

Background: Mutations in GNPTAB cause the lysosomal disorders mucolipidosis II and III αβ.
Results: All reported missense mutations were studied and showed various consequences on its gene product, αβ GlcNAc-1-phosphotransferase.
Conclusion: Domains responsible for catalytic activity and lysosomal hydrolase recognition were identified.
Significance: Analysis of patient mutations provided new insight into the functional domains of αβ GlcNAc-1-phosphotransferase.
Keywords: Enzyme, Genetic Disease, Golgi, Lysosomal Storage Disease, Zebrafish, GlcNAc-1-phosphotransferase, Notch Domains, Mannose 6-phosphate, Mucolipidosis II, Mucolipidosis III αβ
Abstract
UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase tags newly synthesized lysosomal enzymes with mannose 6-phosphate recognition markers, which are required for their targeting to the endolysosomal system. GNPTAB encodes the α and β subunits of GlcNAc-1-phosphotransferase, and mutations in this gene cause the lysosomal storage disorders mucolipidosis II and III αβ. Prior investigation of missense mutations in GNPTAB uncovered amino acids in the N-terminal region and within the DMAP domain involved in Golgi retention of GlcNAc-1-phosphotransferase and its ability to specifically recognize lysosomal hydrolases, respectively. Here, we undertook a comprehensive analysis of the remaining missense mutations in GNPTAB reported in mucolipidosis II and III αβ patients using cell- and zebrafish-based approaches. We show that the Stealth domain harbors the catalytic site, as some mutations in these regions greatly impaired the activity of the enzyme without affecting its Golgi localization and proteolytic processing. We also demonstrate a role for the Notch repeat 1 in lysosomal hydrolase recognition, as missense mutations in conserved cysteine residues in this domain do not affect the catalytic activity but impair mannose phosphorylation of certain lysosomal hydrolases. Rescue experiments using mRNA bearing Notch repeat 1 mutations in GNPTAB-deficient zebrafish revealed selective effects on hydrolase recognition that differ from the DMAP mutation. Finally, the mutant R587P, located in the spacer between Notch 2 and DMAP, was partially rescued by overexpression of the γ subunit, suggesting a role for this region in γ subunit binding. These studies provide new insight into the functions of the different domains of the α and β subunits.
Introduction
UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase (GlcNAc-1-phosphotransferase)3 catalyzes the initial step in the formation of the mannose 6-phosphate targeting signal on newly synthesized lysosomal acid hydrolases. These mannose 6-phosphate residues function as high affinity ligands for binding to mannose 6-phosphate receptors in the trans-Golgi network and subsequent delivery to lysosomes (1). GlcNAc-1-phosphotransferase is an α2β2γ2 heterohexamer that is encoded by two genes (2). The GNPTAB gene encodes the α and β subunits, and the GNPTG gene encodes the γ subunit (3–5). The α and β subunits are synthesized as a catalytically inactive type 3 transmembrane precursor of 1256 amino acids, which undergoes a proteolytic cleavage between Lys-928 and Asp-929 in the Golgi to generate the α and β subunits (6). These subunits are responsible for the specific recognition of lysosomal acid hydrolases and the catalytic function of the transferase (6, 7). The γ subunit is a soluble glycoprotein of 305 amino acids that enhances the phosphorylation of a subset of the lysosomal acid hydrolase substrates (7).
The αβ subunits contain three identifiable domains separated by so-called spacer regions; that is, the Stealth domain, which consists of four regions spread throughout the α and β subunits, the Notch repeats, and a DMAP interaction domain. The Stealth domain is similar to bacterial genes involved in the synthesis of cell wall polysaccharides and, in several instances, has been shown to function as sugar-phosphate transferases (8–11). This domain has been assumed to mediate the catalytic activity of GlcNAc-1-phosphotransferase, although this has not been formally demonstrated. The Notch modules found in Notch receptor family members are thought to regulate the ligand-induced proteolytic cleavage of the Notch receptor by an undetermined mechanism (12). The DMAP interaction domain was initially described as a component of DNA methyltransferase (DNMT1) where it was proposed to function as a protein-protein interaction domain in a repressive transcription complex (13).
Mutations in the GNPTAB gene give rise to the severe lysosomal storage disorder mucolipidosis II (I-cell disease, MIM#252500) and the attenuated mucolipidosis III αβ (pseudo-Hurler polydystrophy, MIM#252600). Mutations in the GNPTG gene cause the least severe phenotype known as mucolipidosis III γ (14).
Among the mutations reported in patients with these disorders are 31 GNPTAB missense mutations. Analysis of the effects of these mutations on the maturation and function of GlcNAc-1-phosphotransferase not only provides insight into the pathogenic effects of these amino acid substitutions but enhances the understanding of the function of the conserved domains of this complex enzyme. We previously reported that the K732N patient mutation in the DMAP interaction domain resulted in impaired binding and decreased phosphorylation of lysosomal acid hydrolases without affecting the catalytic activity toward the simple sugar α-methyl d-mannoside (α-MM) (15). This implicated the DMAP domain as a protein substrate recognition module. We also found that the K4Q and S15Y patient mutations within the N-terminal cytoplasmic tail of the α subunit decreased retention of the catalytically active enzyme in the Golgi complex, indicating a role for these residues in the trafficking of the enzyme (16).
In this study we analyzed the consequences of the remaining missense mutations on GlcNAc-1-phosphotransferase function using a combination of cell- and zebrafish-based studies. Several missense mutations were identified that impact either exit from the ER, proteolytic cleavage in the Golgi, or enzymatic activity. Domains involved in catalytic activity (Stealth) and lysosomal hydrolase recognition (Notch repeat 1) were characterized. Together, these findings highlight the ability of individual disease-causing missense mutations to elicit a broad range of consequences on GlcNAc-1-phosphotransferase function.
EXPERIMENTAL PROCEDURES
DNA Constructs
Human GNPTAB-V5/His in pcDNA6 (a kind gift from Dr. W. Canfield, Genzyme; see also Ref. 15) was modified by QuikChange site-directed mutagenesis (Stratagene) to generate the various missense mutants; see Table 1 for all primer sequences used. The full cDNA sequences were confirmed by DNA sequencing.
TABLE 1.
