

Background: DNA is replicated in vitro by proteins encoded by herpes simplex virus type 1.
Results: Electron microscopic examination of the replication forks reveals looping of the lagging strand.
Conclusion: The trombone model has been demonstrated.
Significance: This provides the first evidence of the trombone mechanism in a eukaryotic viral system.
Keywords: DNA Replication, Electron Microscopy (EM), Gel Electrophoresis, Herpesvirus, Structural Biology, Virology
Abstract
Using purified replication factors encoded by herpes simplex virus type 1 and a 70-base minicircle template, we obtained robust DNA synthesis with leading strand products of >20,000 nucleotides and lagging strand fragments from 600 to 9,000 nucleotides as seen by alkaline gel electrophoresis. ICP8 was crucial for the synthesis on both strands. Visualization of the deproteinized products using electron microscopy revealed long, linear dsDNAs, and in 87%, one end, presumably the end with the 70-base circle, was single-stranded. The remaining 13% had multiple single-stranded segments separated by dsDNA segments 500 to 1,000 nucleotides in length located at one end. These features are diagnostic of the trombone mechanism of replication. Indeed, when the products were examined with the replication proteins bound, a dsDNA loop was frequently associated with the replication complex located at one end of the replicated DNA. Furthermore, the frequency of loops correlated with the fraction of DNA undergoing Okazaki fragment synthesis.
Introduction
In the trombone model of DNA replication, leading and lagging strand synthesis is coupled by looping the lagging strand back to the fork where both leading and lagging strands are synthesized in cycles as short fragments (Okazaki fragments), whose sizes range from ∼500 to 6,000 nucleotides (nt)2 in the well characterized prokaryotic systems (1, 2). A typical trombone cycle is illustrated in Fig. 1A. The average size of the dsDNA portion of the loops at the fork is ∼1 kb in T4 and T7 bacteriophages (3, 4). The replication products resulting from multiple trombone cycles are long, linear dsDNA molecules containing a pattern of ss gaps at the end closest to the fork (Fig. 1B).
FIGURE 1.
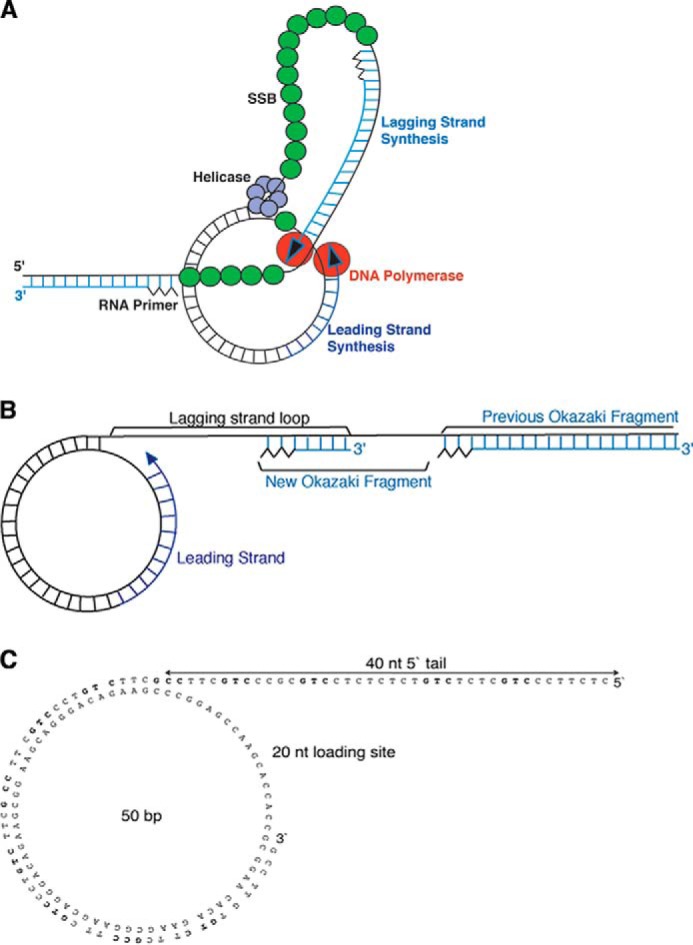
A, scheme for the formation of a trombone loop as initially described by Alberts et al. (1). Leading strand synthesis on the circular DNA is depicted on the right and lagging strand synthesis on the left. DNA polymerase on the leading strand (red sphere, right) is in contact with the DNA polymerase on the lagging strand (red sphere, left), and the coupling generates a DNA loop. DNA helicase represented as a hexamer (purple spheres) unwinds the duplex circular DNA at the fork and generates single-stranded (ss) regions of the loop, which is coated with SSB (green spheres). Lagging strand synthesis is initiated by the synthesis of an RNA primer (black zigzag) and synthesized in (Okazaki) fragments. The ss region covered with SSB between two duplex regions indicates the discontinuous synthesis. B, architecture of the replication products. C, structure and nucleotide sequence of the minicircle template. The minicircle is a partial ds circle with a 40-nt ss 5′ tail and 20-nt ss gap next to the fork. The 70-nt-long strand lacks thymine residues and serves as a template for leading strand synthesis. The 90-nt-long strand lacks adenine residues and contains 12 primase recognition sites (3′-GCC or 3′-GTC) for lagging strand synthesis. Primer initiation sites within the 90-nt strand are shown in bold. Leading and lagging strand synthesis can be identified by measuring the incorporation of [α-32P]dTTP and [α-32P]dATP, respectively.
