diabetes Care 36, S175–S179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Briscoe C. P., Tadayyon M., Andrews J. L., Benson W. G., Chambers J. K., Eilert M. M., Ellis C., Elshourbagy N. A., Goetz A. S., Minnick D. T., Murdock P. R., Sauls H. R., Jr., Shabon U., Spinage L. D., Strum J. C., Szekeres P. G., Tan K. B., Way J. M., Ignar D. M., Wilson S., Muir A. I. (2003) The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 278, 11303–11311 [DOI] [PubMed] [Google Scholar]
- 33. Salehi A., Flodgren E., Nilsson N. E., Jimenez-Feltstrom J., Miyazaki J., Owman C., Olde B. (2005) Free fatty acid receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res. 322, 207–215 [DOI] [PubMed] [Google Scholar]
- 34. Itoh Y., Hinuma S. (2005) GPR40, a free fatty acid receptor on pancreatic beta cells, regulates insulin secretion. Hepatol. Res. 33, 171–173 [DOI] [PubMed] [Google Scholar]
- 35. Wauquier F., Philippe C., Léotoing L., Mercier S., Davicco M. J., Lebecque P., Guicheux J., Pilet P., Miot-Noirault E., Poitout V., Alquier T., Coxam V., Wittrant Y. (2013) The free fatty acid receptor G protein-coupled receptor 40 (GPR40) protects from bone loss through inhibition of osteoclast differentiation. J. Biol. Chem. 288, 6542–6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Del Guerra S., Bugliani M., D'Aleo V., Del Prato S., Boggi U., Mosca F., Filipponi F., Lupi R. (2010) G-protein-coupled receptor 40 (GPR40) expression and its regulation in human pancreatic islets: the role of type 2 diabetes and fatty acids. Nutr. Metab. Cardiovasc Dis. 20, 22–25 [DOI] [PubMed] [Google Scholar]
- 37. Miyauchi E., Ogita T., Miyamoto J., Kawamoto S., Morita H., Ohno H., Suzuki T., Tanabe S. (2013) Bifidobacterium longum alleviates dextran sulfate sodium-induced colitis by suppressing IL-17A response: involvement of intestinal epithelial costimulatory molecules. PLoS One 8, e79735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Itoh S., Yurimoto H. (2003) Contemporaneous formation of chondrules and refractory inclusions in the early solar system. Nature 423, 728–731 [DOI] [PubMed] [Google Scholar]
- 39. Bergamo P., Gogliettino M., Palmieri G., Cocca E., Maurano F., Stefanile R., Balestrieri M., Mazzarella G., David C., Rossi M. (2011) Conjugated linoleic acid protects against gliadin-induced depletion of intestinal defenses. Mol. Nutr. Food Res. 55, S248–S256 [DOI] [PubMed] [Google Scholar]
- 40. Martinasso G., Saracino S., Maggiora M., Oraldi M., Canuto R. A., Muzio G. (2010) Conjugated linoleic acid prevents cell growth and cytokine production induced by TPA in human keratinocytes NCTC 2544. Cancer Lett. 287, 62–66 [DOI] [PubMed] [Google Scholar]
- 41. Amasheh M., Fromm A., Krug S. M., Amasheh S., Andres S., Zeitz M., Fromm M., Schulzke J. D. (2010) TNFα-induced and berberine-antagonized tight junction barrier impairment via tyrosine kinase, Akt and NFκB signaling. J. Cell Sci. 123, 4145–4155 [DOI] [PubMed] [Google Scholar]
- 42. Petecchia L., Sabatini F., Usai C., Caci E., Varesio L., Rossi G. A. (2012) Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab. Invest. 92, 1140–1148 [DOI] [PubMed] [Google Scholar]
- 43. Dai C., Zhao D. H., Jiang M. (2012) VSL#3 probiotics regulate the intestinal epithelial barrier in vivo and in vitro via the p38 and ERK signaling pathways. Int. J. Mol. Med. 29, 202–208 [DOI] [PubMed] [Google Scholar]
- 44. Heller F., Florian P., Bojarski C., Richter J., Christ M., Hillenbrand B., Mankertz J., Gitter A. H., Bürgel N., Fromm M., Zeitz M., Fuss I., Strober W., Schulzke J. D. (2005) Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129, 550–564 [DOI] [PubMed] [Google Scholar]
- 45. Blair S. A., Kane S. V., Clayburgh D. R., Turner J. R. (2006) Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Invest. 86, 191–201 [DOI] [PubMed] [Google Scholar]
- 46. Zamarbide M., Etayo-Labiano I., Ricobaraza A., Martínez-Pinilla E., Aymerich M. S., Luis Lanciego J., Pérez-Mediavilla A., Franco R. (2014) GPR40 activation leads to CREB and ERK phosphorylation in primary cultures of neurons from the mouse CNS and in human neuroblastoma cells. Hippocampus 24, 733–739 [DOI] [PubMed] [Google Scholar]
- 47. Srinivas S. P., Satpathy M., Gallagher P., Larivière E., Van Driessche W. (2004) Adenosine induces dephosphorylation of myosin II regulatory light chain in cultured bovine corneal endothelial cells. Exp. Eye Res. 79, 543–551 [DOI] [PubMed] [Google Scholar]