Primer sequences used in QuikChange mutagenesis
| Amino acid change | Forward primer (5′ → 3′) | Reverse primer (5′ → 3′) |
|---|---|---|
| W81L | GACGTTGTTTACACCTTGGTGAATGGCACAGATCTTG | CAAGATCTGTGCCATTCACCAAGGTGTAAACAACGTC |
| V182D | CCTTCTACCAATGTCTCAGATGTTGTTTTTGACAGTAC | GTACTGTCAAAAACAACATCTGAGACATTGGTAGAAGG |
| D190V | GACAGTACTAAGGTTGTTGAAGATGCCCACTC | GAGTGGGCATCTTCAACAACCTTAGTACTGTC |
| R334Q | CTGAGGTACTCATTGCAATCTATCGAGAGGCATGC | GCATGCCTCTCGATAGATTGCAATGAGTACCTCAG |
| R334L | CTGAGGTACTCATTGCTATCTATCGAGAGGCATGC | GCATGCCTCTCGATAGATAGCAATGAGTACCTCAG |
| I348L | GGGTTCGGAATATTTTCCTTGTCACCAACGGGC | GCCCGTTGGTGACAAGGAAAATATTCCGAACCC |
| F374L | CACACCAGGATGTTCTTCGAAATTTGAGCCACTTG | CAAGTGGCTCAAATTTCGAAGAACATCCTGGTGTG |
| I403T | GGGCTGTCCCAGAAGTTTACTTACCTAAATGATGATG | CATCATCATTTAGGTAAGTAAACTTCTGGGACAGCCC |
| D407A | GTTTATTTACCTAAATGCTGATGTCATGTTTGGGAAGG | CCTTCCCAAACATGACATCAGCATTTAGGTAAATAAAC |
| C442Y | CTGTGCCGAGGGCTACCCAGGTTCCTG | CAGGAACCTGGGTAGCCCTCGGCACAG |
| C461G | GTAATAATTCAGCCGGCGATTGGGATGGTG | CACCATCCCAATCGCCGGCTGAATTATTAC |
| C468S | GGGATGGTGGGGATAGCTCTGGAAACAGTG | CACTGTTTCCAGAGCTATCCCCACCATCCC |
| C505Y | CAGTGTCTCTTACTATAATCAGGGATGTGCG | CGCACATCCCTGATTATAGTAAGAGACACTG |
| C523R | GTTCTGTGACCAAGCACGCAATGTCTTGTCCTGTGG | CCACAGGACAAGACATTGCGTGCTTGGTCACAGAAC |
| R587P | GTGACAATCCAATAATTCCACATGCTTCTATTGCCAAC | GTTGGCAATAGAAGCATGTGGAATTATTGGATTGTCAC |
| A592T | CCAATAATTCGACATGCTTCTATTACCAACAAGTGGAAAACCATCC | GGATGGTTTTCCACTTGTTGGTAATAGAAGCATGTCGAATTATTGG |
| K732N | GGATACAATTTGTCCAATTCAGCCTTGCTGAG | CTCAGCAAGGCTGAATTGGACAAATTGTATCC |
| L785W | GCTTAGGAGTGTCTGAAAGATGGCAGAGGTTGACTTTTCC | GGAAAAGTCAACCTCTGCCATCTTTCAGACACTCCTAAGC |
| Q926P | GCAAAAATACTGGGAGGCCACTAAAAGATACATTTGCAG | CTGCAAATGTATCTTTTAGTGGCCTCCCAGTATTTTTGC |
| A955V | GCGGAAAGTCCCTGTTCACATGCCTCACATG | CATGTGAGGCATGTGAACAGGGACTTTCCGC |
| H956Y | GGAAAGTCCCTGCTTACATGCCTCACATGATTG | CAATCATGTGAGGCATGTAAGCAGGGACTTTCC |
| R986C | GTCATTTCACAAAGTGTGCCATTCTGAGGATATGCAG | CTGCATATCCTCAGAATGGCACACTTTGTGAAATGAC |
| L1001P | CTCTTATTTTTATTATCCCATGAGTGCAGTGCAGCC | GGCTGCACTGCACTCATGGGATAATAAAAATAAGAG |
| D1018G | GTCTTTGATGAAGTTGGTACAGATCAATCTGGTGTC | GACACCAGATTGATCTGTACCAACTTCATCAAAGAC |
| L1054V | GACAGGTCTGGAACACATGGTAATAAATTGCTCAAAAATG | CATTTTTGAGCAATTTATTACCATGTGTTCCAGACCTGTC |
| N1153S | GTTTGCCTGAATGACAGCATTGACCACAATCATAAAG | CTTTATGATTGTGGTCAATGCTGTCATTCAGGCAAAC |
| K1236M | CAGTTAATTGCACTTATGCGGAAGATATTTCCCAG | CTGGGAAATATCTTCCGCATAAGTGCAATTAACTG |
Zebrafish GNPTAB in pCSDest (17) was modified by QuikChange site-directed mutagenesis to generate C447Y and C473S αβ phosphotransferase using the primers 5′-CAA ATT GTG CTG AGG GCT ATC CAG GAT CCT GGA TCA AAG-3′ and 5′-CTT TGA TCC AGG ATC CTG GAT AGC CCT CAG CAC AAT TTG-3′ (C447Y) and primers 5′-GGG ATG GAG GAG ACT CTC AAG GCA GCA GTC G-3′ and 5′-CGA CTG CTG CCT TGA GAG TCT CCT CCA TCC C-3′ (C473S).
Human GNPTG-Myc/FLAG in pCMV6-Entry was obtained from OriGene. GNPTG was amplified using the primers 5′-CCG CCA AGC TTA TGG CGG CGG GGC TG-3′ and 5′-CGC GTC TCG AGT TAC TTA TCG TCG TCA TCC TTG TAA TCG GTA CCC AAA CTC CCA CGC AG-3′ and cloned into pcDNA3.1(+) using the restriction sites HindIII and XhoI.
Phosphotransferase Activity Assays
The activity of each mutant toward α-MM was measured in at least three independent experiments as described in van Meel et al. (16). All mutants were expressed in HEK293 cells by transfection with GeneCellin (BioCellChallenge) or Lipofectamine 3000 (Invitrogen) according to the manufacturers' protocols and assayed ∼48 h after transfection. In brief, the cells were lysed in 1% Triton X-100, PBS containing protease inhibitors (Complete; Roche Diagnostics), and 100 μg of total protein was incubated in 50 mm Tris, pH 7.4, 10 mm MgCl2, 10 mm MnCl2, 2 mg/ml BSA, 2 mm ATP, 75 μm UDP-GlcNAc, and 1 μCi UDP-[3H]GlcNAc with 100 mm α-MM for 1 h at 37 °C. At the end of the incubation period 1 ml of 2 mm Tris, pH 8, was added to the reaction, and the samples were immediately subjected to QAE-Sephadex chromatography.
The activity toward α-iduronidase was determined in the same reaction mixture, which contained 20 μm α-iduronidase (15). The reactions were performed for 4 h at 37 °C and stopped by the addition of 300 μl of 1.5% (w/v) phosphotungstic acid, 0.75 n HCl, and 100 μl of 1 mg/ml BSA. The samples were vortexed and centrifuged at 25,000 × g for 10 min at 4 °C after which the pellets were washed 3 times with 1 ml of the acid mixture and resuspended in 1 ml of 50 mm Tris, pH 11. Incorporated [3H]GlcNAc-P was determined in all cases by scintillation counting.
Western Blotting
SDS-PAGE and immunoblotting with mouse anti-V5 antibody (Invitrogen) or mouse anti-GAPDH (Sigma) was performed as described in van Meel et al. (16) with 10–20 μg of cell lysate. When indicated, 10 μg of cell lysate was treated with 3000 units of endoglycosidase Hf or 750 units of peptide N-glycosidase F for 3 h at 37 °C before SDS-PAGE and immunoblotting as described in van Meel et al. (16).
Immunofluorescence Microscopy
Confocal immunofluorescence microscopy was performed as described in Qian et al. (15). Our preliminary experiments showed that the subcellular localization of WT or mutant phosphotransferase varies to some degree between cells and depends on the level of overexpression and the time point after transfection (early time points show relatively more ER than Golgi staining). Therefore, all mutants were analyzed 2 days after transfection, and sufficient numbers of low to moderate overexpressors (at least 200 cells/mutant) were screened for phosphotransferase localization. All mutants were evaluated in at least two independent experiments.
Rescue Experiments with the γ Subunit
HeLa or HEK293 cells in 12-well plates were transfected with 1 μg of WT or mutant αβ cDNA with or without 90 ng of WT γ cDNA using Lipofectamine 3000 (Invitrogen). Staining for immunofluorescence microscopy, α-MM activity assays, or SDS-PAGE and Western blotting were performed ∼48 h after transfection as described.
Zebrafish Strains and Embryo Maintenance
WT zebrafish were maintained using standard protocols. Embryos were staged according to the criteria established by Kimmel et al. (18). In some cases, 0.003% 1-phenyl-2-thiourea was added to the growth medium to block pigmentation. Handling and euthanasia of fish for all experiments were carried out in compliance with University of Georgia's policies. This protocol was approved by the University of Georgia Institutional Animal Care and Use Committee (permit number A2012 07-037-Y2-A0).
Antisense Morpholino Injection and mRNA Rescue
Expression of GlcNAc-1-phosphotransferase (αβ subunit, gnptab) was inhibited by injection of morpholino oligonucleotides (17). Experiments involving mRNA rescue in the morphant background were performed on embryos sequentially injected with 8 ng of the gnptab splice blocking morpholino oligonucleotides, which was previously validated to yield maximal specific inhibition, and 300 pg of the appropriate mRNA. Based on titration experiments, 300 pg of WT or mutant gnptab RNA yielded the highest degree of phenotypic rescue within MLII embryos. Rescue was assessed by Alcian blue-stained cartilage, normalized cathepsin K activity, and direct GlcNAc-1-phosphotransferase activity measurements. The Message Machine kit (Ambion) was used to synthesize all full-length mRNAs from PCSII+ constructs containing either zebrafish WT gnptab cDNA or the Notch domain mutations corresponding to the two patient missense mutations studied. The PCSII+WT zebrafish gnptab construct was generated as previously described (17) and utilized to generate the C447Y and C473S mutant sequences (see DNA constructs for cloning details).
