

Background: Mutations in Krev interaction trapped 1 (KRIT1) and cerebral cavernous malformations 2 (CCM2) are associated with CCM disease.
Results: The CCM2-KRIT1 interaction is characterized structurally and biochemically.
Conclusion: CCM2 preferentially binds the third NPX(Y/F) motif of KRIT1, and disease-associated mutations destabilize this interaction.
Significance: These data may inform future studies into the biology of CCM disease.
Keywords: Crystal Structure, Crystallography, Genetic Disease, Protein Complex, Protein-Protein Interaction, Cerebral Cavernous Malformations, PTB Domain, Point Mutation
Abstract
Familial cerebral cavernous malformations (CCMs) are predominantly neurovascular lesions and are associated with mutations within the KRIT1, CCM2, and PDCD10 genes. The protein products of KRIT1 and CCM2 (Krev interaction trapped 1 (KRIT1) and cerebral cavernous malformations 2 (CCM2), respectively) directly interact with each other. Disease-associated mutations in KRIT1 and CCM2 mostly result in loss of their protein products, although rare missense point mutations can also occur. From gene sequencing of patients known or suspected to have one or more CCMs, we discover a series of missense point mutations in KRIT1 and CCM2 that result in missense mutations in the CCM2 and KRIT1 proteins. To place these mutations in the context of the molecular level interactions of CCM2 and KRIT1, we map the interaction of KRIT1 and CCM2 and find that the CCM2 phosphotyrosine binding (PTB) domain displays a preference toward the third of the three KRIT1 NPX(Y/F) motifs. We determine the 2.75 Å co-crystal structure of the CCM2 PTB domain with a peptide corresponding to KRIT1NPX(Y/F)3, revealing a Dab-like PTB fold for CCM2 and its interaction with KRIT1NPX(Y/F)3. We find that several disease-associated missense mutations in CCM2 have the potential to interrupt the KRIT1-CCM2 interaction by destabilizing the CCM2 PTB domain and that a KRIT1 mutation also disrupts this interaction. We therefore provide new insights into the architecture of CCM2 and how the CCM complex is disrupted in CCM disease.
Introduction
Cerebral cavernous malformations (CCMs)4 occur in up to 0.5% of the world's population (1) and most commonly present as mulberry-shaped lesions within the neurovasculature. The disease can have severe outcomes, including intracranial hemorrhage, stroke, and other focal neurological deficits (2). CCMs occur in sporadic and genetic forms, with the genetic form of CCMs (familial CCMs) occurring by a Knudsonian mechanism in which a patient inherits a first mutation that is then followed by a “second hit” mutation acquired later in life (3–5). Genetic acquisition of CCM is linked to mutations in KRIT1/CCM1 (6, 7), CCM2/MGC4607/OSM/Malcavernin (8), or PDCD10/CCM3/TFAR15 (9, 10), which encode the protein products Krev interaction trapped 1 (KRIT1), cerebral cavernous malformations 2 (CCM2), or cerebral cavernous malformations 3 (CCM3), respectively. Familial CCM disease is associated with an unusually high proportion of mutations in KRIT1/CCM1, CCM2, and CCM3/PDCD10 that result in truncation or deletion of their protein products (11), but rare missense mutations can also occur (12–14), implying critical importance for the KRIT1, CCM2, and CCM3 proteins.
KRIT1, CCM2, and CCM3 function predominantly as scaffolds (15, 16) and can form a heterotrimeric complex (17). Importantly, the CCM2 interaction with KRIT1 is one of the critical components of this complex. The members of the CCM signaling complex have each been found to interact with a diverse array of protein binding partners, including the small GTPase Rap1 (18–20), the heart of glass receptor (HEG1) (21, 22), integrin cytoplasmic associated protein 1 (ICAP1) (23–26), the serine-threonine kinase MAP3K3 (27), and the E3 ubiquitin ligase SMURF1 (28). Better understanding of the basis for the KRIT1 interaction with CCM2 will allow deconvolution of the functional roles of both KRIT1 and CCM2 in signaling and in CCM disease.
Structurally, CCM2 is predicted to contain an N-terminal phosphotyrosine binding (PTB) domain (8, 13) and was also recently discovered to contain a C-terminal harmonin homology domain (29). KRIT1 contains an N-terminal Nudix domain (25), three Asn-Pro-X-Tyr/Phe (NPX(Y/F)) motifs (24), an ankyrin repeat domain, and a C-terminal FERM (band 4.1, ezrin, radixin, moesin) domain (18) (Fig. 1A). Despite what their name implies, the specificity of PTB domains is not restricted to phosphotyrosine motifs, because many of them can also bind to NPXY or NPXF motifs either in place of or in addition to NPXpY (where pY represents phosphotyrosine). The modes of interaction between the PTB domain and Asn-Pro-X-Tyr/Phe/Tyr(P) (NPX(Y/F/pY)) motifs are similar (30). Previous studies have suggested that the putative CCM2 PTB domain is important for the CCM2 association with KRIT1 (31), and a point mutation in CCM2 PTB domain (F217A) based on canonical interactions between PTB domains and NPX(Y/F/pY) motifs reduces its binding to KRIT1 (32). The interaction site within KRIT1, however, is controversial. Previous studies have suggested either no involvement of the conserved KRIT1 NPX(Y/F) motifs (33) or that the CCM2 PTB domain binds equally to both the second and third of these motifs (34).
FIGURE 1.

Locations of disease-associated missense mutations in the CCM2 and KRIT1 proteins. A, domain schematic of CCM2 (top) and KRIT1 (bottom). For CCM2, the PTB domain and harmonin homology domain (HHD) are indicated. The KRIT1 Nudix domain, ankyrin repeat domain (ARD), and FERM domain, as well as the NPX(Y/F) motifs and RR protein binding site, are indicated. The locations of the mutations listed in Table 1 are denoted by stars and red type, and those explored experimentally are in boldface type. B, crystal structure of KRIT1 Nudix domain in complex with ICAP1 PTB domain (Protein Data Bank code 4DX8) showing the locations of KRIT1 mutations R16C and G101R, both of which are completely conserved through evolution. C, crystal structure of KRIT1 FERM domain in complex with HEG1 (Protein Data Bank code 3U7D) showing the locations of KRIT1 mutations Q473H and L667R.
To investigate the impact of rare missense point mutations in the KRIT1 and CCM2 genes and to further elucidate the structural basis for recruitment of CCM2 to KRIT1, we took a two-pronged approach. We conducted clinical sequencing of patients suspected of having one or more CCM lesions to identify novel missense point mutations and used biochemical and structural studies to probe the interaction between CCM2 and KRIT1. In our clinical sequencing of the three CCM genes in hundreds of affected patients, we identified small numbers of missense mutations amid the much larger group of chain termination mutations. Because many of the mutations we found were within the predicted PTB domain of CCM2, we then went on to investigate the structure of the CCM2 PTB domain and the basis of its interaction with KRIT1. We began by confirming that CCM2 and KRIT1 can interact with one another in cells and that mutation of both the second and third NPX(Y/F) motifs of KRIT1 (KRIT1NPX(Y/F)2 and KRIT1NPX(Y/F)3) reduces this interaction. We found that the CCM2 PTB domain exhibits preferential binding in vitro to KRIT1NPX(Y/F)3 over either KRIT1NPX(Y/F)1 or KRIT1NPX(Y/F)2, indicating that this is the dominant region of KRIT1 that mediates its interaction with CCM2. We then determined the 2.75 Å co-crystal structure of the CCM2 PTB domain in complex with KRIT1NPX(Y/F)3 and validated the crystallographically observed interaction by pull-down. To explore the role of the potential disease-related missense mutations that we had identified within the CCM2 PTB domain and KRIT1NPX(Y/F)3, we tested the impact of these mutations on CCM2, KRIT1, and their interaction. We find that the missense mutations identified in the region of CCM2 that encodes its PTB domain significantly reduce the solubility of the CCM2 protein product and that a missense mutation in KRIT1 that results in a V244L mutation reduces the ability of KRIT1 to interact with CCM2. These results provide a significantly improved understanding of the CCM2-KRIT1 interaction and its disruption in CCM disease.