Although this looping mechanism has been demonstrated in in vitro studies with the T7 proteins by electron microscopy (EM) (5), single molecule methods (6), and EM using T4 factors (3, 7), DNA looping has not been shown to be employed in any eukaryotic system. Indeed, because of the assembly of eukaryotic DNA into chromatin and other differences, the trombone mechanism might be restricted to prokaryotes. The linear dsDNA herpes simplex virus-1 (HSV-1) genome is replicated by a set of six proteins whose activities are well characterized. These consist of a DNA polymerase (UL30), a processivity factor (UL42), a three-subunit helicase-primase (UL5/UL8/UL52), and an ssDNA-binding protein (SSB) (ICP8) (8, 9), and each can be purified to homogeneity following expression in insect cells. Thus, these proteins provide an ideal model system for studies of eukaryotic replication fork structure and dynamics.
The basic architecture of replication forks, whether they involve θ form or rolling circle replication, can be studied using small circular DNAs with displaced forks that provide templates for rolling circle replication. Rolling circle replication has been reconstituted in vitro using HSV-1-infected cell extracts on plasmid DNA substrates (10, 11). However, these studies reported only limited synthesis with a small fraction of molecules engaged in replication, and the rolling circle tails were at most a few times the length of the plasmid circles. A more recent study used a synthetic 70-base minicircle DNA as template to reconstitute replication using purified proteins. This template was initially developed to study T7 replication (4) and was re-engineered by Falkenberg et al. (12) to examine HSV-1 replication. The 70-base-long minicircle they employed was a partial dsDNA circle with a 40-base ss 5′-tail required for efficient UL5/UL8/UL52 binding and unwinding (13) and a 20-base leading strand gap at the fork, providing a site for assembly of the replisome. They observed leading strands up to 10,000 nt and Okazaki fragments of ∼3,000 nt in length. More recently, Stengel and Kuchta (14) modified the DNA sequence of the minicircle template by adding 12 primase recognition sites (three sites of 3′-GCC-5′ and nine sites of 3′-GTC-5′, where 3′G is noncoding) on the lagging strand template, and they utilized higher primase concentrations (Fig. 1C). Using the optimized minicircle, these authors reported leading strand products ∼2,000–10,000 nt and lagging strand products of ∼200–600 nt.
A major step toward understanding HSV-1 replication would be the demonstration of robust rolling circle replication using purified HSV-1 proteins coupled with an EM analysis to determine whether or not the mode of replication follows what has been described for the in vitro T7 and T4 systems. To approach this, we have improved the replication of the 70-base minicircle and used a combination of EM and alkaline gel analysis to examine the replication products. Examination of the fork architecture revealed a rolling circle replication mechanism that involves the formation of lagging strand loops in a nearly identical fashion as seen in the previous phage studies. This provides the first direct evidence for the trombone mechanism in a eukaryotic viral system.
EXPERIMENTAL PROCEDURES
Reagents
Unlabeled ribonucleoside triphosphates (NTP) and deoxyribonucleoside triphosphates (dNTP) were obtained from New England Biolabs. [α-32P]dATP and [α-32P]dTTP were purchased from PerkinElmer Life Sciences. Oligonucleotides were from Eurofins Scientific.
Proteins
The UL5-UL8-UL52 complex was expressed in SF9 insect cells with recombinant baculoviruses encoding UL5, His-tagged UL8, and UL52, which were provided by Dr. Sandra Weller (University of Connecticut). Baculovirus expressing His-tagged UL42 was provided by Dr. Robert Kuchta (University of Colorado, Denver). His-tagged proteins were purified according to the procedures described in Ref. 15, except that we used TALON cobalt affinity chromatography (Clontech). Wild-type UL30 and ICP8 were purified using the chromatography protocols described previously (16, 17).
Minicircle Substrate
A linear 70-nt oligonucleotide (5′-CGGAACACAGAAGCGGAAGCAGGGACAGAAGCGGAAGCAGGGACAGAAGCCCGGAGCCAAGCACCACCG-3′) was converted into a ss circle using CircLigase II ssDNA ligase (Epicenter Biotechnologies) following the manufacturer's protocol. Briefly, 13 pmol of linear oligonucleotide was incubated with 1 μl of 100 units of Circligase in 20 μl of reaction volume at 60 °C overnight. The ssDNA ligase was inactivated by incubation at 80 °C for 10 min. The remaining linear ssDNA and linear adenylated intermediates were removed by treatment with exonuclease I and III at 37 °C for 3 h. The efficiency of the ligation was assessed by electrophoresis of the products on 20% acrylamide, 8 m urea denaturing gel. The minicircle was then annealed to a 90-nt oligonucleotide (5′-CTCTTCCCTGCTCTCTGTCTCTCTCCTGCGCCCTGCTTCCGCTTCTGTCCCTGCTTCCGCTTCCGCTTCTGTCCCTGCTTCCGCTTCTGTGTTCCG-3′), which provides the template strand for lagging strand synthesis (Fig. 1C).
