

Background: NUT midline carcinoma is an aggressive cancer typically caused by the formation of the BRD4-NUT fusion oncoprotein.
Results: BRD4-NUT-stimulated histone hyperacetylation recruits BRD4 and associated transcription factors to activate gene expression.
Conclusion: BRD4-NUT perturbs normal gene expression to trigger oncogenic events in NMC.
Significance: Breaking BRD4-NUT interaction with histone acetyltransferases is an excellent strategy to abrogate the BRD4-NUT oncogenic activities.
Keywords: Acetyltransferase, Bromodomain-containing Protein 4 (BRD4), E1A Binding Protein p300 (P300), Transcription Elongation Factor, Transcription Factor, BRD4-NUT, NUT Midline Carcinoma, Transcriptional Activation
Abstract
NUT midline carcinoma (NMC) is a rare but highly aggressive cancer typically caused by the translocation t(15;19), which results in the formation of the BRD4-NUT fusion oncoprotein. Previous studies have demonstrated that fusion of the NUT protein with the double bromodomains of BRD4 may significantly alter the cellular gene expression profile to contribute to NMC tumorigenesis. However, the mechanistic details of this BRD4-NUT function remain poorly understood. In this study, we examined the NUT function in transcriptional regulation by targeting it to a LacO transgene array integrated in U2OS 2-6-3 cells, which allow us to visualize how NUT alters the in situ gene transcription dynamic. Using this system, we demonstrated that the NUT protein tethered to the LacO locus recruits p300/CREB-binding protein (CBP), induces histone hyperacetylation, and enriches BRD4 to the transgene array chromatin foci. We also discovered that, in BRD4-NUT expressed in NMC cells, the NUT moiety of the fusion protein anchored to chromatin by the double bromodomains also stimulates histone hyperacetylation, which causes BRD4 to bind tighter to chromatin. Consequently, multiple BRD4-interacting factors are recruited to the NUT-associated chromatin locus to activate in situ transgene expression. This gene transcription function was repressed by either expression of a dominant negative inhibitor of the p300-NUT interaction or treatment with (+)-JQ1, which dissociates BRD4 from the LacO chromatin locus. Our data support a model in which BRD4-NUT-stimulated histone hyperacetylation recruits additional BRD4 and interacting partners to support transcriptional activation, which underlies the BRD4-NUT oncogenic mechanism in NMC.
Introduction
NUT midline carcinoma (NMC)2 belongs to a class of highly lethal squamous cell carcinomas occurring in children and adults of all ages (1). It arises from genetic translocations targeting the nuclear protein in testis (NUT) gene on chromosome 15 (2, 3). In the majority of NMC cases, the translocation t(15;19) fuses NUT to the bromodomain-containing protein 4 (BRD4) gene on chromosome 19 (4). BRD4 is expressed normally as long (BRD4 or BRD4L) and short (BRD4S) isoforms with identical N-terminal double bromodomains and an extraterminal domain, whereas the long form encodes an additional C-terminal proline-rich and glutamine-rich region (Fig. 1A). The t(15;19) translocation break point splits BRD4L in half, causing in-frame fusion of the common N-terminal region of BRD4 (amino acids 1–719) with nearly the entire sequence of NUT protein (amino acids 6–1132), leaving expression of BRD4S unperturbed (Fig. 1A) (2). Interestingly, in some of the NMCs lacking a BRD4 rearrangement, NUT is found to be fused to either BRD3, another member of the bromodomain and extra-terminal domain (BET) protein family (4), or NSD3, a BRD4-interacting partner (5). There are also some NMC cases with unknown NUT fusion partner(s) (6).
FIGURE 1.

NUT protein activates in situ gene transcription. A, schematics of the BRD4 isoforms and BRD4-NUT oncogenic fusion. The t(15;19) translocation breakpoint bisects BRD4L at amino acid 719. The N-terminal component contains the double bromodomains (BD), a potential kinase domain, an extraterminal (ET) protein-protein interaction domain, and a serine-rich domain. The C-terminal of BRD4-NUT incorporates almost the entire NUT sequence. The BRD4C region spanning the BRD4 C-terminal amino acids 720–1362 is specifically present in BRD4L but not in BRD4-NUT. B, U2OS 2-6-3 YFP-MS2 stable cells were transfected with LacI-CHERRY-BRD4S or LacI-CHERRY-NUT. 24 h post-transfection, cells were fixed and counterstained with DAPI. LacI-CHERRY fusions allow the LacO locus to be visualized in red, whereas YFP-MS2 marks the sites of active transcription. IF, immunofluorescent staining with indicated antibodies (p300, CBP, AcH4, or BRD4). The RGB intensity profiles show the intensity curve of red, green, and blue signals along the highlighted yellow bars in the Merge panel. Scale bars = 5 μm.
NMCs represent the most lethal subset of squamous cell carcinomas. All of the known t(15;19)-positive carcinomas metastasize rapidly, respond poorly to standard chemotherapeutic strategies, and are extremely aggressive, with a median survival of 6.7 months (3). Therefore, it is important to study the oncogenic mechanisms of these carcinomas to develop novel therapeutic approaches. Moreover, NMC is a valuable model to investigate squamous carcinogenesis because its remarkably simple karyotypes suggest that these translocations targeting the NUT and BET proteins may represent a shortcut to transformation, bypassing the complex process of accumulating genetic mutations normally required for developing invasive squamous cell carcinomas (1).
Given the fact that NUT is expressed normally only in testes, it is reasonable to assume that typical BRD4-NUT NMCs are caused by the oncogenic consequences of unscheduled NUT expression and altered BRD4 function (2). BRD4 normally binds acetylated histones on chromatin through its double bromodomains (7) and plays a central role in cellular growth control (8–15). It facilitates transcriptional activation by recruiting positive transcription elongation factor b (P-TEFb), mediators, and other transcriptional activators (14, 16, 17). BRD4 has been identified as a critical therapeutic target in a number of different cancers (18–20). In these tumor cells, dissociation of BRD4 from chromatin leads to selective inhibition of numerous key oncogenes (17). For NMC tumors, the BRD4-NUT fusion oncoprotein is also tethered to acetylated chromatin by the bromodomains (4, 21, 22). It causes malignancy by blocking NMC differentiation and driving tumor growth (1, 4, 23). However, the molecular mechanisms by which BRD4-NUT drives the highly aggressive NMC tumorigenesis remain elusive.