EXPERIMENTAL PROCEDURES
Sequencing
Nomenclature for sequence variants was taken from the Human Genome Variation Society recommendations. As required, DNA was extracted from the patient specimen using a 5 Prime ArchivePure DNA blood kit. PCR was used to amplify the indicated exons plus additional flanking intronic or other non-coding sequence. After cleaning of the PCR products, cycle sequencing was carried out using the ABI Big Dye Terminator version 3.0 kit. Products were resolved by electrophoresis on an ABI 3730xl capillary sequencer. Sequencing was performed separately in both the forward and reverse directions.
Co-Immunoprecipitation
Full-length human CCM2 cDNA was subcloned into pcDNA3-FLAG vector, and full-length human KRIT1 cDNA was subcloned into pEGFP-C1 vector. Human embryonic kidney 293T cells were grown to 80% confluence in DMEM with 10% FBS, 1 mm sodium pyruvate, 10 units/ml penicillin/streptomycin at 37 °C, and cells were transfected using Lipofectamine (Invitrogen) with both FLAG-CCM2 and GFP-KRIT1 or with the appropriate mutants. Following two washes in PBS, cells were lysed in radioimmune precipitation assay lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% NP50, 0.5% sodium deoxycholate, 0.1% SDS), and soluble lysate was incubated with either anti-FLAG antibody (Sigma, F3165) or anti-GFP antibody (Rockland, 600-101-215) followed by protein A/G-agarose beads. Supernatant and precipitant samples were immunoblotted with anti-FLAG antibody or anti-GFP antibody.
Protein Expression and Purification
Human CCM2 (UniProt Q9BSQ5) cDNA corresponding to amino acid residues 51–228 was subcloned into a modified pET-32 vector with a tobacco etch virus protease-cleavable N-terminal hexahistidine (His6) tag and transformed into Escherichia coli Rosetta (DE3) cells (Novagen). Overnight cultures of these cells were inoculated into Luria Broth medium until A600 = 0.6. Protein expression was induced by the addition of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C overnight. The cells were harvested the next day and were resuspended in lysis buffer (500 mm NaCl, 20 mm imidazole, 20 mm Tris, pH 8.0) supplemented with 1 mm DTT, protease inhibitors, and DNase I. Cell lysis was performed by three cycles of freeze/thaw in a dry ice/ethanol bath followed by sonication. Following clarification, the supernatant was loaded onto a HisTrap HP column (GE Healthcare). His6-CCM2PTB was eluted by lysis buffer supplemented with 500 mm imidazole and dialyzed overnight against 250 mm NaCl, 20 mm Tris, pH 8.0, 1 mm DTT in the presence of tobacco etch virus to remove the His6 tag. Cleaved CCM2PTB was loaded to a Superdex 75 column (GE Healthcare) and eluted as a monodisperse peak in buffer containing 250 mm NaCl, 20 mm Tris, 1 mm DTT, pH 8.0.
Human KRIT1 (UniProt O00522) cDNA corresponding to NPX(Y/F) motifs 1 (residues 186–198) (KRIT1NPX(Y/F)1), 2 (residues 225–237) (KRIT1NPX(Y/F)2), and 3 (residues 244–256) (KRIT1NPX(Y/F)3), was subcloned into pGEX-6p1 (GE Healthcare) as described previously (35). Cells were grown and lysed following a protocol similar to that described above for CCM2. After lysis, the supernatant was incubated with glutathione-Sepharose 4B beads (GE Healthcare) for 1 h and washed three times in pull-down buffer (150 mm NaCl, 20 mm Tris, pH 7.5, 0.1% Triton X-100).
Point mutations were introduced using the QuikChange Lightning mutagenesis kit (Stratagene). The GST-KRIT1NPX(Y/F)3 mutants were treated in the same way as the wild-type protein. For the His6-CCM2PTB mutants, an additional desalting step was added following affinity purification into a final buffer of 150 mm NaCl, 20 mm Tris, pH 8.0, and 0.5 mm tris(2-carboxyethyl)phosphine.
Size Exclusion Chromatography with Multiangle Light Scattering
Purified CCM2PTB at a concentration of 1 mg/ml was mixed with a synthesized and HPLC-purified 13-mer peptide corresponding to KRIT1NPX(Y/F)3 (VDKVVINPYFGLG; KRIT1NPX(Y/F)3) (Tufts University Core Facility) in a 1:1 molar ratio. After incubation on ice for 2 h, the sample was filtered, and 100 μl was injected onto an SRT-300 column (Sepax Technologies) at 0.4 ml/min in a buffer composed of 150 mm NaCl, 20 mm Tris, pH 8.0, 1 mm tris(2-carboxyethyl)phosphine, and 0.02% sodium azide. The column was run on an HPLC system (Agilent Technologies 1260) in-line with a DAWN-HELEOS II multiangle light scattering detector (Wyatt Technology) and Optilab T-rEX differential refractometer (Wyatt Technology). Data were analyzed using the ASTRA 6 software from Wyatt Technology.
Crystallization, Data Collection, and Structure Determination
Purified CCM2PTB was concentrated to 10 mg/ml and mixed with the synthetic KRIT1NPX(Y/F)3 peptide in a 1:1 molar ratio. Following incubation on ice for 2 h, sparse matrix and grid crystallization screening was conducted using the sitting drop method and the Classics, pHClear, PEGs, and JCSG+ screening kits (Qiagen) in 1-μl drops with a 1:1 ratio of protein and precipitant solutions. Crystals grew against 20% (w/v) PEG 6000 and 0.1 m HEPES, pH 7.0, in the pH Clear Suite (Qiagen). For cryoprotection, crystals were transferred to the precipitant condition supplemented with 10% ethylene glycol prior to flash cooling in liquid nitrogen.
Diffraction data were collected at Northeastern Collaborative Access Team beamline 24-ID-E at the Advanced Photon Source and were processed using HKL-2000 (36) to 2.75 Å resolution. An initial structure solution was obtained by molecular replacement using Phaser (37) with an ensemble search model of previously determined PTB domains (Protein Data Bank entries 4JIF, 3SO6, 3G9W, 3HQC, 3DXE, 2EJ8, 2V76, 1J0W, 1WVH, 2CY4, 2DYQ, 1QQG, 1X11, and 1AQC) edited to remove ligands, water molecules, and non-PTB peptide chains. Four copies of CCM2PTB were found per asymmetric unit, and autobuilding was performed using PHENIX (37), which built 531 residues in 14 chains, including residues of KRIT1NPX(Y/F)3. Autobuilding yielded R and Rfree values of 26.3 and 28.9% respectively. Iterative rounds of refinement and model building were conducted using PHENIX (37), REFMAC5 (38) using jelly body refinement, and Coot (39). TLS and NCS were used. The KRIT1 peptide was clearly visible in the electron density, allowing unambiguous determination of its register. The structure displayed good geometry and was validated using MolProbity (40). Crystallographic programs were supported by the SBGrid Consortium (41). The atomic coordinates and structure factors are deposited in the Protein Data Bank with accession code 4WJ7.
Pull-down Experiments
Pull-down experiments were conducted by incubating 67.5 μg of CCM2PTB with 20 μg of GST-KRIT1-loaded glutathione-Sepharose beads for 2 h at 4 °C with agitation. Samples were washed three times with 1 ml of pull-down buffer (150 mm NaCl, 50 mm Tris, pH 7.5, 0.1% Triton) and visualized by SDS-PAGE. Quantification was performed using ImageJ.
Solubility Tests
CCM2 disease mutants (L113P, L115R, L155P, L198R, and L213P) were introduced into His6-CCM2PTB. All of these mutants as well as the wild-type protein were transformed into BL21 cells. Overnight cultures of the cells were inoculated into 100 ml of Luria broth, and protein expression was induced with isopropyl 1-thio-β-d-galactopyranoside when A600 = 0.6. Cells were grown overnight at 18 °C, harvested, and resuspended in 1 ml of a buffer composed of 500 mm NaCl and 20 mm Tris, pH 8.0, supplemented with DTT, protease inhibitors, lysozyme, and DNase I. The resuspended cells were lysed by three cycles of freeze/thaw in a dry ice/ethanol bath followed by sonication. After sonication, 10 μl of each lysate was spun down at 13,000 rpm. The pellets and supernatants were each resuspended to a total volume of 50 μl. One μl of each sample was run on an SDS-PAGE gel and visualized by Coomassie staining.