DNA Synthesis
The replication reaction mixture (20 μl for alkaline-agarose gel analysis and 40 μl for EM) contained 40 nm minicircle, 0.1 μm UL30, 0.1 μm UL42, 0.3 μm UL5/UL8/UL52, 0.8 μm ICP8, dNTPs (100 μm dATP, 100 μm dGTP, 40 μm dCTP, and 40 μm dTTP), NTPs (3.2 mm ATP, 3.2 mm GTP, 0.8 mm CTP, and 0.8 mm UTP), 50 mm Tris·HCl (pH 7.6), 5 mm magnesium acetate, and 0.1 mg/ml BSA. For alkaline-agarose gel analysis, 1 μCi of [α-32P]dTTP was included to monitor the leading strand and 1 μCi of [α-32P]-dATP for lagging strand synthesis. Reaction mixtures were incubated for 30 and 60 min at 37 °C and stopped by 0.1 m EDTA at final concentration prior to alkaline-agarose gel electrophoresis or EM.
Alkaline-Agarose Gel Electrophoresis
Alkaline-agarose loading buffer was added to the stopped reactions to produce a final concentration of 50 mm NaOH, 1 mm EDTA, 4% glycerol, 0.03% bromocresol green, and 0.05% xylene cyanol. The radioactive DNA products were analyzed by electrophoresis on 0.8% alkaline-agarose gels in 50 mm NaOH and 1 mm EDTA. The products were fixed in 7% (w/v) trichloroacetic acid for 30 min and dried prior to phosphorimaging using a Typhoon 9400 scanner. The sizes of radioactively labeled products were determined by comparison with 3′-end-labeled 32P λ-DNA HindIII marker. The bands were quantitated on ImageJ software.
EM
The reactions in parallel with the electrophoresis were incubated and stopped as described above.
To visualize DNA-protein complexes, the samples were treated with 0.6% glutaraldehyde for 5 min at room temperature and chromatographed through 2-ml columns of 6% agarose beads (Agarose Bead Technologies), equilibrated in 10 mm Tris·HCl (pH 7.6) and 0.1 mm EDTA. The samples were then adsorbed to thin carbon supports in the presence of 2 mm spermidine, washed, air-dried, and rotary shadow cast with tungsten as described (18).
To visualize the deproteinized reaction products, samples were treated with proteinase K (100 μg/ml) and SDS (1%) for 60 min at 55 °C and then passed over agarose columns as above. Following the removal of proteins, two methods were used. Deproteinized molecules were either complexed with Escherichia coli SSB (6) and prepared for EM as above or they were directly prepared for EM by spreading on an air-water interface with cytochrome c protein. Briefly, 48.3 μl of DNA in 0.25 m ammonium acetate was mixed with 1.7 μl of 200 μg/ml cytochrome c solution and placed on a piece of parafilm as a drop. After 10 min, a parlodion-coated copper grid was touched to the surface of the drop, washed in 75% ethanol, air-dried, and shadowed with platinum-palladium wire.
Samples were examined using an FEI Tecnai T12 EM at 40 kV. Length measurements were made by capturing the EM images with a Gatan CCD camera and using Digital Micrograph software.
RESULTS
DNA Substrate
We generated the 70-base minicircle previously described (see “Experimental Procedures”; Fig. 1C) and employed it as a DNA template in our experiments. The leading strand template is devoid of thymine residues, and the complementary lagging strand template consequently is devoid of adenine residues. Thus, we detected the size distribution of the leading or lagging strand products by specifically labeling the strands with [α-32P]dTTP or [α-32P]dATP, respectively.
Synthesis of Leading and Lagging Strands on the Minicircle
The replication reactions were catalyzed by six viral proteins purified from insect cells as follows: WT UL30, UL42, UL5/UL8/UL52, and ICP8. DNA synthesis was carried out at 37 °C by incubating the minicircle with these proteins in the presence of dNTPs and NTPs in a buffer containing 2 mm free Mg2+ ion. The addition of creatine phosphate and creating kinase as an energy-regenerating system did not improve the replication. The optimal amounts of each protein were determined by analysis of the replication products using alkaline-agarose gel electrophoresis following incubations of 0–60 min (see under “Experimental Procedures”).
Both the length and amount of synthesis increased over time (Fig. 2, lanes 3–6). The leading strand products exceed 20,000 nt at the 60-min time point, indicating that each active minicircle had been replicated almost 300 times. The length of the lagging strand products ranged from 600 to 9,000 nt after 60 min, shorter than leading strands, as expected. The total amount of the leading strand products seen on the gel was somewhat greater than the lagging strand products. This difference may reflect the fraction of templates consisting of just the circle and a long ss tail, which had not yet transitioned to coupled leading/lagging synthesis. To further examine these reactions, we examined their dependence on ICP8.
FIGURE 2.

Alkaline-agarose gel electrophoresis of the products of DNA synthesis on the minicircle. DNA synthesis was carried out for 30 or 60 min at 37 °C using the minicircle template with mixtures containing either [α-32P]dTTP or [α-32P]dATP to follow leading and lagging strand synthesis, respectively. The amounts of ICP8 are indicated. Products were analyzed using electrophoresis through 0.8% alkaline-agarose gels.
ICP8 Is Essential for Replication
In the absence of ICP8, leading strand synthesis was barely detectable even at 60-min incubations, and the leading strands produced were small, ranging from 200 to 2,000 nt (Fig. 2, lane 1). In addition, lagging strand synthesis was not observed when ICP8 was omitted due to the lack of leading strand templates for lagging strand synthesis (Fig. 2, lane 2). Optimal ICP8 concentration for the synthesis of both strands was found to be 0.8 μm, with higher concentrations impairing the quantity of lagging strand products (Fig. 2, lane 8). Leading strand synthesis, however, was not affected at the higher saturating concentrations of ICP8 (Fig. 2, lane 7).