We and others have shown that the NUT moiety of the BRD4-NUT fusion strongly interacts with and recruits histone acetyltransferases (HATs) to discrete chromatin foci, where it activates the HAT activity to stimulate histone hyperacetylation (21, 22). This leads to recruitment of additional BRD4-NUT/HATs and formation of hyperacetylated chromatin domains. Sequestration of BRD4 and associated transcription factors into these hyperacetylated BRD4-NUT foci causes repression of genes outside of these regions, such as the epithelial differentiation regulator c-FOS and inhibition of cell differentiation (21, 22). On the other hand, BRD4-NUT is recruited to c-MYC, which contributes to oncogenesis by blocking epithelial cell differentiation while promoting proliferation (24). We also demonstrated that BRD4-NUT enriched at the stem cell marker SOX2 strongly stimulates its abnormal activation through a gain-of-function recruitment of p300 by the NUT moiety. The activated SOX2 expression, in turn, drives the aberrant stem cell-like proliferation and the highly aggressive transforming activity of NMC (25). These recent studies demonstrated compelling evidence to support the hypothesis that association of BRD4-NUT with chromatin plays an important role in modulating gene expression activities in NMC. However, the mechanism of BRD4-NUT-induced transcriptional activation is unclear. It is also unknown whether BRD4-NUT activates the expression of other associated genes genome-wide in a similar fashion.
In this study, we performed both biochemical and in situ transcription analyses to investigate NUT protein function in transcriptional regulation. We tethered NUT to a LacO transgene array integrated in U2OS 2-6-3 cells using the LacI-CHERRY-NUT fusion, which allowed us to directly visualize transcriptional activities associated with NUT from individual transcription sites at the levels of DNA and RNA in single cells (25, 26). Using this system, we demonstrated that NUT protein recruits p300/CBP, induces histone hyperacetylation, and enriches additional BRD4 and associated transcription factors at its associated chromatin locus to stimulate in situ gene transcription. Abrogation of p300 recruitment or inhibition of BRD4 binding to these chromatin loci abolishes the in situ gene transcription, providing further support for the role of histone hyperacetylation and BRD4 recruitment in BRD4-NUT-mediated transcriptional activation. Our study therefore reveals the mechanistic details for BRD4-NUT function in transcriptional regulation and will aid future investigation of the NMC oncogenic mechanism.
EXPERIMENTAL PROCEDURES
Recombinant Plasmid Constructs
Plasmids encoding Xpress-tagged BRD4S (pcDNA4c-BRD4S) were generated by cloning the PCR-amplified DNA fragments of BRD4S from the previously described pOZN-BRD4S (27) into the pcDNA4C vector. Plasmids encoding Xpress-tagged NUT (pcDNA4c-NUT) and BRD4-NUT (pcDNA4c-BRD4-NUT) were generated by cloning the PCR-amplified DNA fragments of NUT or BRD4-NUT from the previously described pOZN-BRD4-NUT (22) into the pcDNA4C vector. For the Xpress-tagged H2B-NUT construct (pcDNA4c-H2B-NUT), the H2B-coding DNA fragment was PCR-amplified from the previously described pOZN-H2B-E2TA (28) and subcloned into pcDNA4C-NUT in-frame with the NUT moiety. Constructs of LacI-CHERRY, LacI-CHERRY-BRD4S, LacI-CHERRY-NUT, and LacI-CHERRY-BRD4L have been described in our previous study (25). The LacI-CHERRY-BRD4C construct was generated by cloning the PCR-amplified DNA fragments of BRD4C (encoding BRD4 C-terminal amino acids 720–1362) into the pcDNA4C-LacI-CHERRY plasmid. To generate highly expressed NUT subfragments, DNAs coding for NUT 6–300 and NUT 346–593 were PCR-amplified and cloned into a pOZN vector to generate in-frame fusions of these molecules with an HA-FLAG tag. The HA-FLAG-tagged NUT 6–300 or NUT 346–593 DNA fragments were additionally PCR-amplified and subcloned into a modified pLU vector (a gift from Dr. Susan M. Janicki) (29). All plasmid constructs were verified by DNA sequencing.
Cell Culture, Transfection, and Gene Knockdown
HCC2429 cells (provided by Dr. Thao P. Dang, Vanderbilt University) (30) were maintained in RPMI 1640 medium (Invitrogen) with 10% FBS (Hyclone) and 1% penicillin/streptomycin (Invitrogen). 293T cells were maintained in DMEM (Invitrogen) with 10% FBS and 1% penicillin/streptomycin. U2OS cells were maintained in McCoy's 5A medium (Invitrogen) with 10% FBS and 1% penicillin/streptomycin. The U2OS 2-6-3 and U2OS 2-6-3 YFP-MS2 stable cell lines were a gift from Dr. Susan M. Janicki (26, 29). The U2OS 2-6-3 cell line (26) was maintained in DMEM GlutaMAXTM-I high-glucose (Invitrogen) supplemented with 10% Tet system-approved FBS (Clontech), 1% penicillin/streptomycin, and hygromycin B (100 μg/ml, Clontech). U2OS 2-6-3 YFP-MS2 stable cells (26, 29) were maintained in the same medium supplemented with G418 (400 μg/ml, American Bioanalytical).
For 293T cells, the calcium phosphate transfection method was applied as described previously (27). U2OS cells were transfected with GeneJammer reagent (Agilent Technologies) following the instructions of the manufacturer. For the U2OS 2-6-3 and U2OS 2-6-3 YFP-MS2 stable cell lines, both FuGENE HD (Roche) and GeneJammer (Agilent Technologies) reagents were used for plasmid transfection. HCC2429 cells were transfected with Lipofectamine 2000 (Invitrogen) following the instructions of the manufacturer.
The non-targeting control siRNA (catalog no. D-001210–01) and NUT siRNA (catalog no. D-022211–01) were purchased from Dharmacon (GE Healthcare). For siRNA knockdown, HCC2429 cells were transfected at about 30% confluency using DharmaFECT 2 reagent (GE Healthcare) following the instructions of the manufacturer.