Biolayer Interferometry
The biolayer interferometry technique using the BLItz system (ForteBio) was used to measure binding kinetics for the CCM2PTB interaction with GST-KRIT1NPX(Y/F)1, GST-KRIT1NPX(Y/F)2, and GST-KRIT1NPX(Y/F)3 and the following biotinylated peptides: Biotin-225ADTCIYNPLFGSD237 (KRIT1biotin-NPX(Y/F)2), Biotin-241TNRVDKVVINPYFG254 (KRIT1biotin-NPX(Y/F)3), and Biotin-225ADTCIYNPLFGSDLQYTNRVDKVVINPYFG254 (KRIT1biotin-NPX(Y/F)2-3). Anti-GST biosensors (for the GST fusions) or streptavidin biosensors (for the biotinylated peptides) were hydrated in binding buffer (150 mm NaCl, 50 mm Tris, pH 7.5) for 10 min. For each CCM2PTB concentration (ranging from 2.5 to 415 μm), the following procedure was performed. An initial baseline was collected by immersing the biosensor in binding buffer for 1 min, and then 4 μl of 50 μg/ml KRIT1 was loaded to the biosensor for 5 min. The KRIT1-loaded biosensor was returned to binding buffer for collection of a second baseline for 90 s and then placed in 4 μl of His6-CCM2PTB for a 5-min association step. Finally, the biosensor was returned to binding buffer for a final 2.5-min dissociation step. For each data point, the background binding was also measured. For each CCM2PTB concentration, the difference in the signal (in nm) just prior to the association step and that at the end of the association step was subtracted from the difference in signal for background binding. These values were plotted, and the curves were fit using GraphPad Prism to obtain a dissociation constant.
RESULTS
Discovery of Missense Point Mutations in KRIT1 and CCM2
From 2004 to 2012 at PreventionGenetics, a clinical DNA testing laboratory, 507 full KRIT1 gene sequencing tests and 288 full CCM2 gene sequencing tests were performed on patients suspected by their physicians of having one or more CCMs. Likely causative mutations were identified in 30% of the KRIT1 tests and 12% of the CCM2 tests. The great majority of causative mutations in both genes were chain termination mutations (nonsense, frameshift, or splicing). However, a small number of missense mutations were also identified in these patients (Table 1 and Fig. 1A). Note the preponderance of Leu substitutions in CCM2. None of the missense mutations listed in Table 1 were reported in normal controls except for 1 of 13,000 alleles for KRIT1 R16C. In addition, none of these missense mutations were observed at PreventionGenetics in more than one patient, except for CCM2 L155P, which was observed in several affected members of a large Australian kindred and which we therefore classify as likely pathogenic. The CCM2 L113P mutation was reported in a patient by Riant et al. (12), but the clinical significance of this mutation was listed as unknown. Similarly, the KRIT1 L238F mutation was previously reported in one family (42). Finally, the pathogenic mechanism of the KRIT1 D281G mutation was reported to be through the introduction of a new, alternative splice donor site (14). The clinical significance of the remaining missense mutations listed in Table 1 remains uncertain.
TABLE 1.
Amino acid substitutions found in CCM patients
| KRIT1 (NM_194456.1) | CCM2 (NM_031443.3) |
|---|---|
| R16C (c.46C→T) | L113P (c.338T→C) |
| G101R (c.301G→A) | L115R (c.344T→G) |
| L238F (c.712C→T) | L155P (c.464T→C) |
| V244L (c.730G→T) | L213P (c.638T→C) |
| D281G (c.842A→G) | F270L (c.810C→G) |
| Q473H (c.1419A→C) | |
| L667R (c.2000T→G) |
Two of the KRIT1 mutations are in regions outside of the known functional domains of the KRIT1 protein and therefore may not have a structural or functional impact; KRIT1 residue Leu238 lies between KRIT1NPX(Y/F)2 and KRIT1NPX(Y/F)3 C-terminal to the KRIT1NPX(Y/F)2 binding site for SNX17 (35). Residue Asp281 lies in a predicted flexible region between NPX(Y/F)3 and the N terminus of the KRIT1 ankyrin repeat domain (Fig. 1A). Mutations of R16C and G101R are within surface-exposed regions of the Nudix domain that are not known to be important for KRIT1 function (Fig. 1B). The Q473H mutation is within the F1 lobe of KRIT1 and is one of the residues used in its interaction with HEG1, so this mutation could cause disruption of the HEG1 interaction. The L667R mutation may result in disruption of the hydrophobic core of the F3 lobe of the KRIT1 FERM domain, potentially decreasing the affinity of protein-protein interactions or reducing KRIT1 solubility (Fig. 1C). The V244L mutation in KRIT1 is close to its third NPX(Y/F) motif (Fig. 1A), and it is therefore unclear whether this mutation would affect KRIT1 associations with its binding partners.
Interestingly, the majority of the CCM2 missense mutations fall within its predicted PTB domain (Fig. 1A). Although the predicted CCM2 PTB domain is believed to be critical for the interaction of CCM2 with KRIT1, it has not been structurally characterized; nor has its interaction with KRIT1 been fully mapped. We therefore decided to undertake further studies to investigate the molecular level basis of the CCM2 interaction with KRIT1 to better understand these disease-associated mutations.
CCM2 Interacts with the NPX(Y/F) Region of KRIT1
CCM2 and KRIT1 have been shown to interact with one another by both co-immunoprecipitation (co-IP) (33) and yeast two-hybrid screen (34). Although studies have pointed toward the possible importance of the KRIT1 NPX(Y/F) motifs in interacting with the CCM2 PTB domain (33), a consensus has not been reached on which if any of these motifs are needed to bind to CCM2. To probe the basis for the CCM2-KRIT1 interaction, we began by conducting co-immunoprecipitation experiments for full-length CCM2 and KRIT1. In agreement with previous reports, we observe a robust interaction between the two wild-type proteins (Fig. 2B). We generated mutations in each of the three NPX(Y/F) motifs within full-length KRIT1 (KRIT1NPX(Y/F)1 mutant Y195A, KRIT1Y195A; KRIT1NPX(Y/F)2 mutant F234A, KRIT1F234A; KRIT1NPX(Y/F)3 mutants Y252A/F253A, KRIT1Y252A/F253A) (Fig. 2A), but we did not observe an appreciable effect for any of these mutations individually. It was not surprising that mutation of KRIT1NPX(Y/F)1 did not reduce the interaction with CCM2, because this motif has previously been shown to be used for the KRIT1 association with ICAP1 (25), and competition between ICAP1 and CCM2 for binding to KRIT1 has not been observed previously (33). When we introduced a double mutation in both the second and third motifs (KRIT1F234A/Y252A/F253A), however, we did observe a reduction in co-IP (Fig. 2B), supporting previous data (34) and suggesting that the NPX(Y/F) motifs of KRIT1 are involved in its interaction with the CCM2 PTB domain. It was, however, unclear from this overexpressed co-IP system whether CCM2 shows a preference for KRIT1NPX(Y/F)2, for KRIT1NPX(Y/F)3, or if it binds similarly to both.
FIGURE 2.