Effect of NTPs on the Synthesis of the Leading and Lagging Strands
The coupled synthesis was highly dependent on the concentration of NTPs. In reactions that contained 2.8 mm ATP and 0.8 mm GTP, CTP, and UTP, as described by Stengel and Kuchta (14), to reconstitute HSV-1 replication on the 70-base minicircle, synthesis of Okazaki fragments was initiated only after 45 min of incubation, although 10,000-nt-long leading strands were already present at 30 min. Thus, the synthesis of both strands was not coordinated under these conditions, and there was a long lag period before the onset of lagging strand synthesis. The primer initiation sites contain T and C (3′-GTC or 3′-GCC). Therefore, increasing the concentrations of ATP and GTP involved in diribonucleotide synthesis to 3.2 mm, while holding the concentration of CTP and UTP at 0.8 mm, increased the rate of primer synthesis, and we now obtained the lagging synthesis product at earlier time points (Fig. 2, lane 5).
The collective results from these experiments indicate that coordinated strand synthesis takes place within only a very narrow range of DNA, protein, dNTP, and NTP concentrations. Our optimized conditions are described under “Experimental Procedures.”
Visualization of Deproteinized Replication Intermediates
Using EM, we visualized the products of minicircle replication at 60-min time points. To observe the underlying structure of the DNA products, the samples were deproteinized, and the ss segments were incubated with E. coli SSB protein to extend and thicken them, making it easy to distinguish ssDNA from dsDNA. The limitation of the EM method used here is that the 70-base minicircle itself would only appear as a tiny dot due to the shadow-casting process. Long products generated by rolling circle replication however should be easily visualized. Using EM to examine fields of deproteinized DNA treated with SSB, we identified several different intermediate types summarized in Table 1. Only 7% of the molecules scored (25 of 343) consisted of solely linear ssDNA arising from leading strand synthesis alone. However, 38% appeared as full dsDNA (Fig. 3D, molecule on the right), and 55% consisted of long dsDNA segments joined by one (Fig. 3, A, C, and D) or two ss regions close to one end (Fig. 3B). The DNAs that appeared fully ds could in fact contain short ss gaps that did not bind enough SSB to be easily detected (e.g. ∼50-base gap), and as noted above, the 70-base minicircle at one end of these long dsDNAs would not be distinguished as a small circle. The longest dsDNA products observed (20 kb) were ∼290 times longer than the length of the monomer minicircle (70 base), consistent with the size of replication products as measured by gel analysis (Fig. 2).
TABLE 1.
Distribution of all replication products in different stages in synthesis
The molecules (343) were scored and categorized by the type of molecule and the number of ssDNA gaps.
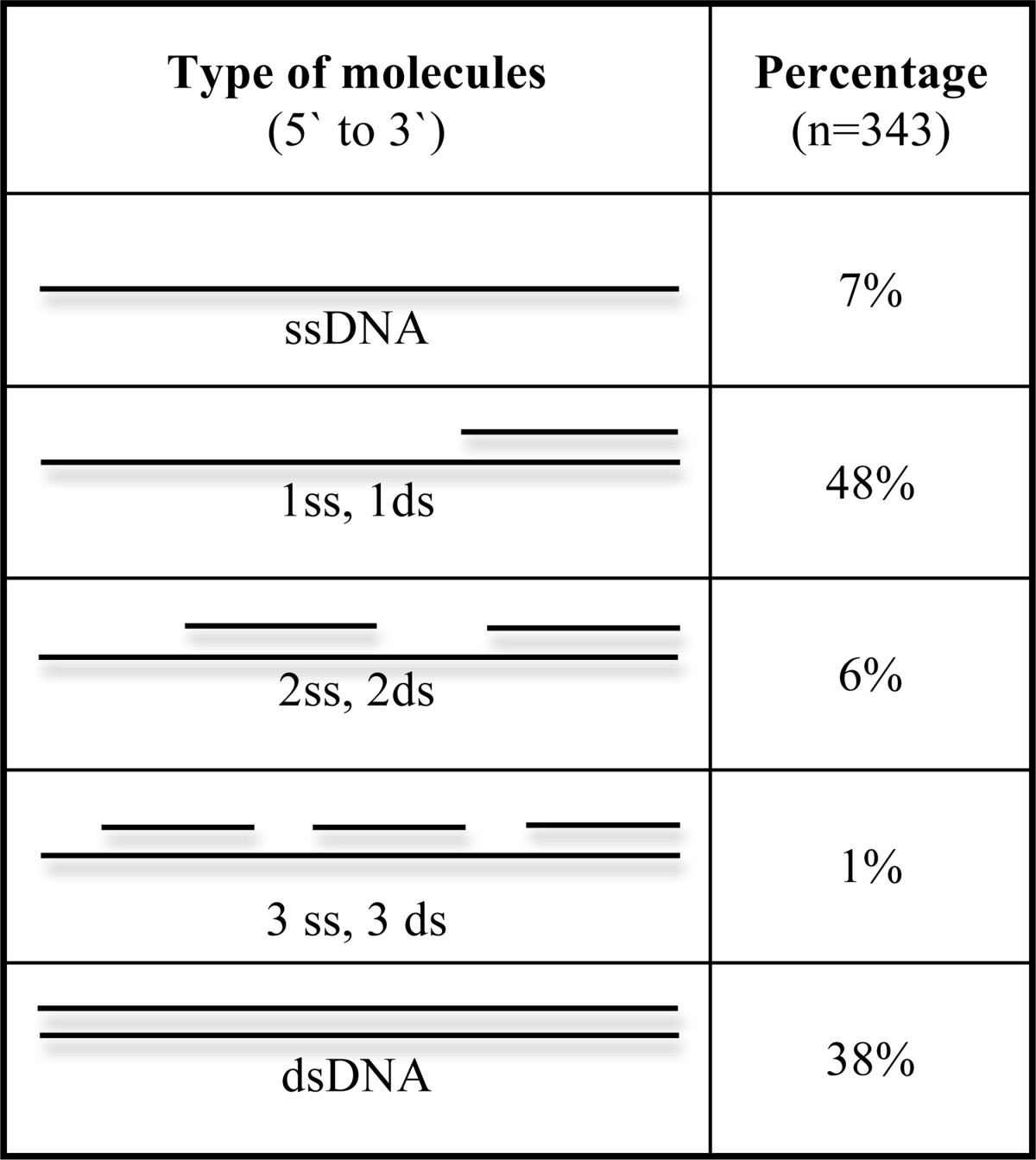
FIGURE 3.