Nuclear Extraction with Different Salt Concentrations
Cells were trypsinized and counted. Around 5 × 106 cells were aliquoted to each tube for nuclear protein extraction using different salt concentrations. Cells were first gently resuspended in 200 μl of buffer A (10 mm Tris-HCl (pH 7.5), 10 mm NaCl, 3 mm MgCl2, 2.5 mm DTT, 1 mm PMSF, and 0.5% Nonidet P-40 supplemented with protease inhibitors). After 10 min of incubation on ice, nuclear pellets were centrifuged at 5000 rpm for 5 min at 4 °C and then resuspended in 100 μl of buffer B (10 mm Tris-HCl (pH 7.5), 3 mm MgCl2, 2.5 mm DTT, 1 mm PMSF, and 0.5% Nonidet P-40 supplemented with protease inhibitors) with different salt concentrations (100–450 mm NaCl). After rotating at 4 °C for 1 h, nuclear extracts were centrifuged at 12,000 rpm for 5–10 min at 4 °C. The supernatants were then boiled in SDS-PAGE sample buffer. The same volume of samples from different salt concentration extracts were loaded onto SDS-PAGE gels, transferred onto PVDF membranes, and immunoblotted with the BRD4 C antibody (recognizing Brd4 amino acids 1313–1362). HRP-linked anti-rabbit IgG (Cell Signaling Technology) was used as the secondary antibody. Western blots were developed using Western Lightning ECL solution (PerkinElmer Life Sciences), and images were captured using a Fuji imaging system.
The intensity of protein bands was quantified using the “Analyze Gels” function of ImageJ software (National Institutes of Health). The relative abundance was calculated by setting the intensity of the BRD4 band from extraction of the highest (450 or 400 mm) NaCl concentration as 1. The curves were fitted by non-linear regression using the “One site-specific binding with Hill slope” equation on GraphPad Prism software (version 5.0).
Salt Treatment and Immunofluorescent Staining
U2OS cells were split onto coverslips 8 h after transfection. At 36 h post-transfection, cells were either fixed directly for 20 min with 4% paraformaldehyde in PBS or pre-extracted on ice for 10 min in CSK buffer (10 mm PIPES (pH 6.8), 30 mm sucrose, 3 mm MgCl2, 1 mm EGTA, 0.5% Triton X-100, and 250 mm NaCl and supplemented with protease inhibitors) before fixing with 4% paraformaldehyde in PBS for 20 min (31).
For other immunofluorescent experiments, cells on coverslips were fixed with 3% paraformaldehyde in PBS for 20 min. Immunofluorescent staining was performed as described previously (27). The following primary antibodies were used: anti-BRD4 C, anti-Xpress (catalog no. R910-25, Invitrogen), anti-p300 (catalog no. 05-257, Millipore), anti-p300 (catalog no. sc-585, Santa Cruz Biotechnology), anti-CBP (catalog no. 06-297, Millipore), anti-AcH4 (catalog no. 06-866, Millipore), anti-Cyclin T1 (catalog no. sc-10750, Santa Cruz Biotechnology), anti-Cdk9 (catalog no. sc-8338, Santa Cruz Biotechnology), anti-MED1 (catalog no. A300-793A, Bethyl Laboratories), and anti-FLAG (catalog no. F-3165, Sigma). Secondary antibodies used were Alexa Fluor 594 goat anti-rabbit IgG (catalog no. A11012, Invitrogen), Alexa Fluor 488 goat anti-mouse IgG (catalog no. A11001, Invitrogen), Alexa Fluor 488 donkey anti-rabbit (catalog no. A21206, Invitrogen), and Alexa Fluor 350 goat anti-mouse (catalog no. A21049, Invitrogen).
All immunofluorescent images were captured using an inverted fluorescence microscope (Olympus IX81) as described previously (32). The scale bars were added using ImageJ software. Profiles of red, green, and blue signals (RGB) for the transgene sites were generated using the “RGB Profile Plot” function of ImageJ software.
Quantitative single-cell image analysis was performed using ImageJ software as described previously (25). All photos used in the analysis were taken with the same exposure and gain settings and exported as 16-bit monochrome images on a scale of 0–4095. The YFP-MS2 intensity was measured at the transgene array regions, which were selected on the basis of the CHERRY signal. YFP-MS2 intensity measurements were normalized by subtracting the background value, which was determined by averaging the measurements from three independent nuclear regions distinct from the transgene array site. YFP-MS2 intensity was measured for more than 50 cells for each sample.
RT-qPCR
Total RNA was isolated using a NucleoSpin RNA II kit (Macherey-Nagel). Reverse transcription was performed using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed using a CFX96 real-time PCR detection system (Bio-Rad) with IQ SYBR Green supermix (Bio-Rad). RT-qPCR Primers used in this study were as follows: reporter 1, 5′ TCA TTA GAT CCT GAG AAC TTC A 3′ (forward) and 5′ TTT TGG CAG AGG GAA AAA GA 3′ (reverse); LacI, 5′ TAA CTA TCC GCT GGA TGA CC 3′ (forward) and LacI, 5′ GCC GAG ACA GAA CTT AAT GG 3′ (reverse). The reporter mRNA level was normalized to the LacI mRNA level, which represents the expression level of LacI-CHERRY constructs.
ChIP
ChIP experiments were performed as previously described (25). Primers used for ChIP samples were as follows: SOX2 promoter, 5′ CCA TAC AGT GCC GTG GGA TG 3′ (forward) and 5′ CAG CTC TTC CTC TTA GCC CC 3′ (reverse); transgene promoter, 5′ TGA AAG TCG TCG ACC GGG TCG AG 3′ (forward) and 5′ GGA TCG GTC CCG GTG TCT TCT ATG 3′ (reverse).
Statistical Analyses
Statistical analysis was performed using unpaired Student's t test of GraphPad Prism software (Version 5.0) to compare the data from the control and experimental groups. A two-tailed p value of <0.05 was considered statistically significant.