Alcian Blue Staining and Quantification of Craniofacial Phenotypes
Embryos were stained with Alcian blue as described previously (17). Analysis of craniofacial structures was performed using the morphometric parameters outlined under “Results.” Stained embryos were photographed on an Olympus SZ-16 dissecting scope outfitted with Q-capture software and a Retiga 2000R color camera. The length of specific cartilage structures was measured using Photoshop Extended measurement tools.
Enzyme Assays and Cation-independent Mannose 6-Phosphate Receptor Affinity Chromatography
The activity assays for GlcNAc-1-phosphotransferase and cathepsin K as well as the determination of the percentage of mannose phosphorylated β-galactosidase activity after affinity chromatography were performed as previously described (15).
RESULTS
Localization of the Missense Mutations
The location of the 31 missense mutations in the αβ subunits is denoted in Fig. 1. The red dots indicate homozygous mutations in MLII patients, the blue dots indicate homozygous mutations in the MLIII αβ patients, and the black dots indicate sites of missense mutations in patients where the second allele has a different missense or other type of mutation. Although the mutations are widely distributed throughout the α and β subunits, they are clearly concentrated within the Stealth domain, with 48% of the mutations (15/31) located in this region despite the fact that it only constitutes 18% of the protein mass. A second concentration occurred in the Notch repeats, which have 16% of the mutations (5/31) but only 5% of the protein mass. On the other hand, the spacer regions, which constitute 57% of the protein mass, only contained 19% of the mutations (6/31). Of note, all the Notch domain mutations occurred in the MLIII αβ patients, whereas the Stealth mutations were present in the MLII and MLIII αβ patients. The remaining mutations are located in the DMAP domain and the cytoplasmic tails (2 and 3 mutations, respectively).
FIGURE 1.
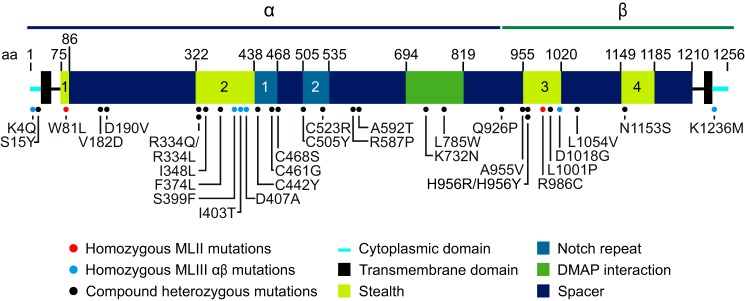
Schematic representation of the domain organization of αβ phosphotransferase and the location of the missense mutations. It should be noted that the precise boundaries of the various domains might vary, as the boundaries shown in this figure were mostly based on sequence alignments. Two of the missense mutations occur at the same residues (334 and 956) but cause different amino acid (aa) changes. H956R was not analyzed in this study, as it likely has the same consequence as H956Y. The patient with C523R was homozygous for this mutation, but the clinical phenotype was not specified (23).
Consequences of the Missense Mutations
To examine the consequences of the missense mutations, plasmids encoding WT and the various mutant GNPTAB cDNAs with C-terminal V5/His tags were generated and transfected into HEK293 cells. Cell lysates were prepared after ∼48 h, and aliquots were subjected to immunoblotting with an anti-V5 antibody to determine αβ precursor expression and monitor the appearance of the mature V5-tagged β subunit. The latter reflects transport of the αβ precursor to the Golgi apparatus where it is cleaved by the site-1 protease to generate the β subunit (19), a process that is required for activation of the enzyme. Additional aliquots of the same extracts were assayed for catalytic activity using α-MM and, in some instances, the acid hydrolase α-iduronidase as acceptors.
Consequences of Stealth Domain Mutations
Analysis by Western blotting showed that similar levels of the αβ precursor were present in all instances, but in seven of the Stealth domain mutants little or no β subunit could be detected. Western blots are shown in Fig. 2A for a subset of the mutants that represent examples of the various defects. The absence or reduction in the β subunit levels indicated that these mutations either prevent the αβ precursor from exiting the ER or, after transport to the Golgi, block the cleavage by the site-1 protease.
FIGURE 2.
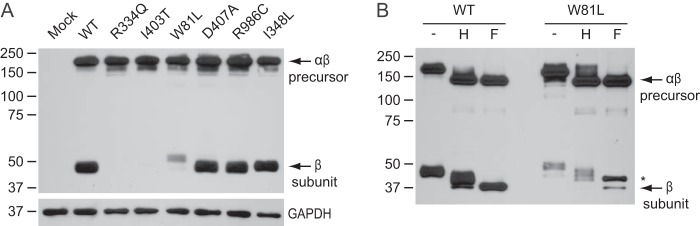
Western blot analysis of a subset of the Stealth domain missense mutants. A shows representative Western blots (anti-V5) of HEK293 cell lysates ∼48 h after transfection with WT or mutant phosphotransferase αβ subunits with a C-terminal V5 tag. The anti-GAPDH control shows that the expression levels of the mutants were similar. B, endoglycosidase Hf (H) and peptide N-glycosidase F (F) treatment of HEK293 cell lysates expressing WT or W81L αβ phosphotransferase. Endoglycosidase Hf cleaved high mannose-type N-linked glycans, whereas peptide N-glycosidase F cleaved both high mannose and complex-type units. The asterisk indicates the slower migrating β subunit of W81L, which contains both high mannose and complex-type N-linked glycans. Note that there is a small amount of β subunit that migrates similar to WT.
To distinguish between these possibilities the subcellular localization of these mutants was examined in HeLa cells by confocal immunofluorescence microscopy along with a WT control. The seven mutants with no or reduced amounts of the β subunit all exhibited increased ER staining (summarized in Table 2 under exit ER). Two of the mutants, R334Q and R334L, showed exclusively ER localization with no detectable Golgi staining (Fig. 3 and Table 2 exit ER −). Western blot analysis showed no detectable levels of the β subunit (Fig. 2A). These two mutations of residue 334 represent the most severe example of mutants that fail to exit the ER. The S399F and I403T mutants also showed strong ER localization. However, in a small population of the cells trace amounts were detected in the Golgi complex (Fig. 3, I403T arrow), and in some experiments the β subunit levels were just above the detection limit in the Western blots, whereas other times it was undetectable (Fig. 2A, I403T). These data indicate that the mutations strongly inhibit ER exit but do not cause a complete block (Table 2, exit ER +/−). A third group of mutants displayed both ER and Golgi localization by immunofluorescence microscopy (Table 2, exit ER +, Fig. 3, W81L). Surprisingly, the β subunit of W81L phosphotransferase showed a slightly slower migration by Western blotting than WT, in addition to a small amount of normally migrating β subunit (Fig. 2A). Treatment with endoglycosidase Hf and peptide N-glycosidase F to analyze the types of N-linked glycans showed a similar effect on the mutant and WT β subunits (Fig. 2B). These data suggest that the differently migrating β subunit has reached the Golgi complex. One of the mutants, L1001P, mostly localized in the Golgi but showed elevated ER staining in a significant population of the cells and thus represents the least impaired of the analyzed mutants (Table 2, exit ER ++; Fig. 3).
TABLE 2.