Visualization of minicircle replication products following deproteinization. A, C, and D, molecules with one ss segment at one end. The white arrows show the location of the ssDNA regions. The molecule shown in C is an example of a replicating minicircle with a long duplex tail whose length exceeds 5 kb. B, product molecule with two ss segments. D, fully duplex DNA without any ss segment (molecule on the right).
The existence of ss segments along the rolling circle tails argues for discontinuous synthesis on the displaced strand. Analysis of the patterns of ss and ds segments present allows one to determine the stage of the replication process for each molecule at the point when the reactions were terminated. Thus, we focused on dsDNA products punctated by one or more ss gaps, as depicted in Fig. 3. Three classes of these molecules were present as follows: dsDNA molecules containing 1–3 ss segments. Independent of the pattern of the ss segments, in all products one end was single-stranded. Scoring 186 partial dsDNAs (Table 2, left column), the majority of the molecules (87%, n = 163) fell into the class with only one ss region adjacent to a long ds tail corresponding to the completed lagging strand (Fig. 3, A, C, and D). The average length of the ss region was 3,800 nt (range 400–7,900 nt). Eleven percent of the partial dsDNAs consisted of molecules with two ss segments as follows: one at an end and the second proximate the first, separated by a dsDNA segment (Fig. 3B). The ss segment located at one end was 2,200-nt long on average (730–7,000 nt), and the distal ss segment was 1,500 nt on average (420–2,700 nt). The duplex region between two ss segments was 1,100 bp on average (150–4,800 bp), and corresponds to the lagging strand synthesis that had occurred on the most recent Okazaki fragment, and the distal segment thus must be the remaining lagging strand template to be synthesized. Therefore, the duplex region and the distal ss segment together determine the length of the Okazaki fragment, and it had an average length of 1,500 bp. The shortest Okazaki fragment was 280 bp, and the longest was 5,600 bp. Thus, the EM profile was similar to the gel profile where the size of lagging strand fragments ranged from 600 to 9,000 nt (Fig. 2, lane 6). A small fraction (2%) had three ssDNA segments (two Okazaki fragments) along their length. The average size of the most recently synthesized Okazaki fragments was ∼890 bp (250–2,700 bp), and the downstream Okazaki fragment was ∼780 bp (540–8,600 bp). A very small fraction of DNA molecules contained more than three ss segments.
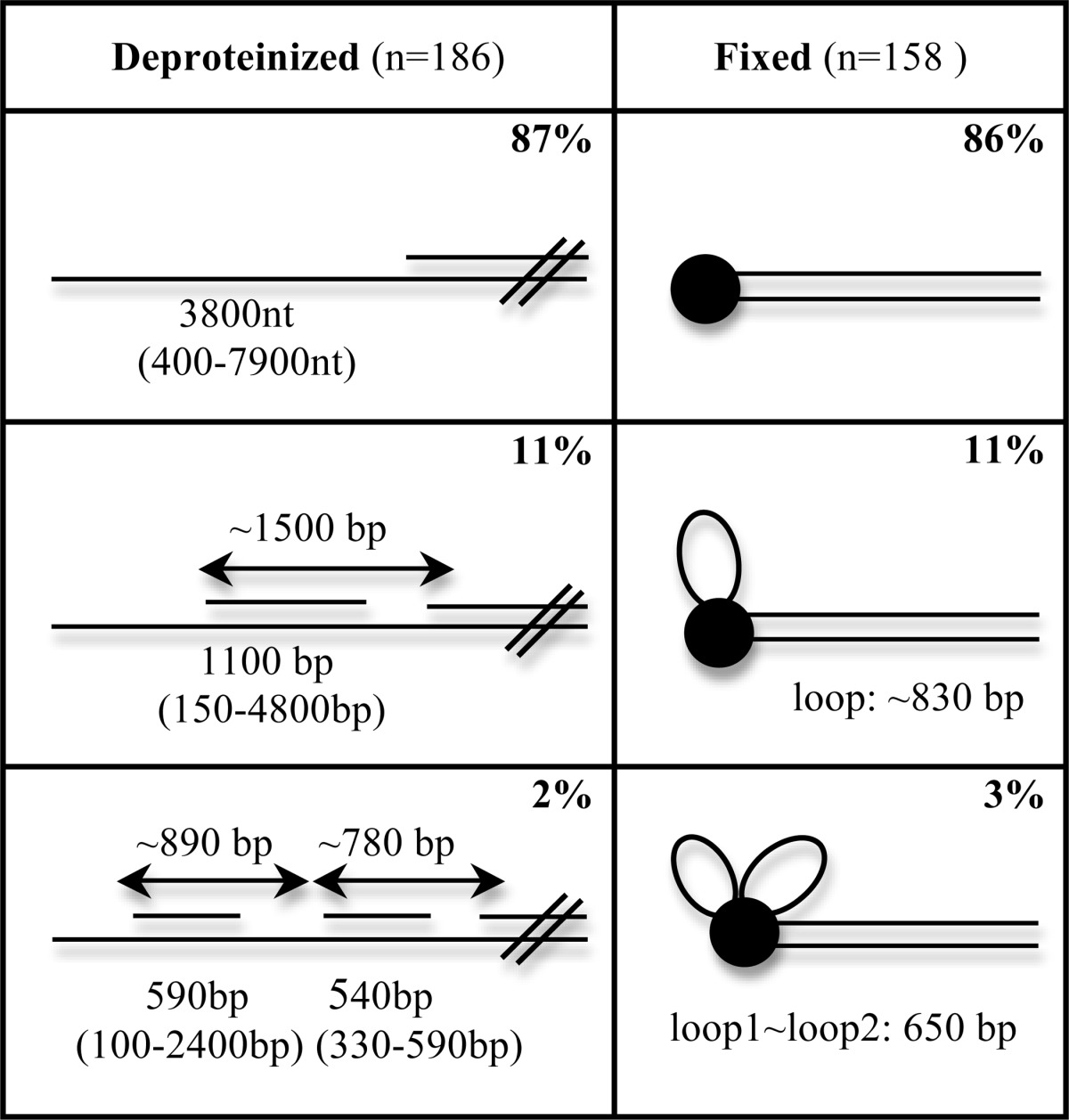
TABLE 2.
Correlation of the pattern of ss segments with the presence of replication loops