RESULTS
NUT Activates in Situ Gene Transcription by Recruiting HATs, Inducing Histone Acetylation and BRD4 Enrichment
We discovered that the BRD4-NUT fusion oncoprotein expressed in NMC cells sequesters transcription factors in hyperacetylated chromatin foci to stimulate some gene expression while repressing the expression of others (22, 25). Further studies suggested that this appears to be a gain of function for NUT (25). To determine how tethering the NUT moiety of BRD4-NUT to cellular chromatin affect in situ gene expression, we performed mechanistic studies by targeting the NUT molecule to a stably integrated LacO transgene array in U2OS 2-6-3 cells using the LacI-CHERRY fusion (26). This system allowed us to visualize in situ recruitment of cellular factors and transcription dynamics in the presence of the NUT molecule (25, 26). In this transgene array system, a transcription unit composed of 256 copies of the lac operator (LacO), 96 copies of the tetracycline response element, a minimal CMV promoter, 24 repeats of the MS2 translational operators (MS2 repeats), and a polyadenylation signal was stably integrated into a euchromatic region of chromosome 1 in the U2OS genome (26). The integrated transgene array in the U2OS 2-6-3 cell line could be visualized by expression of a Lac repressor binding protein (LacI)-CHERRY fusion, whereas the MS2 operator RNA transcribed from this locus was detected by accumulations of the RNA-specific binding protein, MS2 coat-YFP fusion protein (YFP-MS2), at the locus (26). In this study, a LacI-CHERRY-NUT construct was generated to tether the NUT moiety to the LacO transgene locus, whereas a LacI-CHERRY-BRD4S construct was used as a control for detecting the contribution of the BRD4 moiety to the BRD4-NUT function. U2OS 2-6-3 cells stably expressing YFP-MS2 were transfected with these LacI-Cherry constructs. We found that NUT tethered to the LacO array is able to strongly stimulate MS2 operator transcription even in the absence of tetracycline-controlled transactivator, whereas BRD4S did not have the same effect (Fig. 1B and (25)). NUT tethered to the LacO array also efficiently recruits p300 but BRD4S does not have this activity (Fig. 2 and (25)). We have shown previously that, besides p300, other HATs also interact with NUT (22). We therefore examined several other HATs, including p300/CBP-associated factor (P/CAF), Tip60, transcription initiation factor TFIID 250-kDa subunit (TAFII250), MYST, CBP, and ATF-2, and found that the NUT protein also recruits high levels of CBP to the transgene site (Fig. 2). About 80% of the cells transfected with either LacI-CHERRY vector control or LacI-CHERRY-BRD4S also showed a low level of CBP recruited to the transgene site (Fig. 2 and data not shown). However, compared with these controls, LacI-CHERRY-NUT-transfected cells showed a much brighter staining of CBP on the transgene site, suggesting that it is specifically enriched by the NUT moiety (Fig. 2). Likely as a result of the recruitment of p300 and CBP, high levels of acetylated histone H4 (AcH4) and BRD4 were detected at the transgene site only in the LacI-CHERRY-NUT-transfected cells but not in the LacI-CHERRY-BRD4S-positive cells (Fig. 2). To confirm this finding, we performed a ChIP analysis to examine the status of AcH4 and BRD4 recruitment at the transgene locus. Primers targeting the transgene promoter were used in the PCR analysis of the ChIP samples to detect the enrichment of AcH4 or BRD4 at the locus. ChIP results showed that the AcH4 and BRD4 levels at the transgene promoter were increased significantly with LacI-CHERRY-NUT transfection compared with LacI-CHERRY (CO) or LacI-CHERRY-BRD4S transfection (Fig. 3, A and C). It is important to note that although only ∼30% of the U2OS 2-6-3 cells were transfected with the LacI-CHERRY-NUT construct, we were able to see the dramatic effect of LacI-CHERRY-NUT expression on the local AcH4 and BRD4 enrichment at the transgene array chromatin foci, indicating that the increase of AcH4 and BRD4 level at the transgene promoter in LacI-CHERRY-NUT-positive cells compared with control cells could be much more dramatic than the data shown in Fig. 3, A and C. In contrast, LacI-CHERRY-NUT did not change the levels of AcH4 and BRD4 at the SOX2 promoter region (Fig. 3, B and D). Together, these results extend our hypothesis to suggest that the NUT moiety of BRD4-NUT recruits p300/CBP, which induces high levels of histone acetylation, consequently allowing additional BRD4 to bind to surrounding chromatin.
FIGURE 2.

The NUT protein recruits p300 to stimulate local histone H4 acetylation and BRD4 enrichment in the transgene locus. U2OS 2-6-3 cells were transfected with LacI-CHERRY-BRD4S or LacI-CHERRY-NUT. 24 h post-transfection, cells were immunostained with p300, CBP, AcH4, or BRD4 C antibodies and counterstained with DAPI. BRD4 C antibody is specific for the BRD4 long isoform. IF, immunofluorescent staining with indicated antibodies (p300, CBP, AcH4, or BRD4). The RGB intensity profiles show the intensity curve of red, green, and blue signals along the highlighted yellow bars in the Merge panel. Scale bars = 5 μm.
FIGURE 3.

The NUT protein induces local histone H4 hyperacetylation and enriches BRD4 at the transgene locus. U2OS 2-6-3 cells were transfected with LacI-CHERRY (CO), LacI-CHERRY-BRD4S, or LacI-CHERRY-NUT. 24 h post-transfection, cells were subjected to the ChIP assay with normal rabbit IgG (NR IgG), AcH4, or BRD4 C antibodies. ChIP samples were analyzed by qPCR using primers targeting the transgene promoter region (A and C) as well as the SOX2 promoter region (B and D). All values represent the mean ± S.D. of three independent experiments.
BRD4-NUT Causes Tighter BRD4 Binding to NMC Chromatin
Besides the transgene array study, we also examined whether BRD4-NUT expressed in NMC cells affects BRD4 binding to chromatin through stimulating histone acetylation. In the BRD4-NUT-positive NMC lung cancer cell line HCC2429, which carries one intact BRD4 locus and expresses normal BRD4 as well as the BRD4-NUT fusion proteins (22), we noticed that BRD4 extraction efficiency was much lower compared with other BRD4-NUT-negative cell lines. We hypothesized that BRD4 bound more tightly to chromatin in HCC2429 than in the BRD4-NUT-negative cell lines. Indeed, increasing the salt concentration in the extraction buffer from 150 to 400 mm greatly improved recovery of BRD4 from this cell line (Fig. 4A). We were curious whether HCC2429 was unique in having very tightly bound BRD4, so we tested a number of other cell lines using various salt concentrations (Fig. 4, A and B). Nuclear extracts were prepared with 100–450 mm salt concentrations, and extracted BRD4 relative intensities were analyzed. Compared with the BRD4-NUT-negative cell lines, including 293T, C33A, and U2OS cells, it is clear that HCC2429 cells need a higher salt concentration to extract BRD4 from chromatin (Fig. 4, A and B), although the levels of endogenous BRD4 in these cell lines are not dramatically different (Fig. 4A, bottom panel), suggesting that BRD4 is truly binding tighter to chromatin in HCC2429 cells.
FIGURE 4.