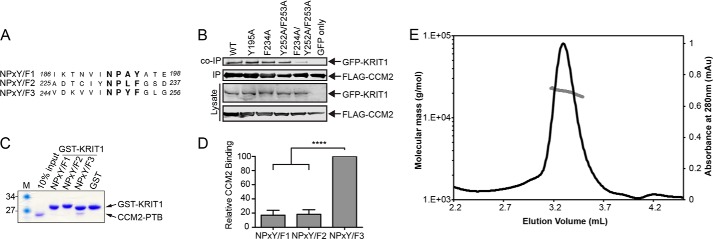
Mutations in KRIT1NPX(Y/F)2 and KRIT1NPX(Y/F)3 reduce the KRIT1 interaction with CCM2 and CCM2 preferentially interacts with KRIT1NPX(Y/F)3in vitro. A, sequence alignment of the three KRIT1 NPX(Y/F) motifs. Residue numbers are indicated. B, co-immunoprecipitation experiments were conducted with FLAG-tagged CCM2 and GFP-tagged KRIT1. Experiments were performed with wild-type CCM2 and either WT KRIT1 or mutants within its NPX(Y/F) motifs (KRIT1NPX(Y/F)1 mutant, Y195A; KRIT1NPX(Y/F)2 mutant, F234A; KRIT1NPX(Y/F)3 mutants, Y252A/F253A; KRIT1NPX(Y/F)2 and KRIT1NPX(Y/F)3 mutants, F234A/Y252A/F253A) using GFP only as a negative control. C, representative pull-down assay. GST fusion KRIT1 NPX(Y/F) motifs were immobilized on beads and incubated with purified CCM2PTB. Coomassie staining is shown. D, quantification of pull-down experiments (n = 5) expressed as a percentage of wild-type CCM2PTB binding within each experiment and averaged across experiments. Error bars, S.E.; ****, p < 0.0001. E, size exclusion chromatography with multiangle light scattering analysis of CCM2PTB-KRIT1NPX(Y/F)3 complex shows that it elutes as a peak of molar mass 20.8 kDa ± 1.1%.
CCM2 Exhibits Binding Preference for KRIT1NPX(Y/F)3 Motif in Vitro
We therefore decided to explore whether purified CCM2 PTB domain exhibits a binding preference to any of the KRIT1 NPX(Y/F) motifs in an in vitro system with purified proteins by conducting pull-down experiments. We expressed and purified the PTB domain of CCM2 encoding residues 51–228 (CCM2PTB) and designed GST fusion constructs corresponding to each of the three NPX(Y/F) motifs in KRIT1: GST-KRIT1NPX(Y/F)1, GST-KRIT1NPX(Y/F)2, and GST-KRIT1NPX(Y/F)3 (Fig. 2A). We expressed and purified the GST-KRIT1 NPX(Y/F) motif proteins and bound them to glutathione-Sepharose beads. We then tested the ability of purified CCM2PTB to interact with each of the KRIT1 NPX(Y/F) motifs. We found that CCM2PTB directly interacts with KRIT1NPX(Y/F)3 and that this interaction is stronger than with either KRIT1NPX(Y/F)1 or KRIT1NPX(Y/F)2 (Fig. 2, C and D). These are the first data to suggest that CCM2 exhibits a preference among the KRIT1 NPX(Y/F) motifs.
Structural Basis for the CCM2 Interaction with KRIT1
The structure of the predicted CCM2 PTB domain, as well as its mode of binding to KRIT1, was previously unknown. Based on our pull-down experiments, we therefore set out to determine the structure of CCM2PTB in complex with a 13-residue peptide corresponding to KRIT1NPX(Y/F)3 (244VDKVVINPYFGLG256). We analyzed this complex by size exclusion chromatography with multiangle light scattering and found that it elutes as a monodisperse peak with a molecular mass of 20.8 kDa ± 1.1%, consistent with a 1:1 CCM2PTB-KRIT1NPX(Y/F)3 complex with an expected molecular mass of 20.8 kDa (Fig. 2E). We conducted co-crystallization trials and obtained a 2.75 Å resolution (I/σI is 1.2 at 2.75 Å and 2.75 at 2.96 Å resolution) data set that allowed structure determination by the molecular replacement method. Four copies of the CCM2PTB-KRIT1NPX(Y/F)3 complex are observed per asymmetric unit (Tables 2 and 3). The peptide chains for CCM2PTB and the KRIT1NPX(Y/F)3 peptide were first built using automated procedures and subsequently refined by manual model building. Because the register of each of the chains was clear, this allowed for complete assignment of built residues (Fig. 3B). We find that CCM2PTB adopts a Dab-like PH/PTB fold that interacts with KRIT1NPX(Y/F)3 in 1:1 stoichiometry (Fig. 3A). As predicted by structure-based sequence alignment (43), CCM2PTB has the highest structural homology to ICAP1, a protein that binds both integrin β1 cytoplasmic tail and KRIT1NPX(Y/F)1 (25, 26) (root mean square deviation 2.5 Å over 131 residues and a z score of 17.7 calculated by the Dali server (44)). Based on our crystal structure, CCM2PTB comprises residues Ser60–Tyr221 with a 32-residue flexible loop (loop β6/β7) inserted between strands β6 and β7 (residues Asp160–Glu191), for which we do not observe density in any of the four copies of CCM2PTB (Fig. 3A). Like other PH/PTB fold domains, CCM2PTB comprises a C-terminal α-helix and seven β-strands arranged in two anti-parallel β-sheets with strand β1 a constituent of both β-sheets (β1-β7-β6-β5 and β1-β2-β3-β4). Similar to other Dab-like PTB domains, CCM2PTB contains an additional α-helix, α1, inserted between strands β1 and β2, and a 310 helix, which we term helix α1′, connecting strands β4 and β5. We do not observe a basic patch at the PH fold phosphoinositide headgroup binding site located between loops β1/β2, β3/β4, and β6/β7 (30) or the Dab-like headgroup binding site (30). Our structure therefore places the PTB domain of CCM2 into the Dab-like family of NPX(Y/F/pY) motif-binding PTB domains and suggests that it does not interact with phosphoinositide headgroups. Both the surface of CCM2 that interacts with KRIT1 and the sequence of KRIT1NPX(Y/F)3 itself are well conserved over evolution (Fig. 3, C and D). Interestingly, there is reduced sequence conservation in the 32-residue flexible β6/β7 loop of CCM2, and serine residues (Ser164, Ser166, and Ser168) in this loop have previously been found to be phosphorylated (45).
TABLE 2.
Data collection and refinement statistics for the CCM2PTB-KRIT1NPx(Y/F)3 complex
Parentheses indicate highest resolution shell, and square brackets indicate statistics for a shell where I/σ(I) is 2.75.
| Data collection | |
| Space group | P6522 |
| X-ray source | APS 24-ID-E |
| Cell dimensions, a, b, c (Å) | 110.9, 110.9, 315.2 |
| α, β, γ (degrees) | 90, 90, 120 |
| Wavelength (Å) | 0.97926 |
| Resolution range (Å) | 30–2.75 (2.85–2.75) [3.1–2.96] |
| No. of unique reflections | 30,768 |
| Degrees of data (degrees) | 180 |
| Completeness (%) | 100 (100) [100] |
| Rpim (%) | 4.0 (106.5) [41.9] |
| I/σ(I) | 44 (1.2) [2.75] |
| Redundancy | 17.9 (19.0) [18.6] |
| Wilson B-factor (Å2) | 93.7 |
| Refinement | |
| Resolution range (Å) | 29.9–2.75 (2.83–2.75) |
| Rfactor (%) | 22.0 (36.1) |
| Free Rfactor (%) | 25.0 (44.2) |
| No. of free R reflections | 1809 (106) |
| Free R reflections (%) | 6.5 (7.4) |
| Residue range built | CCM2: A/55–221, B/60–221, C/61–220, D/63–217 |
| KRIT1: W/244–254, X/244–255, Y/244–254, Z/244–255 | |
| No. of non-hydrogen protein atoms | 4428 |
| No. of water molecules | 10 |
| Model quality | |
| Root mean square deviation bond lengths (Å) | 0.004 |
| Root mean square deviation bond angles (degrees) | 0.868 |
| Overall B (all atoms) | 102.7 |
| B (protein) | CCM2: A/86.2, B/89.4, C/96.0, D/143.0 |
| KRIT1: W/76.8, X/71.3, Y/104, Z/161.6 | |
| B (water) | 69.1 |
| Ramachandran plot (%): favored/allowed/outliers | 96.9/3.1/0.0 |
| MolProbity score/Percentile | 2.08/98% |
TABLE 3.