Consequences of GNPTAB missense mutations
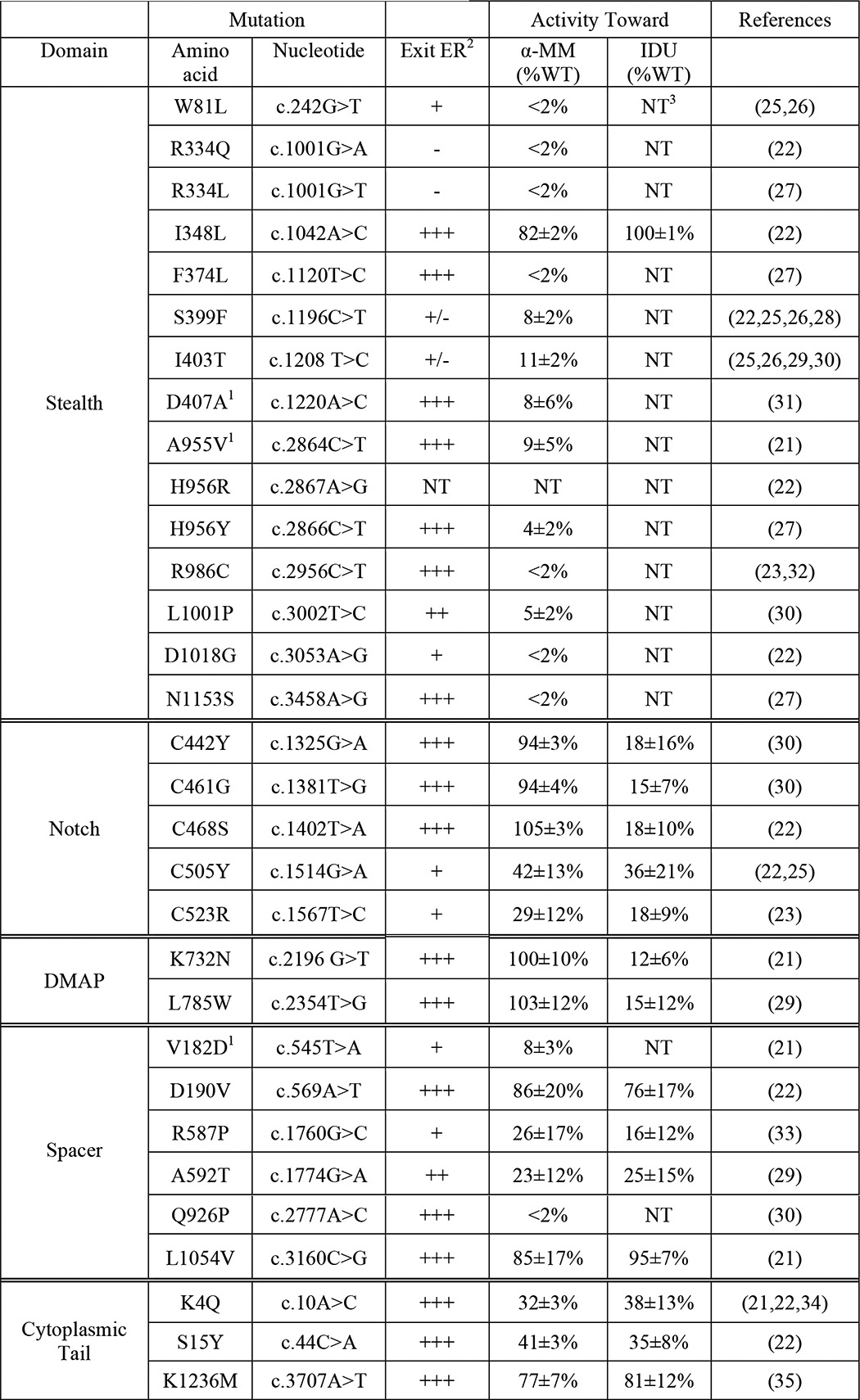
1 These mutations were reported with an additional, novel mutation on the same allele (D407A and A663G; A955V and K928R; V182D and Q205P). However, our analysis only showed defects with the listed mutations. The additional changes are likely single nucleotide polymorphisms.
2 Exit ER was evaluated by confocal immunofluorescence microscopy (co-localization with Golgi marker GM130) and Western blot analysis (level of processed β subunit relative to WT β subunit). In almost all cases the results were in agreement. A592T αβ phosphotransferase localized predominantly in the Golgi complex. However, the β subunit level was lower than WT in all experiments. Therefore, this mutant is likely somewhat retained in the ER.
3 NT, not tested.
FIGURE 3.

Subcellular localization of αβ phosphotransferase Stealth domain mutants by confocal immunofluorescence microscopy in HeLa cells. αβ phosphotransferase was detected with an antibody to the α subunit (green) and GM130 (red) marks the cis-Golgi complex. The arrow in the I403T panels indicates Golgi localization. Scale bars, 10 μm.
The enzymatic activity of WT and GNPTAB mutants toward α-MM was measured next (Table 2). As expected, the mutants R334Q and R334L that failed to exit the ER had no detectable activity toward α-MM (<2% of WT). The mutants that showed partial exit from the ER exhibited either low levels of activity (S399F, I403T, L1001P) or undetectable activity (W81L, D1018G). As the latter three mutants had both ER and Golgi localization, it is possible that their mutations impair the catalytic activity of the molecules that reach the Golgi.
Interestingly, six mutants in the Stealth domain that trafficked to the Golgi (Table 2 and Fig. 3, D407A and R986C) and underwent proteolytic cleavage had little or no enzymatic activity (Fig. 2A). This indicates that the mutations likely involve residues that participate in the catalytic function of the transferase. Finally, one of the Stealth domain mutants, I348L, behaved similarly to WT in all the assays.
Consequences of DMAP Domain Mutations
We previously reported that the DMAP interaction domain mutation K732N has full activity toward α-MM but impaired ability to phosphorylate lysosomal acid hydrolases (15). Here we show that a second DMAP mutant (L785W) behaves in an analogous fashion, with 103 ± 12% of WT activity toward α-MM and only 15 ± 12% of WT activity toward the lysosomal hydrolase α-iduronidase (Table 2).
Consequences of Notch Repeat Mutations
Interestingly, all the Notch mutations involve cysteine residues, and all block the formation of a different disulfide bond (Fig. 4A). However, only the mutations in Notch repeat 2 resulted in partial ER retention (Fig. 4B, C505Y and C523R) and reduced β subunit levels (Fig. 4C). The three Notch 1 mutants were efficiently delivered to the Golgi complex (Fig. 4B, C442Y, C461G, and C468S), and the αβ precursors underwent proteolytic cleavage (Fig. 4C). The Notch 1 mutants were fully active toward α-MM (Fig. 4D), documenting that the catalytic function of these mutants was intact. However, they exhibited decreased activity toward α-iduronidase (Fig. 4E), indicating an impairment in the selective recognition of acid hydrolases as substrates. The catalytic activity of the Notch 2 mutants was significantly reduced (Fig. 4D), consistent with the partially impaired ER exit. The activity toward α-iduronidase was similarly decreased (Fig. 4E). Unfortunately, this assay was not sensitive enough to determine whether the portion of the mutant enzyme that was catalytically active was impaired in the recognition of this lysosomal enzyme. Therefore, it remains to be established whether the Notch repeat 2 has a role in substrate recognition.
FIGURE 4.

Missense mutations in phosphotransferase Notch repeat 1 impair recognition of α-iduronidase. A, alignment of the Notch repeats 1 and 2 in human αβ phosphotransferase (PT) and the Lin12-Notch repeats (LNR) of human Notch 1. Indicated in bold are the cysteines that are mutated in MLIII αβ patients, and the different colors show disulfide bond formation. B, confocal immunofluorescence microscopy shows normal Golgi localization for the Notch 1 mutants (C442Y, C461G, and C468S) but partial Golgi/ER localization for the Notch 2 mutants (C505Y and C523R). Scale bars, 10 μm. C, HEK293 cell lysates expressing WT or mutant αβ phosphotransferase were subjected to SDS-PAGE and anti-V5 immunoblotting ∼48 h after transfection. Although the Notch 1 mutant β subunit levels were comparable with WT, the Notch 2 mutants showed reduced levels of β subunit, which is consistent with impaired ER exit. The anti-GAPDH control shows that the expression levels of the mutants were similar. D and E, although the activity toward α-MM (D) is normal with the Notch 1 mutants, the activity toward α-iduronidase (IDU, E) was significantly decreased (*, p < 0.05, Student's t test). The activity of the Notch 2 mutants C505Y and C523R was reduced for both α-MM (**, p < 0.01) and α-iduronidase. The values show the activity as a percentage of WT enzyme activity (averages ± S.E.) of at least three independent experiments, measured in duplicate.