Left column shows EM analysis of deproteinized molecules with ss segments shown in Fig. 2. The percentage of molecules with 1–3 ssDNA segments in a population of 186 molecules is shown at the right. All molecules were single-stranded at one end. The average and range of lengths of the ss segments are shown below the DNA. The lengths of Okazaki fragments are reported above the DNA. Right column, EM analysis of molecules with HSV-1 proteins fixed on the DNA with glutaraldehyde. Examples are shown in Fig. 3. Molecules with proteins (158) (black spheres) were scored, and the percentage of molecules with no loop, 1 loop, and 2 loops is shown at the right. The average of dsDNA loops is shown below the DNA.
Visualization of DNA-Protein Replication Complexes
In the coupled replication reactions catalyzed by the purified T4 and T7 proteins, the lagging strand is folded into a trombone loop at the fork (Fig. 1A). These loops can be observed by EM only if the proteins are fixed in place. Otherwise, they will be released upon removal of the proteins from the DNA (Fig. 1B). To examine the architecture of the replication fork with the HSV-1 proteins present, we fixed the DNA-protein complexes with glutaraldehyde. Of 258 molecules scored, 60% (n = 158) of the linear duplex DNAs contained a protein complex at one end (Fig. 4). This replication-associated complex at the end of the ds tail was observed as a very large electron-dense mass. When ferritin (440 kDa) was mounted along side the replication complexes and the projected areas of the ferritin and replication complexes compared in the micrographs, the replication complexes had areas varying greatly and often 5–10 times that of ferritin. This variability and their irregular shape made any mass estimates unreliable. If the replication complex contains one molecule each of UL5-UL8-UL52 (293 kDa), UL30 (136 kDa), UL42 (62 kDa), and ICP8 (128 kDa), this sums to 619 kDa. With an additional UL30-UL42 complex on the lagging strand and a second copy of UL5-UL8-UL52 (13), the mass could be as large as 1,110 kDa. However, we must assume that multiple copies of ICP8 are present and in different amounts from one complex to the next. This would be expected because the length of ss segments at the DNA ends varied from 400 to 7,900 nt (Table 2, left column) and would result in complexes of variable size, with some quite large.
FIGURE 4.
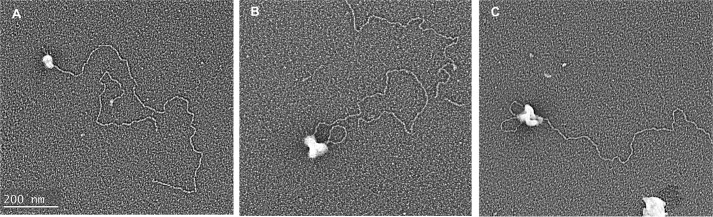
Visualization of minicircle replication products in protein-bound molecules. Replication intermediates without a loop (A), with one loop (B), and with two loops (C). White balls represent protein complexes.
Interestingly, 40% of the replication products did not have any visible protein bound despite fixation with glutaraldehyde, suggesting that the replisome had dissociated from the replication fork. In addition, this percentage (40%) corresponds to the fraction of fully duplex samples without any ss region as seen in the deproteinized molecules (42%) (Fig. 3D).