Data collection statistics by resolution shell
| Resolution bins |
No. of reflections | Average redundancy | Average I | Average error | Rmerge | Rpim | |
|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||
| 30.0 | 5.91 | 3390 | 16.50 | 299.4 | 6.8 | 0.037 | 0.009 |
| 5.91 | 4.70 | 3163 | 16.64 | 103.7 | 3.3 | 0.093 | 0.023 |
| 4.70 | 4.11 | 3091 | 17.24 | 124.7 | 4.0 | 0.098 | 0.024 |
| 4.11 | 3.73 | 3084 | 17.66 | 59.5 | 2.3 | 0.158 | 0.038 |
| 3.73 | 3.46 | 3046 | 17.98 | 40.7 | 2.4 | 0.250 | 0.060 |
| 3.46 | 3.26 | 3034 | 18.15 | 25.9 | 2.4 | 0.411 | 0.099 |
| 3.26 | 3.10 | 3026 | 18.46 | 14.7 | 2.3 | 0.734 | 0.174 |
| 3.10 | 2.96 | 3027 | 18.64 | 6.6 | 2.4 | 1.771 | 0.419 |
| 2.96 | 2.85 | 3001 | 18.78 | 4.2 | 2.4 | 3.196 | 0.753 |
| 2.85 | 2.75 | 2958 | 18.98 | 2.9 | 2.4 | 4.543 | 1.065 |
| All reflections | 30,820 | 17.88 | 71.3 | 3.1 | 0.165 | 0.040 | |
FIGURE 3.

Co-crystal structure of CCM2 PTB domain in complex with the KRIT1NPX(Y/F)3. A, overall schematic format view of the CCM2PTB-KRIT1NPX(Y/F)3 co-crystal structure. The CCM2 PTB domain is shown in green with secondary structure elements labeled. KRIT1 is shown in yellow. N and C termini are indicated. Dotted line, disordered loop β6-β7. Structural figures were generated using CCP4mg (50). B, stereoview of electron density map for CCM2 PTB domain in complex with KRIT1NPX(Y/F)3. The simulated annealing omit map contoured at 1.5 σ shown in purple was calculated using Phenix. The CCM2 PTB domain is shown in green stick format, and KRIT1NPX(Y/F)3 is shown in yellow stick format. C, conservation of CCM2 PTB domain calculated based on analysis conducted using the Consurf server (51). CCM2PTB is shown in a surface representation. Dark blue, complete conservation; lighter shades, less conservation. The KRIT1 peptide is depicted in a yellow stick format. The structure shown in same orientation as in A. D, conservation of KRIT1NPX(Y/F)3 motif over 29 species. Yellow triangles indicate residues involved in the interaction with the CCM2 PTB domain. A red star indicates residues that are mutated in CCM disease. Residues mutated in this study are indicated by blue stars. The Uniprot or GenBankTM accession number is followed by the Latin name and label in parentheses: O00522, Homo sapiens (Human); Q6S5J6, Mus musculus (Mouse); Q6TNJ1, Bos taurus (Cow); B5DF47, Rattus norvegicus (Rat); B8JIZ5, Danio rerio (Zebrafish); F7I3T9, Callithrix jacchus (Marmoset); G3X3N7, Sarcophilus harrisii (Tasmanian-devil); G1RZ72, Nomascus leucogenys (Gibbon); F7CJC5, Xenopus tropicalis (Frog); G1N921, Meleagris gallopavo (Turkey); E2RAA5, Canis familiaris (Dog); F6TVZ5, Macaca mulatta (Macaque); F7CJF5, Monodelphis domestica (Opossum); G3QHP7 Gorilla gorilla (Gorilla); G1SM20, Oryctolagus cuniculus (Rabbit); F1SFD0, Sus scrofa (Pig); G3TAH0, Loxodonta africana (Elephant); G1KRC6, Anolis carolinensis (Chameleon); G3NJI7, Gasterosteus aculeatus (Stickleback); G1PWA1, Myotis lucifugus (Bat); C0H9F0, Salmo salar (Atlantic-Salmon); XP_001166592, Pan troglodytes (Chimpanzee); XP_002925462, Ailuropoda melanoleuca (Giant-panda); XP_001491579, Equus caballus (Horse); XP_003475143, Cavia porcellus (Guinea-pig); XP_003496996, Cricetulus griseus (Hamster); XP_002196126, Taeniopygia guttata (Zebra-finch); NP_001026144, Gallus gallus (Chicken); XP_003452511, Oreochromis niloticus (Nile-tilapia).
We clearly observe binding of a KRIT1NPX(Y/F)3 peptide to each of the four CCM2PTB molecules in the asymmetric unit, with KRIT1 residues Val244–Leu254 built in each copy (Fig. 4A). The mode of binding we observe is similar to other structures of canonical PTB domains in complex with NPX(Y/F) motifs (30). A β-strand is formed by KRIT1 residues Lys246–Ile249 that hydrogen-bonds to β5 of CCM2PTB, extending the β1-β7-β6-β5 anti-parallel β-sheet. This KRIT1 β-strand also stacks against helix α2 of CCM2PTB (Fig. 4B). At the C terminus of KRIT1NPX(Y/F)3, the NPYF motif forms a type III turn and packs against helix α1′. Multiple hydrophobic and hydrogen-bonding contacts are made between KRIT1 and CCM2. Of note, KRIT1 residue Val248 packs snugly against CCM2 helix α2 between residues Cys210 and Phe217; KRIT1 residues Val244 and Val247 pack against the turn N-terminal to CCM2 helix α1 comprising residues Pro82, Ser83, and Ser84; and KRIT1 residue Phe253 fits between the CCM2 310 helix α1′ and the N terminus of strand β5 (Fig. 4, B and C). The interface between CCM2PTB and KRIT1NPX(Y/F)3 comprises eight or nine hydrogen bonds for each of the four copies and buries an average of 1359 Å2 (between 576 and 637 Å2 for the four copies of CCM2PTB and between 700 and 802 Å2 for the four copies of KRIT1NPX(Y/F)3) as calculated by PDBsum (46).
FIGURE 4.

Molecular interactions between the CCM2 PTB domain and KRIT1NPX(Y/F)3. A, superposition of the four copies of the CCM2 PTB domain with KRIT1NPX(Y/F)3 found in the asymmetric unit. Chains A–D are CCM2 PTB molecules, and chains W–Z are the KRIT1 peptides to which they bind. B, close-up of KRIT1 interactions with CCM2. Stick format is used for KRIT1 and CCM2 residues discussed in this work. Residues labeled in blue are mutated in this study, and the underlined label indicates CCM disease-associated mutation. C, schematic map of interactions between KRIT1 and CCM2. Dashed red lines, hydrogen bonds; blue lines, non-bonded contacts. Secondary structure elements are labeled. Blue labels, residues mutated in this study; underlined label, CCM disease-associated mutation. All interactions shown are observed in at least three of the four copies in the asymmetric unit, with the exception of KRIT1 residue Pro251, which makes contacts with CCM2 in only one copy.