Notch Mutants Partially Rescue MLII Zebrafish
To gain further insight into the consequences of the Notch domain mutations, we turned to the zebrafish MLII model to examine their effects in an intact animal system. In this model the expression of GlcNAc-1-phosphotransferase is inhibited by injection of embryos with morpholinos that target GNPTAB mRNA, resulting in a phenotype with features similar to MLII (17). These MLII zebrafish embryos can be rescued by co-injection with WT GNPTAB mRNA resistant to the morpholino. Alternatively, mRNAs bearing mutations can be evaluated for their ability to prevent the phenotype. We focused on the Notch 1 C442Y and C468S mutants, as they exhibit WT activity toward α-MM but impaired phosphorylation of α-iduronidase in the in vitro assays. The C442 and C468 residues correspond to Cys-447 and Cys-473 in the zebrafish GlcNAc-1-phosphotransferase. Rescued embryos were collected at 3 days post fertilization and analyzed for the activity and mannose phosphorylation of relevant enzymes. Consistent with the cellular assays, neither mutation affected the catalytic activity of GlcNAc-1-phosphotransferase as comparable activity was observed in embryos rescued with WT or mutant mRNA (Fig. 5A). The inability of either mRNA to restore WT levels of GlcNAc-1-phosphotransferase activity likely reflects the low efficiency of GNPTAB mRNA expression due to its large size. Nonetheless, the level of GlcNAc-1-phosphotransferase activity achieved in embryos rescued with WT mRNA is sufficient for complete biochemical and phenotypic rescue (15, 17). Analysis of cathepsin K activity in C447Y and C473S rescued embryos showed a minor, but not statistically significant, increase in total activity, indicating that the elevated activity of this protease within the MLII background can be effectively reduced despite the presence of the Notch domain mutants (Fig. 5B). However, reduced mannose phosphorylation of the glycosidase β-galactosidase was still noted in C447Y and C473S rescued embryos but not those rescued with WT mRNA (Fig. 5C). This finding supports a role for the Notch domain in lysosomal hydrolase recognition.
FIGURE 5.
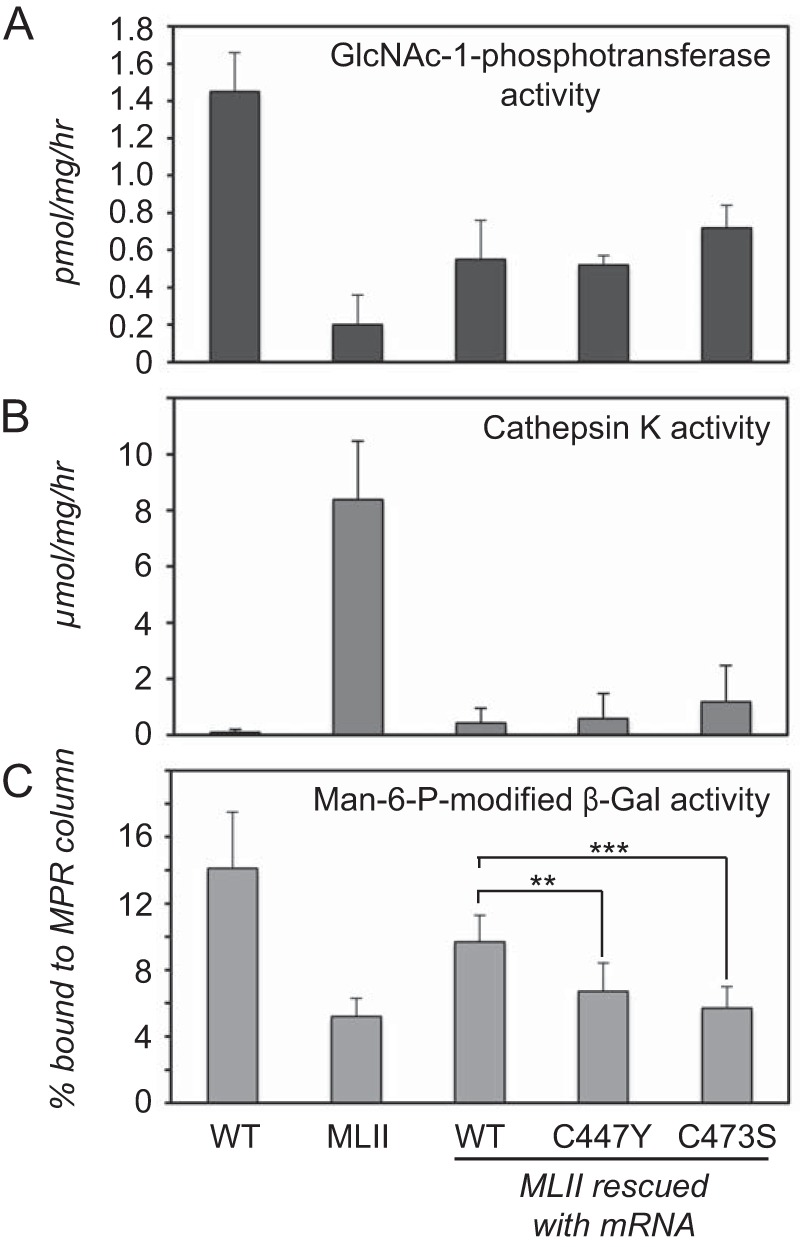
The Notch domain mutations selectively impact lysosomal hydrolase recognition in zebrafish embryos. A, direct measurement of GlcNAc-1-phopshotransferase activity in 3 days post fertilization lysates revealed comparable activity in embryos rescued with WT, C447Y, and C473S mRNA (n = 2). B, cathepsin K activity was reduced to near WT levels in all rescued embryos (n = 5). C, determination of the amount of mannose 6-phosphate modified β-galactosidase activity using cation-independent mannose 6-phosphate receptor (MPR) affinity chromatography demonstrated that C447Y and C473S (but not WT) rescued embryos had β-galactosidase mannose phosphorylation levels that were comparable to MLII embryos (n = 4). p < 0.001. Man-6-P, mannose 6-phosphate.
To determine whether the C447Y and C473S mutants were able to restore the abnormal tissue development seen with the MLII embryos, craniofacial phenotypes were assessed in Alcian blue-stained embryos (Fig. 6A). MLII zebrafish typically exhibit 1) short flattened jaws with Meckel's (M) cartilages that do not protrude beyond the eyes, 2) decreased distance between the Meckel's and ceratohyal (CH) structures, 3) a straightened angle of articulation between the ceratohyal elements, and 4) reduced Alcian blue staining of posterior structures (17, 20). Therefore, these criteria (outlined in Fig. 6B) were used to initially assess phenotypic rescue. Although 94% of the embryos injected with WT mRNA corrected all four abnormalities, embryos rescued with C447Y and C473S mRNAs only corrected these features 84 and 80% of the time, respectively. Furthermore, embryos rescued with the mutant mRNA appeared visually different from embryos rescued with WT mRNA. Therefore, we quantitatively assessed the degree of phenotypic rescue by measuring the length of the M, palatoquadrate (PQ) and CH structures (Fig. 6C). Collectively, these analyses showed that all measurement parameters were improved compared with MLII embryos. Of note, in the case of C447Y the M to CH length was significantly shorter compared with WT mRNA rescued zebrafish, the CH angle was significantly larger, and in both C447Y and C473S mRNA rescued zebrafish the CH length was significantly shorter relative to WT mRNA rescued (p < 0.05, two-tailed Student's t test). The other parameters were not significantly altered in the Notch mutant mRNA rescued zebrafish compared with those rescued with WT mRNA.
FIGURE 6.

Notch mutants partially rescue the cartilage phenotypes in MLII zebrafish. A, representative Alcian blue stains of WT, MLII, and rescued embryos. The percentage of embryos with the representative staining pattern is shown. B, schematic representation of four different parameters, including the angle, length, and position of multiple cartilage elements, quantified for all embryos in each condition. PF, pectoral fin; CB, cartilage. C, measurements were made in at least 25 embryos for each condition. The results of this quantification support partial rescue when Notch mutant mRNA is used; see “Results” for details. All parameters evaluated in the MLII zebrafish were significantly altered compared with WT zebrafish (p < 0.001). The parameters in the rescued zebrafish (WT, C447Y, and C473S) were all significantly improved compared with the MLII zebrafish (p < 0.01). PQ, palatoquadrate.
Consequences of Spacer and Cytoplasmic Tail Mutations
The six mutations in the spacer regions showed various consequences. The V182D and R587P mutants exhibited considerable ER staining with some Golgi localization (Fig. 7B and Fig. 8A). The A592T mutant showed predominant Golgi localization (Fig. 7B) with only a small reduction in the level of the β subunit. However, the activity toward α-MM was relatively more reduced (23 ± 12% of WT), consistent with some loss of catalytic activity.
FIGURE 7.