Presence of Replication Loops
Among the fixed complexes scored bearing a replication complex (n = 158), 86% did not contain a loop (Fig. 4A), 11% contained one ds loop (Fig. 4B), and 3% contained two ds loops (Fig. 4C). For the molecules with one ds loop, the length of the loops varied from 440 to 1,800 bp, with a mean length of 830 bp (Table 2, right column). For the molecules bearing two ds loops, the size of each loop in any particular molecule was very similar (around 650 bp), and calculation of all the molecules showed that the sizes of the loops ranged from 330 to 1,500 bp. Comparison of the fixed (Table 2, right column) and deproteinized (left column) molecules revealed a correlation between the fraction of molecules with one single ds loop (11%) and molecules having one Okazaki fragment (11%). There was also a good correlation between the frequency of molecules containing two ds loops (3%) and molecules with two Okazaki fragments (2%). Consistent with the correlation between the number of ds loops and the number of Okazaki fragments, the frequency of species without loops (86%) corresponds to the fraction of the molecules with only one ss segment at one end (87%). All of the replication loops were associated with protein complexes bound to the end of the molecules, and none were observed internally along the dsDNA.
DISCUSSION
In this study, we carried out replication in vitro using the HSV-1 six core proteins and a 70-base minicircle template. Applying our improved conditions, the reactions were followed using alkaline gel electrophoresis and EM. Although previous efforts using these proteins and circular templates have been reported, our results provide the first evidence of highly robust replication coupled with a structural analysis of the replication intermediates. We show that coupled leading-lagging synthesis occurs, generating linear dsDNAs up to 20 kb in length resulting from ∼300 rounds of replication from the minicircle. A pattern of ds and ss segments near one end of the DNA was typical of the synthesis of Okazaki fragments whose sizes were similar to those generated in reactions catalyzed by the T7 and T4 proteins. Present at one end of the DNA products were large replisomes frequently featuring a DNA loop whose size reflected the growing Okazaki fragment. This is the first direct evidence that the trombone mechanism can operate in a eukaryotic viral replication system.
The minicircle DNA used here provides many advantages. Compared with large plasmids, it allows the use of a high concentration of forks in the reactions, and the sequence bias on the strands makes it possible to differentiate synthesis on the two strands independently. However, its small size could potentially impose steric constraints on loading the replication components and places it below the limit of our EM methods to visualize it as a circle, and thus we were only able to observe the long linear tails generated by its replication. Thus, we had to assume that the minicircle was located at the end of the long linear DNA that contained the pattern of ss and ds segments (as seen on the deproteinized molecules) or the replisome and trombone loops (in the fixed DNA-protein complexes).
Using the minicircle as a template and the HSV-1 factors, leading and lagging strand DNA synthesis was previously reported (12, 14). However, our results differ in significant ways. First, we were able to achieve a higher level of replication with leading strand products exceeding 20,000 nt and Okazaki fragments of up to 9,000 nt. Second, although the other groups used 10 mm Mg2+, the optimal concentration in our experiments was 2 mm free Mg2+. The most important difference, however, was that the two previous studies reported that ICP8 was not required in their reactions, although in our experiments ICP8 was crucial for the synthesis on both strands. ICP8 presumably stimulates synthesis on the leading strand by preventing reannealing of ssDNA unwound by UL5/UL8/UL52. In addition, the direct interaction between ICP8, UL8, and UL52 has been reported recently (19). The collaboration of ICP8 with the subunits of the helicase/primase might be required for the coupled synthesis. Lagging strand synthesis was also ICP8-dependent with high ICP8 concentrations being inhibitory. In the presence of excess ICP8, UL30 may have difficulty displacing ICP8 from the nascent ssDNA thus limiting synthesis.
The results described here for HSV-1 are very similar to what we and others have described for reactions using the T7 and T4 factors. The distribution of replicating HSV-1 molecules with 1–3 ss segments was similar to the pattern observed in the T4 (3) and T7 studies (4). The fraction of rolling circle tails with one ss segment was 43% for T4, 79% for T7, and 87% for HSV-1, and the percentage of duplex DNA molecules containing two ss segments was 43% for T4, 21% for T7, and 11% for HSV-1. There were also rolling circle tails with three ss segments observed for T4 (8%) and HSV-1 (2%) but none reported for T7. The size of the Okazaki fragments was ∼2,000 nt (1,000–6,000 nt) in the T4 (20) and T7 (4, 5, 21, 22) studies and was ∼1,500 nt (600–9,000 nt) in the work here. Similar to the prokaryotic systems, 13% of the replicating molecules contained DNA loops associated with the replication complex. The fraction of replicating molecules containing one ds loop was 43% in T4 (3), 50% in T7 (4), and 11% for HSV-1. Eight percent of molecules contained two loops in T4 work and 3% in HSV-1, although none were observed in the T7 studies.
The results described here provide an important milestone in the efforts of many laboratories over nearly 2 decades to demonstrate robust coupled leading and lagging strand replication in vitro using the highly purified core HSV-1 replication factors. Building on the work of others, we have achieved active replication and present an analysis of the fork architecture showing that the lagging strand is indeed looped back during Okazaki fragment synthesis. These results will provide a platform for future studies employing EM, single molecule methods, and other approaches to further investigate the kinetics of fork movement. Furthermore, employing plasmids containing the lytic origin of replication, oriS, and the UL9 origin-binding protein, it should be possible to approach the goal of reconstituting origin-dependent replication using purified HSV-1 factors.
Acknowledgment
We thank Dr. Sandra Weller (University of Connecticut) for discussion and advice.
This work was supported, in whole or in part, by National Institutes of Health Grants GM31819 and CA19014.