Structure-guided Mutagenesis to Disrupt CCM2PTB-KRIT1NPX(Y/F)3 Interaction
We conducted biolayer interferometry to measure the affinity of the interaction between CCM2PTB and GST-KRIT1NPX(Y/F)3 and observed a KD of 13.0 ± 2.5 μm (Fig. 5A). This affinity value is similar to measurements previously observed for PTB domain interactions with NPX(Y/F) motifs (47–49). We were not able to observe binding for CCM2PTB to GST-KRIT1NPX(Y/F)1 or GST-KRIT1NPX(Y/F)2 (Fig. 5A and Table 4). Longer GST fusions encompassing both KRIT1 NPX(Y/F)2 and NPX(Y/F)3 were severely proteolyzed upon expression in E. coli, so to compare the affinity of CCM2PTB for the entire NPX(Y/F)2-3 region of KRIT1 with the individual NPX(Y/F)2 and NPX(Y/F)3 motifs, we synthesized biotinylated KRIT1 peptides encompassing NPX(Y/F)2, NPX(Y/F)3, or NPX(Y/F)2 and NPX(Y/F)3 (KRIT1Biotin-NPX(Y/F)2, KRIT1Biotin-NPX(Y/F)3, and KRIT1Biotin-NPX(Y/F)2-3, respectively). We again used the biolayer interferometry method to measure CCM2PTB affinities for these biotinylated synthesized KRIT1 peptides. We found that although we could not detect a measurable interaction between CCM2PTB and KRIT1Biotin-NPX(Y/F)2, the affinities of CCM2PTB for binding to either KRIT1Biotin-NPX(Y/F)3 or KRIT1Biotin-NPX(Y/F)2-3 were very similar (15.5 ± 4.0 and 14.5 ± 2.9 μm, respectively) and also similar to that measured for the GST-KRIT1NPX(Y/F)3 protein expressed in E. coli (Table 4). To validate the structurally defined interaction between CCM2PTB and KRIT1NPX(Y/F)3, we used a pull-down assay where GST-fused KRIT1 peptide was bound to glutathione-Sepharose beads and purified CCM2PTB was added. Based on the crystal structure, we then introduced point mutations that we predicted would disrupt the CCM2PTB-KRIT1NPX(Y/F)3 interaction (indicated with blue labels in Fig. 4B). In KRIT1, we introduced a mutation of V248D to interrupt the hydrophobic interaction with CCM2 helix α2. We also mutated the NPYF motif to NPAA (Y252A/F253A) as we had done for the full-length protein because mutation of the NPX(Y/F) motifs is often used to interrupt their interactions with PTB domains. These mutations significantly impaired the ability of CCM2PTB to pull down with GST-KRIT1NPX(Y/F)3 (Fig. 5, B and C). In CCM2, we introduced three sets of mutations: C210D, I136A/I139A, and F217A. Cys210 is located within CCM2 helix α2 and oriented toward the KRIT1 binding site, so we hypothesized that its mutation would interrupt the CCM2-KRIT1 interaction. Ile136 and Ile139 are located at the N and C termini of the 310 helix α1′ that interacts with KRIT1NPX(Y/F)3, so we expected that this double alanine mutation would destabilize the helix and consequent binding to KRIT1. Phe217 is at the C terminus of helix α2 stacking against KRIT1 residue Asn250, and its mutation has previously been shown to reduce the ability of CCM2 to bind KRIT1 (32). Each of these mutations significantly impaired the ability of CCM2PTB to pull down with GST-KRIT1NPX(Y/F)3 (Fig. 5, D and E). Introduction of the crystallographically defined mutations C210D and I136A/I139A into full-length CCM2 also interrupted the interaction by co-IP (Fig. 5F). Therefore, because a single point mutation in the PTB domain of CCM2 disrupts binding to KRIT1, we conclude that CCM2 contains a single binding site for KRIT1.
FIGURE 5.

Structure-guided interruption of the CCM2 interaction with KRIT1. A, binding curve for CCM2PTB interaction with GST-KRIT1NPX(Y/F)3 (circles) measured by BLItz. KD = 13 μm ± 2.5. Binding for CCM2PTB interaction with GST-KRIT1NPX(Y/F)1 (triangle) and GST-KRIT1NPX(Y/F)2 (diamond) is shown at 285 μm. No measurable binding was observed for either of these constructs. B, representative experiment showing CCM2PTB pull-down by GST-KRIT1NPX(Y/F)3 and by GST-KRIT1NPX(Y/F)3 mutants predicted to interrupt the interaction. KRIT1 mutants Y252A/F253A and V248D are shown. C, quantification of pull-down experiments from B (n = 5) expressed as a percentage of wild-type CCM2PTB binding within each experiment and averaged across experiments. Error bars, S.E.; ****, p < 0.0001. D, representative pull-down for CCM2PTB pull-down by GST-KRIT1NPX(Y/F)3 showing the effect of mutations in CCM2PTB on binding to KRIT1. CCM2 mutations CCM2F217A (F217A), CCM2C210D (C210D), and CCM2I136A/I139A (II/AA) are shown. E, quantification of pull-down experiments from D (n = 3) expressed as percentage of wild-type CCM2PTB binding within each experiment and averaged across experiments. Error bars, S.E.; ****, p < 0.0001. F, co-IP using GFP-tagged wild-type full-length KRIT1 and FLAG-tagged full-length CCM2 as wild type or with mutations within the PTB domain (I136A/I139A and C210D) and FLAG only as a negative control.
TABLE 4.
Binding affinity measurements of CCM2PTB for KRIT1 NPX(Y/F) motifs. Biolayer interferometry affinity measurements for purified CCM2PTB binding to immobilized KRIT1 NPX(Y/F) motifs. KRIT1 samples were either E. coli expressed GST fusion proteins or N-terminally biotinylated synthesized peptides. n.b., no measurable interaction
| KRIT1 immobilized | Binding affinity for CCM2PTB |
|
|---|---|---|
| KD | 95% confidence interval | |
| GST-NPX(Y/F)1 | n.b. | n.b. |
| GST-NPX(Y/F)2 | n.b. | n.b. |
| GST-NPX(Y/F)3 | 13.0 ± 2.5 μm | 7.8 to 18.1 μm |
| Biotin-NPX(Y/F)2 | n.b. | n.b. |
| Biotin-NPX(Y/F)3 | 15.5 ± 4.0 μm | 6.9 to 24.1 μm |
| Biotin-NPX(Y/F)2-3 | 14.5 ± 2.9 μm | 8.0 to 21.0 μm |
Missense Point Mutations Destabilize the CCM2 PTB Domain
Having shown through biochemical and biophysical assays that KRIT1NPX(Y/F)3 is the predominant site of interaction and having determined the structure of the CCM2 PTB domain in complex with the KRIT1NPX(Y/F)3 peptide, we then asked where the CCM2 disease-associated mutations identified here and elsewhere map to the structure of the PTB domain and whether they affect KRIT1 binding. In addition to the L113P, L115R, L155P, and L213P point mutations identified through our sequencing efforts, other studies have independently found a L198R (13, 33) as well as additional L113P (12) mutations in CCM2 in patient populations. The L198R mutation has been shown to reduce the interaction with KRIT1 (33), but the molecular basis for this is unknown. The effect of the L113P mutation has not yet been analyzed. To better understand the impact of missense mutations in CCM2 and KRIT1, we mapped CCM2 mutations L113P, L115R, L155P, L198R, and L213P and KRIT1 mutation V244L onto our co-crystal structure. Each of these CCM2 leucine residues is located within the hydrophobic core of CCM2PTB, where they interact with one another to form a tight hydrophobic cluster (Fig. 6A). Based on the structural mapping, we postulated that introduction of the CCM2 mutations could negatively affect protein stability. We therefore generated L113P, L115R, L155P, L198R, and L213P point mutations in our His6-CCM2PTB construct and tested their solubility. We found that introduction of each of these mutations resulted in significantly reduced solubility upon overexpression in E. coli (Fig. 6B), suggesting a negative impact on the structural integrity of CCM2PTB. In contrast, KRIT1 residue Val244 packs tightly into a pocket formed by the CCM2 PTB domain N terminus of helix α1, Ser83, and strand β5 (Fig. 6A). To test the impact of the KRIT1 mutant V244L, we introduced the mutation into our GST-KRIT1NPX(Y/F)3 construct and conducted pull-down assays with CCM2PTB. We found that the KRIT1 V244L mutation causes a significant reduction in the ability of KRIT1NPX(Y/F)3 to pull down CCM2 (Fig. 6, C and D), and we were not able to detect binding for this interaction by biolayer interferometry (data not shown). These results suggest that these disease-associated missense mutations in CCM2 or KRIT1 either disrupt protein solubility and stability or destabilize the binding between CCM2 and KRIT1.
FIGURE 6.

Missense point mutations destabilize CCM2 PTB domain and weaken its interaction with KRIT1. A, disease-associated point mutations within the CCM2 PTB domain mapped onto the structure. Top, close-up view of CCM2 disease-related mutations (L113P, L115R, L155P, L198R, and L213P), all within the hydrophobic core of the PTB domain. Bottom, disease-related mutation V244L in KRIT1 N-terminal to the third NPX(Y/F) motif. B, solubility tests for missense mutations discovered in CCM disease. CCM2PTB is expressed in both the soluble fraction and insoluble fractions, but the CCM disease-associated mutants (L113P, L115R, L155P, L198R, and L213P) are mostly found in the insoluble fraction. M, marker. C, representative KRIT1 V244L pull-down assay. D, quantification of pull-down experiments from B (n = 5) expressed as a percentage of wild-type CCM2PTB binding within each experiment and averaged across experiments. Error bars, S.E.; ****, p < 0.0001.