- 48. Wang K., Han G., Dou Y., Wang Y., Liu G., Wang R., Xiao H., Li X., Hou C., Shen B., Guo R., Li Y., Shi Y., Chen G. (2012) Opposite role of tumor necrosis factor receptors in dextran sulfate sodium-induced colitis in mice. PLoS One 7, e52924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mizoguchi E., Mizoguchi A., Takedatsu H., Cario E., de Jong Y. P., Ooi C. J., Xavier R. J., Terhorst C., Podolsky D. K., Bhan A. K. (2002) Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology 122, 134–144 [DOI] [PubMed] [Google Scholar]
- 50. Ayyadurai S., Charania M. A., Xiao B., Viennois E., Merlin D. (2013) PepT1 expressed in immune cells has an important role in promoting the immune response during experimentally induced colitis. Lab. Invest. 93, 888–899 [DOI] [PubMed] [Google Scholar]
- 51. Wang Y., Han G., Chen Y., Wang K., Liu G., Wang R., Xiao H., Li X., Hou C., Shen B., Guo R., Li Y., Chen G. (2013) Protective role of tumor necrosis factor (TNF) receptors in chronic intestinal inflammation: TNFR1 ablation boosts systemic inflammatory response. Lab. Invest. 93, 1024–1035 [DOI] [PubMed] [Google Scholar]
- 52. Zenhom M., Hyder A., Kraus-Stojanowic I., Auinger A., Roeder T., Schrezenmeir J. (2011) PPARγ-dependent peptidoglycan recognition protein 3 (PGlyRP3) expression regulates proinflammatory cytokines by microbial and dietary fatty acids. Immunobiology 216, 715–724 [DOI] [PubMed] [Google Scholar]
- 53. Oh D. Y., Talukdar S., Bae E. J., Imamura T., Morinaga H., Fan W., Li P., Lu W. J., Watkins S. M., Olefsky J. M. (2010) GPR120 is an ω-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith P. M., Howitt M. R., Panikov N., Michaud M., Gallini C. A., Bohlooly-Y M., Glickman J. N., Garrett W. S. (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hirasawa A., Tsumaya K., Awaji T., Katsuma S., Adachi T., Yamada M., Sugimoto Y., Miyazaki S., Tsujimoto G. (2005) Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 11, 90–94 [DOI] [PubMed] [Google Scholar]
- 56. Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S., Ogi K., Hosoya M., Tanaka Y., Uejima H., Tanaka H., Maruyama M., Satoh R., Okubo S., Kizawa H., Komatsu H., Matsumura F., Noguchi Y., Shinohara T., Hinuma S., Fujisawa Y., Fujino M. (2003) Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 422, 173–176 [DOI] [PubMed] [Google Scholar]
- 57. Fujita T., Matsuoka T., Honda T., Kabashima K., Hirata T., Narumiya S. (2011) A GPR40 agonist GW9508 suppresses CCL5, CCL17, and CXCL10 induction in keratinocytes and attenuates cutaneous immune inflammation. J. Invest. Dermatol. 131, 1660–1667 [DOI] [PubMed] [Google Scholar]
- 58. Duca F. A., Swartz T. D., Sakar Y., Covasa M. (2012) Increased oral detection, but decreased intestinal signaling for fats in mice lacking gut microbiota. PLoS One 7, e39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Furusawa Y., Obata Y., Fukuda S., Endo T. A., Nakato G., Takahashi D., Nakanishi Y., Uetake C., Kato K., Kato T., Takahashi M., Fukuda N. N., Murakami S., Miyauchi E., Hino S., Atarashi K., Onawa S., Fujimura Y., Lockett T., Clarke J. M., Topping D. L., Tomita M., Hori S., Ohara O., Morita T., Koseki H., Kikuchi J., Honda K., Hase K., Ohno H. (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 [DOI] [PubMed] [Google Scholar]
- 60. Sina C., Gavrilova O., Förster M., Till A., Derer S., Hildebrand F., Raabe B., Chalaris A., Scheller J., Rehmann A., Franke A., Ott S., Häsler R., Nikolaus S., Fölsch U. R., Rose-John S., Jiang H. P., Li J., Schreiber S., Rosenstiel P. (2009) G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J. Immunol. 183, 7514–7522 [DOI] [PubMed] [Google Scholar]
- 61. Kimura I., Ozawa K., Inoue D., Imamura T., Kimura K., Maeda T., Terasawa K., Kashihara D., Hirano K., Tani T., Takahashi T., Miyauchi S., Shioi G., Inoue H., Tsujimoto G. (2013) The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 4, 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Samuel B. S., Shaito A., Motoike T., Rey F. E., Backhed F., Manchester J. K., Hammer R. E., Williams S. C., Crowley J., Yanagisawa M., Gordon J. I. (2008) Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. U.S.A. 105, 16767–16772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Inoue D., Kimura I., Wakabayashi M., Tsumoto H., Ozawa K., Hara T., Takei Y., Hirasawa A., Ishihama Y., Tsujimoto G. (2012) Short-chain fatty acid receptor GPR41-mediated activation of sympathetic neurons involves synapsin 2b phosphorylation. FEBS Lett. 586, 1547–1554 [DOI] [PubMed] [Google Scholar]