This article was selected as a Paper of the Week.
- nt
- nucleotide(s)
- SSB
- single-stranded DNA-binding protein
- ss
- single strand.
REFERENCES
- 1. Alberts B. M., Barry J., Bedinger P., Formosa T., Jongeneel C. V., Kreuzer K. N. (1983) Studies on DNA replication in the bacteriophage T4 in vitro system. Cold Spring Harb. Symp. Quant. Biol. 47, 655–668 [DOI] [PubMed] [Google Scholar]
- 2. Okazaki T., Okazaki R. (1969) Mechanism of DNA chain growth. IV. Direction of synthesis of T4 short DNA chains as revealed by exonucleolytic degradation. Proc. Natl. Acad. Sci. U.S.A. 64, 1242–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chastain P. D., 2nd, Makhov A. M., Nossal N. G., Griffith J. (2003) Architecture of the replication complex and DNA loops at the fork generated by the bacteriophage T4 proteins. J. Biol. Chem. 278, 21276–21285 [DOI] [PubMed] [Google Scholar]
- 4. Lee J., Chastain P. D., 2nd, Kusakabe T., Griffith J. D., Richardson C. C. (1998) Coordinated leading and lagging strand DNA synthesis on a minicircular template. Mol. Cell 1, 1001–1010 [DOI] [PubMed] [Google Scholar]
- 5. Park K., Debyser Z., Tabor S., Richardson C. C., Griffith J. D. (1998) Formation of a DNA loop at the replication fork generated by bacteriophage T7 replication proteins. J. Biol. Chem. 273, 5260–5270 [DOI] [PubMed] [Google Scholar]
- 6. Hamdan S. M., Loparo J. J., Takahashi M., Richardson C. C., van Oijen A. M. (2009) Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 457, 336–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nossal N. G., Makhov A. M., Chastain P. D., 2nd, Jones C. E., Griffith J. D. (2007) Architecture of the bacteriophage T4 replication complex revealed with nanoscale biopointers. J. Biol. Chem. 282, 1098–1108 [DOI] [PubMed] [Google Scholar]
- 8. Boehmer P. E., Lehman I. R. (1997) Herpes simplex virus DNA replication. Annu. Rev. Biochem. 66, 347–384 [DOI] [PubMed] [Google Scholar]
- 9. Weller S. K., Coen D. M. (2012) Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 4, a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skaliter R., Lehman I. R. (1994) Rolling circle DNA replication in vitro by a complex of herpes simplex virus type 1-encoded enzymes. Proc. Natl. Acad. Sci. U.S.A. 91, 10665–10669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Skaliter R., Makhov A. M., Griffith J. D., Lehman I. R. (1996) Rolling circle DNA replication by extracts of herpes simplex virus type 1-infected human cells. J. Virol. 70, 1132–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falkenberg M., Lehman I. R., Elias P. (2000) Leading and lagging strand DNA synthesis in vitro by a reconstituted herpes simplex virus type 1 replisome. Proc. Natl. Acad. Sci. U.S.A. 97, 3896–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Y., Bai P., Mackay S., Korza G., Carson J. H., Kuchta R. D., Weller S. K. (2011) Herpes simplex virus type 1 helicase-primase: DNA binding and consequent protein oligomerization and primase activation. J. Virol. 85, 968–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stengel G., Kuchta R. D. (2011) Coordinated leading and lagging strand DNA synthesis by using the herpes simplex virus 1 replication complex and minicircle DNA templates. J. Virol. 85, 957–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramirez-Aguilar K. A., Low-Nam N. A., Kuchta R. D. (2002) Key role of template sequence for primer synthesis by the herpes simplex virus 1 helicase-primase. Biochemistry 41, 14569–14579 [DOI] [PubMed] [Google Scholar]
- 16. Hernandez T. R., Lehman I. R. (1990) Functional interaction between the herpes simplex-1 DNA polymerase and UL42 protein. J. Biol. Chem. 265, 11227–11232 [PubMed] [Google Scholar]
- 17. Boehmer P. E., Lehman I. R. (1993) Herpes simplex virus type 1 ICP8: helix-destabilizing properties. J. Virol. 67, 711–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Griffith J. D., Christiansen G. (1978) Electron microscope visualization of chromatin and other DNA-protein complexes. Annu. Rev. Biophys. Bioeng 7, 19–35 [DOI] [PubMed] [Google Scholar]
- 19. Muylaert I., Zhao Z., Elias P. (2014) UL52 primase interactions in the herpes simplex virus 1 helicase-primase are affected by antiviral compounds and mutations causing drug resistance. J. Biol. Chem. 289, 32583–32592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chastain P. D., 2nd, Makhov A. M., Nossal N. G., Griffith J. D. (2000) Analysis of the Okazaki fragment distributions along single long DNAs replicated by the bacteriophage T4 proteins. Mol. Cell 6, 803–814 [DOI] [PubMed] [Google Scholar]
- 21. Debyser Z., Tabor S., Richardson C. C. (1994) Coordination of leading and lagging strand DNA synthesis at the replication fork of bacteriophage T7. Cell 77, 157–166 [DOI] [PubMed] [Google Scholar]
- 22. Lee J., Chastain P. D., 2nd, Griffith J. D., Richardson C. C. (2002) Lagging strand synthesis in coordinated DNA synthesis by bacteriophage T7 replication proteins. J. Mol. Biol. 316, 19–34 [DOI] [PubMed] [Google Scholar]