DISCUSSION
The disease cerebral cavernous malformations is closely linked to mutations in the KRIT1, CCM2, and PDCD10/CCM3 genes and their resulting proteins KRIT1, CCM2, and CCM3. We investigated the impact of germ line mutations in patients who harbor multiple CCM lesions and discovered missense mutations in both KRIT1 and CCM2, with many of the CCM2 missense mutations clustering within its PTB domain. Because the molecular basis for how CCM2 and KRIT1 interact was not known and was the subject of controversy, we mapped the interaction, determined its structural basis, and validated it using in vitro assays. We then conducted further analysis of the CCM2 and KRIT1 missense mutations in the context of these new data, finding that many of these disease-associated mutations have a negative impact on folding of the CCM2 PTB domain or affect its associated with KRIT1. Our study therefore provides a significant improvement in our understanding of the molecular level impact of CCM-associated mutations.
In the in vitro system we used to investigate the interactions of the purified CCM2 PTB domain with each of the individual KRIT1 NPX(Y/F) motifs, we see a clear preference of CCM2 for binding to KRIT1NPX(Y/F)3 and cannot detect binding of the CCM2 PTB domain to either isolated KRIT1NPX(Y/F)1 or KRIT1NPX(Y/F)2 (Fig. 2, C and D). The affinity measured for CCM2 PTB interaction with KRIT1NPX(Y/F)3 is within the range previously observed for PTB interactions with NPX(Y/F) motifs (47–49) and is also similar to the affinity we measured with KRIT1NPX(Y/F)2-3. Additionally, our structure-directed point mutations within CCM2 that interrupt the interaction with KRIT1 show that CCM2 utilizes a single binding site within its PTB domain to interact with KRIT1. We therefore conclude that the predominant interaction between KRIT1 and CCM2 is by the CCM2 PTB domain binding to KRIT1NPX(Y/F)3 via a canonical PTB-NPX(Y/F) motif binding site. In the overexpression system we use in Fig. 2B, however, we observe that to interrupt KRIT1 interaction with CCM2 by co-immunoprecipitation, there seems to be a requirement to simultaneously mutate both the KRIT1NPX(Y/F)2 and KRIT1NPX(Y/F)3 motifs. This is in accordance with previous yeast two-hybrid studies suggesting that CCM2 has no clear preference among the KRIT1 NPX(Y/F) motifs (34) and could be an artifact of this overexpression system. Although our biochemical studies do not suggest a role for the KRIT1NPX(Y/F)2 in this interaction, they do not rule out the possibility of weaker secondary KRIT1 interaction sites for CCM2, so further studies to explore the role of a potential secondary interaction of CCM2 with KRIT1 could be merited.
Our structural analysis provides the first atomic resolution picture of the N-terminal PTB domain of CCM2, confirming previous predictions that CCM2 contains a PH/PTB domain fold in the Dab-like subfamily. Among other PTB domains, the CCM2 PTB domain has highest structural similarity to that of ICAP1 (25). In contrast to previous predictions, we do not observe canonical or non-canonical phosphoinositide binding sites on the CCM2 PTB structure (43), a finding that may impact ongoing research into potential membrane recruitment of CCM2. This structure additionally shows that the CCM2 PTB domain interacts with the third NPX(Y/F) motif of KRIT1 via a canonical PTB-NPX(Y/F) interaction. It is also possible that the CCM2 PTB domain interacts with other CCM2 binding partners, such as SMURF1 (28). If other proteins can bind at this site using a similar mode of binding, it will also be interesting to determine whether there is competition among CCM2 binding partners.
Interestingly, a number of the patient-derived mutations identified in our large scale sequencing effort result in missense mutation of leucine amino acids within the hydrophobic ore of the CCM2 PTB domain to either proline or arginine. Our solubility tests show that these mutations destabilize the CCM2 PTB domain, potentially implying that these mutations act as pseudononsense mutations. In contrast, the large scale sequencing of KRIT1 revealed a missense mutation, V244L, within the CCM2-binding region N-terminal of KRIT1NPX(Y/F)3, a location that had not previously been discovered to be mutated in CCM disease. We find that Val244 is important for the interaction of KRIT1 and CCM2, and even a conservative mutation to leucine results in weakening the interaction between CCM2 and KRIT1.
Taken together, our results provide a molecular level understanding of how the CCM complex is formed around the CCM2-KRIT1 interaction. The results also suggest that proper folding of CCM2 is lost for some of the missense mutations found in CCM disease patients. Therefore, we anticipate that our data will allow for an improved understanding of the cellular function of the CCM2-KRIT1 interaction and other CCM2 PTB domain interactions, providing the foundation for future studies into the mechanisms by which mutations in KRIT1 and CCM2 might lead to CCM disease.
Acknowledgments
We thank Xiaofeng Li, Wang Min, David Calderwood, and Sven-Eric Jordt as well as the Northeastern Collaborative Access Team at the Advanced Photon Source.
This work was supported, in whole or in part, by National Institutes of Health Grant NS085078 (to T. J. B.) and National Institutes of Health Training Grant GM007324 (to O. S. F.).
- CCM
- cerebral cavernous malformation
- PTB
- phosphotyrosine binding
- FERM
- band 4.1, ezrin, radixin, moesin
- IP
- immunoprecipitation
- PH
- pleckstrin homology.
REFERENCES
- 1. Otten P., Pizzolato G. P., Rilliet B., Berney J. (1989) [131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies]. Neurochirurgie 35, 82–83, 128–131 [PubMed] [Google Scholar]
- 2. Fischer A., Zalvide J., Faurobert E., Albiges-Rizo C., Tournier-Lasserve E. (2013) Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol. Med. 19, 302–308 [DOI] [PubMed] [Google Scholar]
- 3. Akers A. L., Johnson E., Steinberg G. K., Zabramski J. M., Marchuk D. A. (2009) Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis. Hum. Mol. Genet. 18, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pagenstecher A., Stahl S., Sure U., Felbor U. (2009) A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum. Mol. Genet. 18, 911–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gault J., Shenkar R., Recksiek P., Awad I. A. (2005) Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke 36, 872–874 [DOI] [PubMed] [Google Scholar]
- 6. Sahoo T., Johnson E. W., Thomas J. W., Kuehl P. M., Jones T. L., Dokken C. G., Touchman J. W., Gallione C. J., Lee-Lin S. Q., Kosofsky B., Kurth J. H., Louis D. N., Mettler G., Morrison L., Gil-Nagel A., Rich S. S., Zabramski J. M., Boguski M. S., Green E. D., Marchuk D. A. (1999) Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet. 8, 2325–2333 [DOI] [PubMed] [Google Scholar]
- 7. Laberge-le Couteulx S., Jung H. H., Labauge P., Houtteville J. P., Lescoat C., Cecillon M., Marechal E., Joutel A., Bach J. F., Tournier-Lasserve E. (1999) Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat. Genet. 23, 189–193 [DOI] [PubMed] [Google Scholar]
- 8. Liquori C. L., Berg M. J., Siegel A. M., Huang E., Zawistowski J. S., Stoffer T., Verlaan D., Balogun F., Hughes L., Leedom T. P., Plummer N. W., Cannella M., Maglione V., Squitieri F., Johnson E. W., Rouleau G. A., Ptacek L., Marchuk D. A. (2003) Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 73, 1459–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guclu B., Ozturk A. K., Pricola K. L., Bilguvar K., Shin D., O'Roak B. J., Gunel M. (2005) Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery 57, 1008–1013 [DOI] [PubMed] [Google Scholar]
- 10. Bergametti F., Denier C., Labauge P., Arnoult M., Boetto S., Clanet M., Coubes P., Echenne B., Ibrahim R., Irthum B., Jacquet G., Lonjon M., Moreau J. J., Neau J. P., Parker F., Tremoulet M., Tournier-Lasserve E. (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 76, 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stahl S., Gaetzner S., Voss K., Brackertz B., Schleider E., Sürücü O., Kunze E., Netzer C., Korenke C., Finckh U., Habek M., Poljakovic Z., Elbracht M., Rudnik-Schöneborn S., Bertalanffy H., Sure U., Felbor U. (2008) Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum. Mutat. 29, 709–717 [DOI] [PubMed] [Google Scholar]