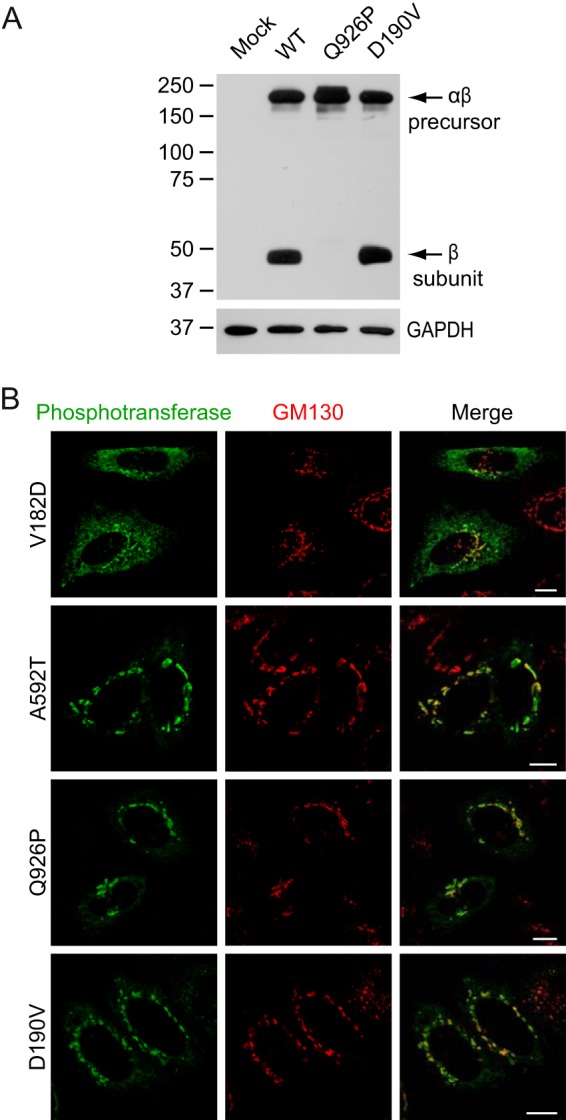
Consequences of mutations in the spacer regions of αβ phosphotransferase. A, Western blots (anti-V5) of HEK293 cell lysates ∼48 h after transfection with WT or mutant phosphotransferase αβ subunits with a C-terminal V5 tag. B, confocal immunofluorescence microscopy shows the subcellular localization of several spacer mutants in HeLa cells. Note that the V182D mutant shows partial ER localization (anti-α in green) and overlaps with the cis-Golgi marker GM130 (red). A592T αβ phosphotransferase has a predominant Golgi localization. Scale bars, 10 μm.
FIGURE 8.
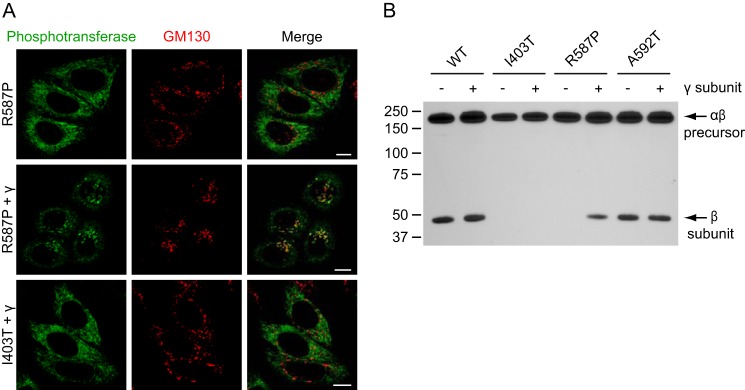
Overexpression of the γ subunit rescues the Golgi localization of R587P αβ phosphotransferase. A, confocal immunofluorescence microscopy of HeLa cells shows predominant ER localization of R587P αβ phosphotransferase (anti-α in green, upper panels) and significant co-localization with GM130 (red) in the Golgi complex after co-expression of the γ subunit (middle panels). Overexpression of the γ subunit had no effect on the localization of I403T αβ phosphotransferase (lower panels). Scale bars, 10 μm. B, HeLa cell lysates expressing WT, I403T, R587P, or A592T αβ phosphotransferase with or without overexpressed WT γ subunit were subjected to SDS-PAGE and anti-V5 immunoblotting 48h after transfection (representative blot of three independent experiments is shown). Overexpression of the γ subunit of phosphotransferase increased the level of the β subunit in the case of R587P but had no effect on the β subunit levels of WT, I403T, or A592T αβ phosphotransferase.
Of particular interest was the mutant enzyme with the Q926P substitution, which is located just 2 residues upstream of the site-1 protease cleavage site between Lys-928 and Asp-929. The expressed protein had no detectable β subunit or activity toward α-MM (Fig. 7A, Table 2). Immunofluorescence staining revealed a typical Golgi pattern (Fig. 7B), indicating that the mutant protein is transported to the Golgi but is not cleaved by the site-1 protease. Finally, the D190V and L1054V mutants exhibited minimal or no abnormalities in each of the cell-based assays (Fig. 7, Table 2), as did a mutation in the C-terminal cytoplasmic tail region (K1236M).
Overexpression of the γ Subunit Rescues R587P αβ Phosphotransferase
As preliminary experiments suggested that the spacer region between Notch repeat 2 and the DMAP interaction domain (residues 535–694) is involved in binding of the γ subunit, we next studied the effect of overexpressed WT γ subunit on the localization of the mutants in this region, R587P and A592T. This caused the R587P mutant to shift from a predominant ER localization to a predominant Golgi localization (Fig. 8A). There was no effect on the A592T mutant, which already showed a predominant Golgi localization without overexpressed γ subunit (Fig. 7B) or on the I403T (Fig. 8A), W81L, R334Q, or S399F mutants. Consistent with the enhanced Golgi localization of R587P when the γ subunit was overexpressed, Western blot analysis showed an increase in the level of the β subunit, whereas WT, I403T, and A592T did not show significant changes in the levels of this subunit (Fig. 8B). In addition, co-expression of the γ subunit increased the activity of R587P αβ phosphotransferase toward α-MM ∼3.5-fold. These results show that the function of the mutant enzyme can be partially rescued by the γ subunit of phosphotransferase.
DISCUSSION
The analysis of the GNPTAB missense mutations presented here advances our understanding of how the various amino acid substitutions affect the transport, activation, and catalytic function of the α and β subunits of GlcNAc-1-phosphotransferase. Moreover, these studies provide new insight into the function of previously uncharacterized domains of this enzyme. Our findings (summarized in Fig. 9) indicate four different ways in which the missense mutations studied affect GlcNAc-1-phosphotransferase function: 1) impaired exit from the ER, most likely due to misfolding, 2) loss of catalytic activity toward simple sugar substrates (this includes one mutation that prevents cleavage by the site-1 protease), 3) decreased binding and mannose phosphorylation of lysosomal acid hydrolases, and 4) reduced Golgi retention. In addition, several missense mutations were identified that have no deleterious consequences on GlcNAc-1-phosphotransferase function in the assays performed.
FIGURE 9.

Overview of the consequences of the GNPTAB missense mutations. Each mutant was classified based on its main defect. It should be noted that W81L, L1001P, and D1018G αβ phosphotransferase show partially impaired ER exit and undetectable to very low activity toward α-MM, and therefore, these mutations may impair both ER exit and catalytic activity. ER exit of A592T may be slightly impaired; however, the main defect is in the catalytic activity. IDU, α-iduronidase.
Although many of the substitutions in the Stealth domain result in retention in the ER, six of the mutants reach the Golgi and undergo proteolytic processing but have no or minimal catalytic activity toward α-MM. These residues, distributed across the four Stealth regions of the enzyme, appear to selectively impair the catalytic function of the transferase. It is not clear whether these residues constitute the amino acids responsible for transfer of GlcNAc-P to the 6-OH of the mannose acceptor, binding of UDP-GlcNAc, and mannose residues or both. These findings provide the first direct experimental evidence to support the longstanding hypothesis that the Stealth domain mediates the catalytic function of GlcNAc-1-phosphotransferase.
We also demonstrated a role for a second conserved domain (Notch) of the enzyme in the recognition of lysosomal hydrolases. Each of the three cysteine mutations in Notch repeat 1 retained full activity of the enzyme toward α-MM but exhibited significantly decreased activity toward the acid hydrolase α-iduronidase. Furthermore, two of these mutants, C442Y and C468S (C447Y and C473S in zebrafish), failed to restore β-galactosidase phosphorylation in the zebrafish MLII model. Taken together, these findings implicate the Notch repeat 1 in the recognition of acid hydrolases. In contrast to the Notch 1 mutants, the cysteine mutations in Notch repeat 2 resulted in partial retention in the ER. At this point it is unclear whether these mutations also affect the ability of the enzyme that reaches the Golgi to phosphorylate lysosomal enzyme substrates.