Expression of BRD4-NUT increases the binding of BRD4 to chromatin. A, nuclear proteins from four different cell lines (293T, C33A, U2OS, and HCC2429) were extracted with different NaCl concentrations (Conc., 100–450 mm). Nuclear extracts were immunoblotted with the BRD4 C antibody. The concentration of nuclear proteins extracted with 450 mm NaCl buffer was determined by Bradford assay, and 20 μg of proteins was immunoblotted with BRD4 C and GAPDH antibodies to compare endogenous BRD4 levels in these cell lines. B, the intensity of BRD4 bands was quantified using ImageJ. The relative abundance of extracted BRD4 was calculated by setting the intensity of the BRD4 band from 450 mm NaCl extraction as 1. The curves were fit using GraphPad Prism software. C, HCC2429 cells were transfected with a non-targeting control (CO) or NUT siRNA. 36 h post-transfection, nuclear proteins were extracted with different NaCl concentrations (100–400 mm) and immunoblotted with the BRD4 C antibody. The concentration of nuclear proteins extracted with 400 mm NaCl buffer was determined by Bradford assay, and 20 μg of proteins was also immunoblotted with NUT, BRD4 C, and GAPDH antibodies to confirm the NUT knockdown efficiency. D, same as B, except the intensity of the BRD4 band from 400 mm NaCl extraction was set as 1. E, 293T cells were transfected with vector control (V) or constructs expressing Xpress-tagged BRD4S, NUT, BRD4-NUT, or H2B-NUT. 24 h post-transfection, whole cell lysates were immunoblotted with Xpress, BRD4 C, and GAPDH antibodies. The concentration of cell lysates was determined by Bradford assay, and 20 μg of proteins was loaded for each lane. Asterisks indicate the Xpress-tagged proteins. F, 293T cells were transfected as in E. 24 h post-transfection, nuclear proteins were extracted with different NaCl concentrations (150–400 mm) and immunoblotted with the BRD4 C antibody. G, as described in D.
To test whether BRD4-NUT causes strong binding of BRD4 to chromatin in HCC2429 cells, we knocked down BRD4-NUT using NUT siRNA and then performed BRD4 extraction using different salt concentrations (Fig. 4, C and D). Consistent with our data published previously (see Fig. 4A in Ref. 25), we repeatedly observed a reduced BRD4 protein level in BRD4-NUT knockdown cells (Fig. 4C, bottom panel). It is possible that BRD4-NUT-induced tighter BRD4 binding to chromatin contributes to its stabilization. However, although less BRD4 is present in the BRD4-NUT knockdown cells, BRD4 was more efficiently extracted from the chromatin in these cells compared with the non-targeting control siRNA-treated cells (Fig. 4, C and D). This result strongly suggests that BRD4-NUT causes BRD4 to bind tighter to the chromatin.
To further confirm that the NUT moiety of the fusion protein is inducing the stronger binding of BRD4 to chromatin, we expressed BRD4S, NUT, or BRD4-NUT in 293T cells (Fig. 4E) and then examined the extraction efficiency of endogenous BRD4 using different salt concentrations (Fig. 4, F and G). 293T cells expressing BRD4-NUT need a higher salt concentration to extract most of the BRD4 in the nucleus. However, BRD4S- or NUT-transfected cells show an extraction pattern similar to the vector control-transfected cells (Fig. 4, F and G), suggesting that tethering NUT protein to chromatin is required for increasing the binding affinity of BRD4 to chromatin. To confirm this, we made a construct encoding H2B-NUT fusion in which the NUT moiety could be tethered to the chromatin by H2B. As expected, expression of H2B-NUT increased the salt concentration needed to extract BRD4, similar to the observation made in the cells expressing BRD4-NUT (Fig. 4, E–G). Notably, the endogenous BRD4 level in 293T cells was not dramatically changed by transfection of any of these constructs (Fig. 4E).
Visualization Of The Tighter BRD4 Binding to Chromatin Induced by the NUT Protein Tethered to Chromatin
As reported in our previous study (22), the NUT moiety of BRD4-NUT interacts with and stimulates p300 catalytic activity, which consequently causes a high level of acetylation on nearby histones and recruitment of additional BRD4 molecules. We hypothesized that this may be the mechanism by which BRD4-NUT enhances the binding affinity of BRD4 to chromatin. To directly test whether increased histone acetylation makes BRD4 bind tighter to chromatin, we examined the BRD4 extraction efficiency in 293T cells treated with DMSO or a histone deacetylase inhibitor, trichostatin A (TSA), which induces histone hyperacetylation as detected by immunofluorescence staining (data not shown). The nuclear extracts were prepared from these cells using different salt concentrations. TSA treatment dramatically increased the salt concentration needed to extract BRD4 from chromatin (Fig. 5 and Ref. 7). This result further supports our hypothesis that histone hyperacetylation, which could be induced by chromatin-bound NUT protein, is sufficient to cause tighter BRD4 binding to chromatin.
FIGURE 5.
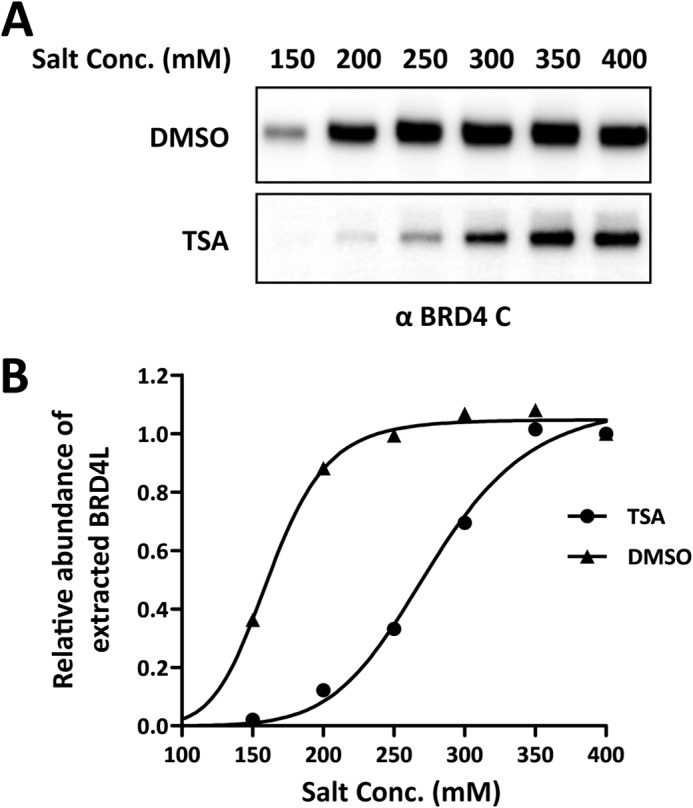
BRD4 is harder to extract upon TSA treatment. A, 293T cells were treated with 100 ng/ml TSA for 12 h. Nuclear proteins were extracted with different NaCl concentrations (Conc., 150–400 mm) and immunoblotted with the BRD4 C antibody. DMSO, dimethyl sulfoxide. B, the intensity of BRD4 bands was quantified using ImageJ. The relative abundance of the extracted BRD4 in the nuclear extracts was calculated by setting the intensity of the BRD4 band from 400 mm NaCl extraction as 1. The curves were fit using GraphPad Prism software.