- 12. Riant F., Cecillon M., Saugier-Veber P., Tournier-Lasserve E. (2013) CCM molecular screening in a diagnosis context: novel unclassified variants leading to abnormal splicing and importance of large deletions. Neurogenetics 14, 133–141 [DOI] [PubMed] [Google Scholar]
- 13. Denier C., Goutagny S., Labauge P., Krivosic V., Arnoult M., Cousin A., Benabid A. L., Comoy J., Frerebeau P., Gilbert B., Houtteville J. P., Jan M., Lapierre F., Loiseau H., Menei P., Mercier P., Moreau J. J., Nivelon-Chevallier A., Parker F., Redondo A. M., Scarabin J. M., Tremoulet M., Zerah M., Maciazek J., Tournier-Lasserve E. (2004) Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 74, 326–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mondéjar R., Solano F., Rubio R., Delgado M., Pérez-Sempere A., González-Meneses A., Vendrell T., Izquierdo G., Martinez-Mir A., Lucas M. (2014) Mutation prevalence of cerebral cavernous malformation genes in Spanish patients. PLoS One 9, e86286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Draheim K. M., Fisher O. S., Boggon T. J., Calderwood D. A. (2014) Cerebral cavernous malformation proteins at a glance. J. Cell Sci. 127, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fisher O. S., Boggon T. J. (2014) Signaling pathways and the cerebral cavernous malformations proteins: lessons from structural biology. Cell Mol. Life Sci. 71, 1881–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Voss K., Stahl S., Schleider E., Ullrich S., Nickel J., Mueller T. D., Felbor U. (2007) CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8, 249–256 [DOI] [PubMed] [Google Scholar]
- 18. Serebriiskii I., Estojak J., Sonoda G., Testa J. R., Golemis E. A. (1997) Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene 15, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 19. Li X., Zhang R., Draheim K. M., Liu W., Calderwood D. A., Boggon T. J. (2012) Structural basis for small G protein effector interaction of Ras-related protein 1 (Rap1) and adaptor protein Krev interaction trapped 1 (KRIT1). J. Biol. Chem. 287, 22317–22327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gingras A. R., Puzon-McLaughlin W., Ginsberg M. H. (2013) The structure of the ternary complex of Krev interaction trapped 1 (KRIT1) bound to both the Rap1 GTPase and the Heart of Glass (HEG1) cytoplasmic tail. J. Biol. Chem. 288, 23639–23649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gingras A. R., Liu J. J., Ginsberg M. H. (2012) Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J. Cell Biol. 199, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kleaveland B., Zheng X., Liu J. J., Blum Y., Tung J. J., Zou Z., Sweeney S. M., Chen M., Guo L., Lu M. M., Zhou D., Kitajewski J., Affolter M., Ginsberg M. H., Kahn M. L. (2009) Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat. Med. 15, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zawistowski J. S., Serebriiskii I. G., Lee M. F., Golemis E. A., Marchuk D. A. (2002) KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis. Hum. Mol. Genet. 11, 389–396 [DOI] [PubMed] [Google Scholar]
- 24. Zhang J., Clatterbuck R. E., Rigamonti D., Chang D. D., Dietz H. C. (2001) Interaction between krit1 and icap1α infers perturbation of integrin β1-mediated angiogenesis in the pathogenesis of cerebral cavernous malformation. Hum. Mol. Genet. 10, 2953–2960 [DOI] [PubMed] [Google Scholar]
- 25. Liu W., Draheim K. M., Zhang R., Calderwood D. A., Boggon T. J. (2013) Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 49, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu W., Boggon T. J. (2013) Cocrystal structure of the ICAP1 PTB domain in complex with a KRIT1 peptide. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 494–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uhlik M. T., Abell A. N., Johnson N. L., Sun W., Cuevas B. D., Lobel-Rice K. E., Horne E. A., Dell'Acqua M. L., Johnson G. L. (2003) Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5, 1104–1110 [DOI] [PubMed] [Google Scholar]
- 28. Crose L. E., Hilder T. L., Sciaky N., Johnson G. L. (2009) Cerebral cavernous malformation 2 protein promotes Smad ubiquitin regulatory factor 1-mediated RhoA degradation in endothelial cells. J. Biol. Chem. 284, 13301–13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fisher O. S., Zhang R., Li X., Murphy J. W., Demeler B., Boggon T. J. (2013) Structural studies of cerebral cavernous malformations 2 (CCM2) reveal a folded helical domain at its C-terminus. FEBS Lett. 587, 272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. DiNitto J. P., Lambright D. G. (2006) Membrane and juxtamembrane targeting by PH and PTB domains. Biochim. Biophys. Acta 1761, 850–867 [DOI] [PubMed] [Google Scholar]
- 31. Li X., Zhang R., Zhang H., He Y., Ji W., Min W., Boggon T. J. (2010) Crystal structure of CCM3, a cerebral cavernous malformation protein critical for vascular integrity. J. Biol. Chem. 285, 24099–24107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hilder T. L., Malone M. H., Bencharit S., Colicelli J., Haystead T. A., Johnson G. L., Wu C. C. (2007) Proteomic identification of the cerebral cavernous malformation signaling complex. J. Proteome Res. 6, 4343–4355 [DOI] [PubMed] [Google Scholar]
- 33. Zawistowski J. S., Stalheim L., Uhlik M. T., Abell A. N., Ancrile B. B., Johnson G. L., Marchuk D. A. (2005) CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum. Mol. Genet. 14, 2521–2531 [DOI] [PubMed] [Google Scholar]
- 34. Zhang J., Rigamonti D., Dietz H. C., Clatterbuck R. E. (2007) Interaction between krit1 and malcavernin: implications for the pathogenesis of cerebral cavernous malformations. Neurosurgery 60, 353–359 [DOI] [PubMed] [Google Scholar]
- 35. Stiegler A. L., Zhang R., Liu W., Boggon T. J. (2014) Structural determinants for binding of sorting nexin 17 (SNX17) to the cytoplasmic adaptor protein Krev interaction trapped 1 (KRIT1). J. Biol. Chem. 289, 25362–25373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., Sliz P. (2013) Cutting edge: collaboration gets the most out of software. eLife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Domingues F., Gasparetto E. L., Andrade R., Noro F., Eiras A., Gault J., Correia C. E., de Souza J. M. (2008) Familial cerebral cavernous malformations: Rio de Janeiro study and review of the recommendations for management. Arq. Neuropsiquiatr. 66, 795–799 [DOI] [PubMed] [Google Scholar]
- 43. Uhlik M. T., Temple B., Bencharit S., Kimple A. J., Siderovski D. P., Johnson G. L. (2005) Structural and evolutionary division of phosphotyrosine binding (PTB) domains. J. Mol. Biol. 345, 1–20 [DOI] [PubMed] [Google Scholar]
- 44. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim J., Sherman N. E., Fox J. W., Ginsberg M. H. (2011) Phosphorylation sites in the cerebral cavernous malformations complex. J. Cell Sci. 124, 3929–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laskowski R. A. (2009) PDBsum new things. Nucleic Acids Res. 37, D355–D359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dvir H., Shah M., Girardi E., Guo L., Farquhar M. G., Zajonc D. M. (2012) Atomic structure of the autosomal recessive hypercholesterolemia phosphotyrosine-binding domain in complex with the LDL-receptor tail. Proc. Natl. Acad. Sci. U.S.A. 109, 6916–6921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Z., Lee C. H., Mandiyan V., Borg J. P., Margolis B., Schlessinger J., Kuriyan J. (1997) Sequence-specific recognition of the internalization motif of the Alzheimer's amyloid precursor protein by the X11 PTB domain. EMBO J. 16, 6141–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shi N., Ye S., Bartlam M., Yang M., Wu J., Liu Y., Sun F., Han X., Peng X., Qiang B., Yuan J., Rao Z. (2004) Structural basis for the specific recognition of RET by the Dok1 phosphotyrosine binding domain. J. Biol. Chem. 279, 4962–4969 [DOI] [PubMed] [Google Scholar]
- 50. McNicholas S., Potterton E., Wilson K. S., Noble M. E. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 67, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]