We previously reported a role for the DMAP interaction domain in the recognition of lysosomal enzymes (15). This conclusion was based on the finding that the DMAP mutant K732N showed normal catalytic activity (α-MM measurement) but impaired activity toward the lysosomal enzymes cathepsin D and α-iduronidase. In addition, expression of K732N in the MLII zebrafish model failed to rescue the craniofacial phenotype or reduce the elevated cathepsin K activity that is associated with abnormal cartilage development. Thus, the impact of the DMAP mutation differs from the effects of the Notch 1 mutants, which showed partial rescue of the phenotypic abnormalities and a reduction in cathepsin K activity back to WT levels. Nonetheless both mutants exhibited reduced mannose phosphorylation of β-galactosidase in our zebrafish studies supporting a role for these domains in substrate recognition. It is possible that these domains mediate the binding of distinct sets of lysosomal enzymes to ensure the phosphorylation of a broad spectrum of substrates. Alternatively, they could act cooperatively to ensure the fidelity of mannose phosphorylation.
Most studies that reported new mutations in MLII and III αβ patients used bioinformatics tools to predict the consequences of these mutations. Although in the majority of the cases these predictions were in agreement with our observations, we found one damaging mutation that was predicted to be benign (A955V) and one mutation that was predicted to be damaging but showed no defects (Q205P) (21). Of interest are the mutations that involve the consensus sequence for the site-1 protease, (R/K)X(hydrophobic)Z, with X representing any amino acid and Z preferentially Leu or Thr but excluding Val, Pro, Glu, Asp, or Cys (19). In αβ phosphotransferase, site-1 protease cleavage occurs after Lys-928, and alanine scanning showed that residues Arg-925, Leu-927, and Lys-928 were most critical for this cleavage (19). Based on the consensus sequence (predicting any amino acid at residue 926), we did not expect that the patient mutation Q926P would affect site-1 protease cleavage. However, although this mutant localized normally to the Golgi complex, the enzyme was inactive, and no β subunit was formed. These data show that a Pro at residue 926 is unfavorable for cleavage. On the other hand, substitution of Lys-928 with Arg (21) is tolerated (data not shown).
Several of the missense mutations were reported in a homozygous state in either MLII or MLIII αβ patients. In general, the severity of these mutations correlated well with the reported clinical phenotypes in the patients. The mutations W81L and R986C that resulted in the severe MLII phenotype showed undetectable catalytic activity (<2% of WT) in our assay. The homozygous mutations that occurred in MLIII αβ patients were found to be less detrimental, as the residual activities were 8–11%. The exceptions were D1018G, which is associated with an MLIII αβ phenotype, although more severe than seen in classical MLIII αβ patients (reported as MLIII intermediate in Cathey et al. (22)), as it showed undetectable catalytic activity, and K1236M, which was normal in our assays. In addition, C523R was homozygous in a patient with either MLII or III αβ (23). Our data predict an attenuated clinical phenotype in this patient, as there was substantial residual catalytic activity (29 ± 12%).
De Pace et al. (24) previously reported expression studies of four of the missense mutants analyzed here (W81L, S399F, R986C, and K1236M). Our results are in agreement with their finding that mutation of the residues Trp-81 and Ser-399 impacts ER exit. Moreover, by measuring the catalytic activity of the mutants, our study suggests that the W81L mutation also possibly impacts the activity of the portion of the enzyme that makes it to Golgi complex. In addition, we established that the R986C mutation completely impairs the catalytic activity. Although our assays revealed the consequences of the great majority of the missense mutations, no abnormalities were found in the case of K1236M αβ phosphotransferase. Likewise, three other mutants, D190V, I348L, and L1054V, showed little or no impairment in their biosynthesis, Golgi localization, or enzymatic activity toward α-MM and α-iduronidase. Thus it is unclear at this point how these mutations give rise to the MLII/MLIII αβ phenotype. Expression of mRNA bearing these mutations within the MLII zebrafish background may lend insight into how these mutations alter GlcNAc-1-phosphotransferase function in a physiological context.
To our surprise, overexpression of the γ subunit of phosphotransferase had a great impact on the ER exit of the R587P αβ mutant. One possible explanation for this rescue is that the γ subunit binding site is located in this spacer region, close to R587P. This interaction might stabilize this region and favor correct folding of the enzyme. The endogenous γ subunit levels may not be sufficient to mediate this rescue effect, possibly because R587P has a modest effect on γ subunit binding. In addition, these results suggest that the αβ and γ subunits assemble in the ER and are transported to the Golgi as a complex.
Having confirmed that the Stealth domain mediates the catalytic function of the enzyme and demonstrating that the Notch and DMAP domains are involved in the selective recognition of acid hydrolase substrates, it is of interest to relate these findings to the evolution of mammalian GlcNAc-1-phosphotransferase. There is now direct evidence that the bacterial Stealth proteins function as sugar-phosphate transferases in the synthesis of cell wall polysaccharides (8–10). In contrast to GlcNAc-1-phosphotransferase, these reactions do not involve the recognition of a protein determinant. Importantly, the bacterial Stealth proteins lack the Notch and DMAP domains. Thus an attractive hypothesis is that the mammalian enzyme acquired these domains, as it evolved to selectively recognize and phosphorylate lysosomal acid hydrolases. The bacterial transferases also lack the γ subunit. We, therefore, hypothesize that the binding site for γ would be in the region of the αβ precursor that contains the Notch and DMAP domains. We are currently investigating this possibility.
Acknowledgments
We thank our colleagues for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants CA-008759 (to S. K.) and GM-086524-7 (to R. S.). This work was also supported by grants from the Yash Gandhi Foundation (to S. K. and R. S.) and the National Mucopolysaccharidosis Society/International Society for Mannosidosis and Related Disorders (to H. F.-S.).
- GlcNAc-1-phosphotransferase
- UDP-GlcNAc:lysosomal enzyme GlcNAc-1-phosphotransferase
- α-MM
- α-methyl d-mannoside
- ER
- endoplasmic reticulum
- M
- Meckel's cartilage
- CH
- ceratohyal.