To directly visualize the effect of chromatin-associated NUT on BRD4 binding to chromatin at the single-cell level, we performed immunofluorescent staining of U2OS cells expressing BRD4S, BRD4-NUT, or H2B-NUT after extraction with a buffer containing 250 mm NaCl. In the control cells transfected with an empty vector, BRD4 was completely removed after salt treatment (Fig. 6). However, after salt treatment of the cells expressing BRD4-NUT or H2B-NUT, BRD4 remained strongly bound to chromatin, showing staining signals as bright as in non-treated cells. In BRD4S-positive cells either with or without salt extraction, we observed a lower level of BRD4 compared with untreated BRD4S-negative cells (Fig. 6). This is likely because the high level of BRD4S functions as a dominant-negative inhibitor to dissociate endogenous BRD4 from chromatin, but it can also interact with BRD4 through the bromodomains to retain some BRD4 in the nucleus (32). The data so far suggest that the NUT moiety tethered to chromatin either through the BRD4 double bromodomains or H2B cause tighter binding of BRD4 to chromatin. We were not able to examine the BRD4 signal in NUT-expressing cells after salt extraction because the NUT protein not associated with chromatin was completely removed after salt extraction (data not shown).
FIGURE 6.

Visualization of the tighter BRD4 chromatin binding induced by the NUT protein tethered to chromatin. U2OS cells were transfected with vector control (V) or constructs for expressing Xpress-tagged BRD4S, BRD4-NUT, or H2B-NUT. 36 h post-transfection, coverslips were collected and either fixed immediately (Non-treated) or pre-extracted in a buffer containing 250 mm NaCl prior to fixation (Salt-treated). The cells were then stained with Xpress (green) and Brd4 C (red) antibodies and counterstained with DAPI. Representative images from three experiments are presented. Scale bars = 10 μm.
NUT-induced Histone Hyperacetylation Allows Further Recruitment of BRD4-interacting Factors to Activate in Situ Gene Transcription
To further understand whether BRD4 recruitment contributes to the in situ transgene activation in U2OS 2-6-3 cells (Figs. 1B and 2), we tested whether BRD4 recruits its known interacting transcription factors, such as P-TEFb, which contains Cdk9 and Cyclin T1 subunits, and mediator of RNA polymerase II transcription subunit 1 (MED1), to the LacO locus. Indeed, the endogenous proteins of these BRD4-interacting factors were all recruited to the transgene foci in LacI-CHERRY-NUT-transfected cells but not in LacI-CHERRY-BRD4S-transfected cells (Fig. 7A). Quantification analysis showed that a high percentage (>80%) of cells show in situ recruitment of these transcription factors as well as activation of MS2 operator RNA transcription (Fig. 7B), demonstrating the mechanistic details that support the transgene activation induced by NUT protein.
FIGURE 7.

The NUT protein enriches the BRD4-interacting partners P-TEFb and MED1 at the transgene locus to stimulate in situ transcription activation. A, U2OS 2-6-3 cells were transfected with LacI-CHERRY-BRD4S or LacI-CHERRY-NUT. 24 h post-transfection, cells were immunostained with Cdk9, Cyclin T1, or MED1 antibodies and counterstained with DAPI. IF, immunofluorescent staining with indicated antibodies (Cdk9, cyclin T1, or MED1). The RGB intensity profiles show the intensity curve of red, green, and blue signals along the highlighted yellow bars in the Merge panel. Scale bars = 5 μm. B, U2OS 2-6-3 cells were transfected with LacI-CHERRY-NUT. 24 h post-transfection, cells were immunostained with p300, CBP, AcH4, BRD4 C, Cdk9, Cyclin T1, or MED1 antibodies and counterstained with DAPI. The percentage of cells with the indicated cellular factors recruited to the transgene locus was calculated from more than 100 positively transfected cells. For quantification of MS2 operator RNA transcription activation, U2OS 2-6-3 YFP-MS2 stable cells were transfected with LacI-CHERRY-NUT. 24 h post-transfection, cells were fixed and counterstained with DAPI. The percentage of cells with the YFP-MS2 signal accumulated at the LacO transgene locus was calculated from more than 100 positively transfected cells. Values represent the means ± S.D. of three independent experiments.
NUT 346–593 Functions as a Dominant-Negative Inhibitor to Block p300 Recruitment and to Inhibit in Situ Transgene Transcription Activation
The NUT moiety of BRD4-NUT has been shown to interact with p300 and to stimulate its catalytic activity (21). Therefore, p300 likely plays a critical role in NUT-mediated in situ transcriptional activation. The p300 binding site has been mapped to the NUT region spanning amino acids 346–593 (21). We therefore tested whether this region may be used as a dominant-negative inhibitor to prevent p300 recruitment. Remarkably, coexpression of the NUT 346–593 domain with LacI-CHERRY-NUT significantly inhibited p300 recruitment to the transgene site (Fig. 8A). Quantification analysis showed that nearly 100% of the cells transfected with a control vector or the construct encoding the NUT 6–300 domain, which does not bind p300 (21), displayed strong p300 enrichment at the LacI-CHERRY-NUT-bound transgene site (Fig. 8A). In contrast, only 32.4% of the LacI-CHERRY-NUT-positive cells coexpressing NUT 346–593 still showed clear p300 recruitment, 38.9% of the cells showed a weakly recruited p300 signal, and 28.7% had no detectable p300 signal at the transgene array. NUT 346–593 expressed in HCC2429 cells also efficiently diffused the p300 signal, which normally accumulates in the BRD4-NUT foci (Fig. 8B) (25). These results demonstrated that NUT 346–593 functions as a dominant-negative inhibitor to block the NUT and p300 interaction. Importantly, in U2OS 2-6-3 cells stably expressing YFP-MS2, coexpressing LacI-CHERRY-NUT and NUT 346–593 caused the transcription level of MS2 operators to be significantly reduced compared with the cells cotransfected with the empty vector or the NUT 6–300 construct (Fig. 8, C and D). Because NUT 346–593 not only blocks p300 recruitment but also inhibits the transcription of the transgene array, these results suggest that p300 recruitment and the downstream events are essential for NUT-mediated transcriptional activation.
FIGURE 8.

Blocking NUT-p300 interaction using a NUT 346–593 dominant-negative inhibitor prevents p300 recruitment and represses NUT-mediated transgene activation. A, U2OS 2-6-3 cells were transfected with LacI-CHERRY-NUT together with either the empty vector (V) or constructs encoding FLAG-tagged NUT 6–300 or the NUT 346–593 fragment. Cells were fixed 24 h after transfection and immunostained with p300 (green) and FLAG (blue) antibodies. The RGB intensity profiles in the right panel show the intensity curve of red, green, and blue signals along the highlighted yellow bars in the Merge panel. Scale bars = 5 μm. B, HCC2429 cells were transfected with constructs encoding FLAG-tagged NUT 6–300 or NUT 346–593. 24 h post-transfection, cells were fixed and immunostained with p300 (green) and FLAG (red) and counterstained with DAPI. Scale bars = 10 μm. C, U2OS 2-6-3 YFP-MS2 stable cells were transfected with LacI-CHERRY-NUT together with either the empty vector or constructs encoding FLAG-tagged NUT 6–300 or the NUT 346–593 fragment. Cells were fixed 24 h after transfection and immunostained with the FLAG antibody. Scale bars = 5 μm. D, quantitative single-cell image analysis of the YFP-MS2 intensity at the transgene array locus. The average YFP-MS2 intensity of the transgene array was quantified for more than 50 cells in each sample transfected in C using ImageJ. Data represent the mean ± S.D. ***, p < 0.001.
Recruitment of BRD4 Plays a Supporting Role in BRD4-NUT-mediated in Situ Transgene Activation
Our data so far show that BRD4-NUT can stimulate transcriptional activation by recruiting HATs through the NUT moiety and that BRD4 is tightly bound to chromatin in the presence of BRD4-NUT. We hypothesized that the BRD4-NUT transcriptional activation of the transgene array in U2OS 2-6-3 cells was mediated by BRD4 binding of acetylated histones at the transgene array through its bromodomains. We therefore abrogated the interaction of BRD4 with acetylated histones on chromatin using a small molecule inhibitor, (+)-JQ1 (23), to test whether, in the absence of BRD4, recruitment of other factors such as p300 by the NUT protein to the transgene array could still activate transcription. Titration analysis showed that 3 μm (+)-JQ1 was sufficient to completely dissociate BRD4 from the transgene site in most of the U2OS 2-6-3 cells, with only 8% of LacI-CHERRY-NUT-positive cells retaining weak BRD4 staining at the transgene locus (Fig. 9A). When BRD4 was dissociated by (+)-JQ1, the P-TEFb subunits Cdk9 and Cyclin T1 were also dissociated from the transgene site in most cases, leaving around 17% and 15% of LacI-CHERRY-NUT-positive cells still showing weak staining of Cdk9 or Cyclin T1, respectively, at the transgene locus (Fig. 9A). Dissociation of BRD4 could only partially inhibit MED1 recruitment to the transgene site, with around 60% of LacI-CHERRY-NUT-positive cells still showing MED1 recruitment at the LacO locus (Fig. 9A), indicating that MED1 may also be recruited to the LacO transgene locus through BRD4-independent mechanisms. In contrast to the BRD4-interacting factors, p300 recruitment was observed in almost 100% of the LacI-CHERRY-NUT-positive cells treated with either (+)-JQ1 or its inactive stereoisomer, (-)-JQ1, indicating that p300 acts upstream of BRD4 and, therefore, is not affected by BRD4 dissociation (Fig. 9A). These results support the role of BRD4 in the recruitment of P-TEFb and, to a lesser extent, MED1, to NUT-bound chromatin.
FIGURE 9.
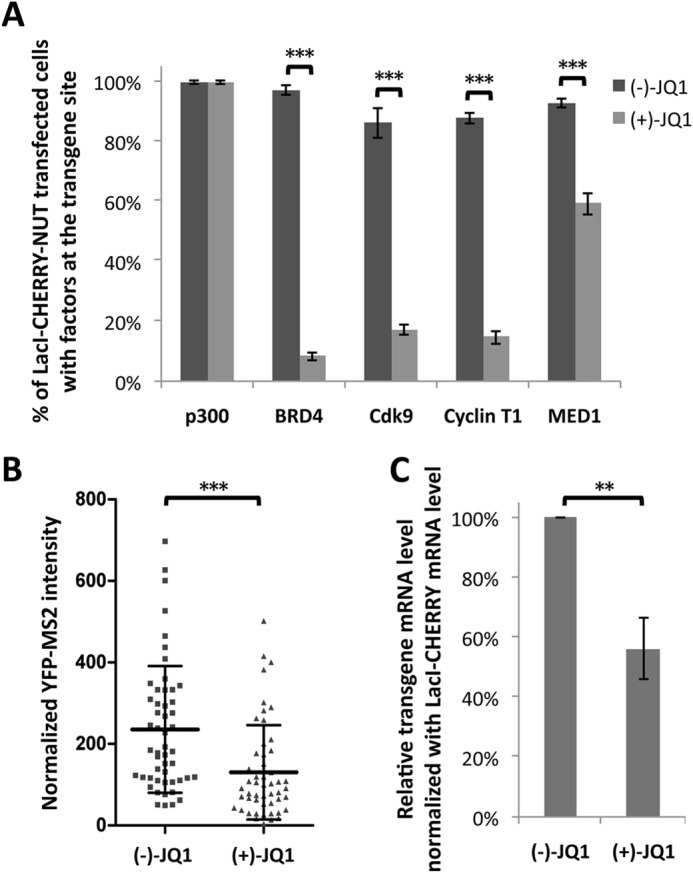
Recruitment of BRD4 contributes to NUT-mediated transcriptional activation. A, U2OS 2-6-3 cells transfected with LacI-CHERRY-NUT were treated with 3 μm (−)-JQ1 or (+)-JQ1. 8 h after adding JQ1, cells were fixed and immunostained with p300, BRD4 C, Cdk9, Cyclin T1, or MED1 antibodies. The percentage of cells with these factors recruited to the transgene locus was calculated from more than 100 positively transfected cells. Values represent the mean ± S.D. of three independent experiments. B, U2OS 2-6-3 YFP-MS2 stable cells were transfected with LacI-CHERRY-NUT and treated with 3 μm (−)-JQ1 or (+)-JQ1. 8 h after adding JQ1, cells were fixed and subjected to single-cell image analysis of the YFP-MS2 intensity at the transgene array locus. The average YFP-MS2 intensity of the transgene array was quantified from more than 50 cells in each sample using ImageJ. Data represent the mean ± S.D. of all cells examined. C, U2OS 2-6-3 YFP-MS2 stable cells were treated as in B. The mRNA levels of the transgene reporter were measured by RT-qPCR and normalized to the LacI mRNA levels. Values represent the mean ± S.D. of three independent experiments. **, p < 0.01; ***, p < 0.001.