REFERENCES
- 1. Ghosh P., Dahms N. M., Kornfeld S. (2003) Mannose 6-phosphate receptors: new twists in the tale. Nat. Rev. Mol. Cell Biol. 4, 202–212 [DOI] [PubMed] [Google Scholar]
- 2. Bao M., Booth J. L., Elmendorf B. J., Canfield W. M. (1996) Bovine UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. I. Purification and subunit structure. J. Biol. Chem. 271, 31437–31445 [DOI] [PubMed] [Google Scholar]
- 3. Kudo M., Bao M., D'Souza A., Ying F., Pan H., Roe B. A., Canfield W. M. (2005) The α- and β-subunits of the human UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cDNA. J. Biol. Chem. 280, 36141–36149 [DOI] [PubMed] [Google Scholar]
- 4. Raas-Rothschild A., Cormier-Daire V., Bao M., Genin E., Salomon R., Brewer K., Zeigler M., Mandel H., Toth S., Roe B., Munnich A., Canfield W. M. (2000) Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). J. Clin. Invest. 105, 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tiede S., Storch S., Lübke T., Henrissat B., Bargal R., Raas-Rothschild A., Braulke T. (2005) Mucolipidosis II is caused by mutations in GNPTA encoding the α/β GlcNAc-1-phosphotransferase. Nat. Med. 11, 1109–1112 [DOI] [PubMed] [Google Scholar]
- 6. Kudo M., Canfield W. M. (2006) Structural requirements for efficient processing and activation of recombinant human UDP-N-acetylglucosamine:lysosomal-enzyme-N-acetylglucosamine-1-phosphotransferase. J. Biol. Chem. 281, 11761–11768 [DOI] [PubMed] [Google Scholar]
- 7. Qian Y., Lee I., Lee W. S., Qian M., Kudo M., Canfield W. M., Lobel P., Kornfeld S. (2010) Functions of the α, β, and γ subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J. Biol. Chem. 285, 3360–3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fiebig T., Freiberger F., Pinto V., Romano M. R., Black A., Litschko C., Bethe A., Yashunsky D., Adamo R., Nikolaev A., Berti F., Gerardy-Schahn R. (2014) Molecular cloning and functional characterization of components of the capsule biosynthesis complex of Neisseria meningitidis serogroup A: toward in vitro vaccine production. J. Biol. Chem. 289, 19395–19407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Muindi K. M., McCarthy P. C., Wang T., Vionnet J., Battistel M., Jankowska E., Vann W. F. (2014) Characterization of the meningococcal serogroup X capsule N-acetylglucosamine-1-phosphotransferase. Glycobiology 24, 139–149 [DOI] [PubMed] [Google Scholar]
- 10. Reilly M. C., Levery S. B., Castle S. A., Klutts J. S., Doering T. L. (2009) A novel xylosylphosphotransferase activity discovered in Cryptococcus neoformans. J. Biol. Chem. 284, 36118–36127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sperisen P., Schmid C. D., Bucher P., Zilian O. (2005) Stealth proteins: in silico identification of a novel protein family rendering bacterial pathogens invisible to host immune defense. PLoS Comput. Biol. 1, e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vardar D., North C. L., Sanchez-Irizarry C., Aster J. C., Blacklow S. C. (2003) Nuclear magnetic resonance structure of a prototype Lin12-Notch repeat module from human Notch1. Biochemistry 42, 7061–7067 [DOI] [PubMed] [Google Scholar]
- 13. Rountree M. R., Bachman K. E., Baylin S. B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277 [DOI] [PubMed] [Google Scholar]
- 14. Braulke T., Raas-Rothschild A., Kornfeld S. (2013) The online metabolic and molecular bases of inherited disease. in I-cell Disease and Pseudo-Hurler Polydystrophy: Disorders of lysosomal Enzyme Phosphorylation and Localization (Valle D., Vogelstein B., Kinzler K., Antonarakis S., Ballabio A., Gibson K., Mitchell G., eds.) McGraw-Hill Inc., New York [Google Scholar]
- 15. Qian Y., Flanagan-Steet H., van Meel E., Steet R., Kornfeld S. A. (2013) The DMAP interaction domain of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase is a substrate recognition module. Proc. Natl. Acad. Sci. U.S.A. 110, 10246–10251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Meel E., Qian Y., Kornfeld S. A. (2014) Mislocalization of phosphotransferase as a cause of mucolipidosis III αβ. Proc. Natl. Acad. Sci. U.S.A. 111, 3532–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flanagan-Steet H., Sias C., Steet R. (2009) Altered chondrocyte differentiation and extracellular matrix homeostasis in a zebrafish model for mucolipidosis II. Am. J. Pathol. 175, 2063–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- 19. Marschner K., Kollmann K., Schweizer M., Braulke T., Pohl S. (2011) A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science 333, 87–90 [DOI] [PubMed] [Google Scholar]
- 20. Petrey A. C., Flanagan-Steet H., Johnson S., Fan X., De la Rosa M., Haskins M. E., Nairn A. V., Moremen K. W., Steet R. (2012) Excessive activity of cathepsin K is associated with cartilage defects in a zebrafish model of mucolipidosis II. Dis. Model Mech. 5, 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zarghooni M., Dittakavi S. S. (2009) Molecular analysis of cell lines from patients with mucolipidosis II and mucolipidosis III. Am. J. Med. Genet. A 149A, 2753–2761 [DOI] [PubMed] [Google Scholar]
- 22. Cathey S. S., Leroy J. G., Wood T., Eaves K., Simensen R. J., Kudo M., Stevenson R. E., Friez M. J. (2010) Phenotype and genotype in mucolipidoses II and III α/β: a study of 61 probands. J. Med. Genet. 47, 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cobos P. N., Steglich C., Santer R., Lukacs Z., Gal A. (2014) Dried blood spots allow targeted screening to diagnose mucopolysaccharidosis and mucolipidosis. JIMD Rep. 15, 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Pace R., Coutinho M. F., Koch-Nolte F., Haag F., Prata M. J., Alves S., Braulke T., Pohl S. (2014) Mucolipidosis II-related mutations inhibit the exit from the endoplasmic reticulum and proteolytic cleavage of GlcNAc-1-phosphotransferase precursor protein (GNPTAB). Hum. Mutat. 35, 368–376 [DOI] [PubMed] [Google Scholar]
- 25. Cury G. K., Matte U., Artigalás O., Alegra T., Velho R. V., Sperb F., Burin M. G., Ribeiro E. M., Lourenço C. M., Kim C. A., Valadares E. R., Galera M. F., Acosta A. X., Schwartz I. V. (2013) Mucolipidosis II and III α/β in brazil: analysis of the GNPTAB gene. Gene 524, 59–64 [DOI] [PubMed] [Google Scholar]
- 26. Encarnação M., Lacerda L., Costa R., Prata M. J., Coutinho M. F., Ribeiro H., Lopes L., Pineda M., Ignatius J., Galvez H., Mustonen A., Vieira P., Lima M. R., Alves S. (2009) Molecular analysis of the GNPTAB and GNPTG genes in 13 patients with mucolipidosis type II or type III: identification of eight novel mutations. Clin. Genet. 76, 76–84 [DOI] [PubMed] [Google Scholar]
- 27. Otomo T., Muramatsu T., Yorifuji T., Okuyama T., Nakabayashi H., Fukao T., Ohura T., Yoshino M., Tanaka A., Okamoto N., Inui K., Ozono K., Sakai N. (2009) Mucolipidosis II and III α/β: mutation analysis of 40 Japanese patients showed genotype-phenotype correlation. J. Hum. Genet. 54, 145–151 [DOI] [PubMed] [Google Scholar]
- 28. Bargal R., Zeigler M., Abu-Libdeh B., Zuri V., Mandel H., Ben Neriah Z., Stewart F., Elcioglu N., Hindi T., Le Merrer M., Bach G., Raas-Rothschild A. (2006) When Mucolipidosis III meets Mucolipidosis II: GNPTA gene mutations in 24 patients. Mol. Genet. Metab. 88, 359–363 [DOI] [PubMed] [Google Scholar]
- 29. Fernández-Marmiesse A., Morey M., Pineda M., Eiris J., Couce M. L., Castro-Gago M., Fraga J. M., Lacerda L., Gouveia S., Pérez-Poyato M. S., Armstrong J., Castiñeiras D., Cocho J. A. (2014) Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet. J. Rare Dis. 9, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tappino B., Chuzhanova N. A., Regis S., Dardis A., Corsolini F., Stroppiano M., Tonoli E., Beccari T., Rosano C., Mucha J., Blanco M., Szlago M., Di Rocco M., Cooper D. N., Filocamo M. (2009) Molecular characterization of 22 novel UDP-N-acetylglucosamine-1-phosphate transferase α- and β-subunit (GNPTAB) gene mutations causing mucolipidosis types IIα/β and IIIα/β in 46 patients. Hum. Mutat. 30, E956–E973 [DOI] [PubMed] [Google Scholar]
- 31. Tiede S., Muschol N., Reutter G., Cantz M., Ullrich K., Braulke T. (2005) Missense mutations in N-acetylglucosamine-1-phosphotransferase α/β subunit gene in a patient with mucolipidosis III and a mild clinical phenotype. Am. J. Med. Genet. A 137A, 235–240 [DOI] [PubMed] [Google Scholar]
- 32. Coutinho M. F., Santos Lda S., Girisha K. M., Satyamoorthy K., Lacerda L., Prata M. J., Alves S. (2012) Mucolipidosis type II α/β with a homozygous missense mutation in the GNPTAB gene. Am. J. Med. Genet. A 158A, 1225–1228 [DOI] [PubMed] [Google Scholar]
- 33. Lam C. W., Yan M. S., Li C. K., Lau K. C., Tong S. F., Tang H. Y. (2007) DNA-based diagnosis of mucolipidosis type IIIA and mucopolysacchariodisis type VI in a Chinese family: a chance of 1 in 7.6 trillion. Clin. Chim. Acta 376, 250–252 [DOI] [PubMed] [Google Scholar]
- 34. Kudo M., Brem M. S., Canfield W. M. (2006) Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase α/β -subunits precursor gene. Am. J. Hum. Genet. 78, 451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tiede S., Cantz M., Spranger J., Braulke T. (2006) Missense mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTA) in a patient with mucolipidosis II induces changes in the size and cellular distribution of GNPTG. Hum. Mutat. 27, 830–831 [DOI] [PubMed] [Google Scholar]