Further studies in U2OS 2-6-3 cells stably expressing YFP-MS2 showed that the transcription of the transgene reporter induced by LacI-CHERRY-NUT, as quantified by both YFP-MS2 accumulation at the transgene site and mRNA level detected by RT-qPCR, was significantly repressed upon treatment with (+)-JQ1 compared with the cells treated with (−)-JQ1 (Fig. 9, B and C). This inhibition was observed in cells treated with 3 μm JQ1 for 8 h to avoid indirect effects on cell proliferation and/or the cell cycle progression, indicating a direct (+)-JQ1 inhibition effect on the transgene transcription.
To further confirm the supporting role of BRD4 in NUT-induced transcriptional activation, we analyzed LacI-CHERRY fusions with either BRD4L or BRD4C, which encode the BRD4 C-terminal amino acid 720 to 1362 that are present specifically in BRD4L but not in BRD4-NUT (Fig. 1A), for their abilities to recruit interacting partners to activate transgene expression. Quantification of immunofluorescently stained cells indicated that, as expected, neither LacI-CHERRY-BRD4C nor LacI-CHERRY-BRD4L could recruit p300 or induce H4 acetylation at the transgene locus. However, more than 90% of LacI-CHERRY-BRD4C- and LacI-CHERRY-BRD4L-transfected cells strongly recruited Cdk9 and Cyclin T1 to the transgene site, whereas only about 20% of positively transfected cells showed MED1 signal accumulated at the transgene locus (Fig. 10A). This agrees with our conclusion from the (+)-JQ1 experiment that BRD4 plays a major role in the recruitment of P-TEFb and a partial role in the recruitment of MED1 to the transgene site. These results show that tethering BRD4, or just the C-terminal region of BRD4, to the transgene array is sufficient to recruit P-TEFb. In addition, both BRD4L and BRD4C could weakly activate the transgene transcription when they were tethered to the LacO locus using the LacI-CHERRY fusion proteins (Fig. 10B). In fact, quantification analysis showed 43% LacI-CHERRY-BRD4C-positive cells and 20% of LacI-CHERRY-BRD4L-positive cells with weak YFP-MS2 accumulation at the transgene array locus (Fig. 10C). The lower percentage with the BRD4L construct was probably due to the fact that BRD4L is expressed at a lower level compared with the smaller BRD4C molecule (data not shown). RT-qPCR analysis further confirmed that BRD4C or BRD4L tethered to the LacO array by LacI-CHERRY fusions weakly induced transgene transcription (data not shown). These results, together with the JQ1 data described above, strongly support the hypothesis that recruitment of BRD4 contributes to the BRD4-NUT-induced in situ gene transcriptional activation.
FIGURE 10.

BRD4 and BRD4C recruit interacting factors to the transgene foci and slightly activate in situ gene transcription. A, U2OS 2-6-3 cells were transfected with LacI-CHERRY control, LacI-CHERRY-BRD4C, or LacI-CHERRY-BRD4L. 24 h post-transfection, cells were immunostained with p300, AcH4, Cdk9, Cyclin T1, or MED1 antibodies and counterstained with DAPI. The percentage of cells with the indicated cellular factors recruited to the transgene foci was calculated from more than 100 positively transfected cells. Values represent the mean ± S.D. of three independent experiments. B, U2OS 2-6-3 YFP-MS2 stable cells were transfected with LacI-CHERRY control, LacI-CHERRY-BRD4C, or LacI-CHERRY-BRD4L. 24 h post-transfection, cells were fixed and counterstained with DAPI. The RGB intensity profiles show the intensity curve of red, green, and blue signals along the highlighted yellow bars in the Merge panel. Scale bars = 5 μm. C, More than 100 positively transfected cells from B were quantified to calculate the percentage of cells with YFP-MS2 accumulated at the transgene foci. Values represent the mean ± S.D. of three independent experiments.
DISCUSSION
Previous studies from our group and others showed that the NUT moiety of the BRD4-NUT fusion protein strongly interacts with and recruits HATs to induce histone hyperacetylation in discrete chromatin domains (21, 22, 25). This effect causes sequestration of BRD4 and associated transcriptional factors into the hyperacetylated chromatin foci and repression of global transcription (21, 22). BRD4-NUT has also been shown to enrich at specific genes, such as c-MYC and SOX2, to stimulate gene expression (24, 25). However, it is unclear how BRD4-NUT affects the transcription of other genes with which it associates at the whole-genome level.
In this study, we examined the transcriptional regulation function of NUT protein using the well established transgene array in U2OS 2-6-3 cells (26), which provides a powerful tool to obtain an integrative view of in situ transcriptional dynamics in single living cells. By tethering the NUT protein to the integrated transgene array, we were able to recapitulate the NUT chromatin interaction observed in punctuate “NUT foci” in the nuclei of NMC cells and to visualize the in situ transcription regulation function of NUT in live cells. Using this system, we demonstrated that NUT protein in the NUT focus stimulates the transgene expression by recruiting HATs such as p300/CBP, which induces regional histone hyperacetylation and enrichment of BRD4 and associated transcription activators, including P-TEFb and MED1. As a consequence of the exceptional ability of NUT to induce histone hyperacetylation and recruit BRD4 cofactors, NUT expression leads to a much more robust stimulation of the transgene activation than BRD4L and BRD4C molecules (compare the YFP-MS2 data in Figs. 7B and 10C, and also compare the YFP-MS2 signal in Figs. 1B and 10B). Building on these observations and on published studies (21, 22, 25), we proposed a working model for the BRD4-NUT oncogenic function in NMCs (Fig. 11). In this model, BRD4-NUT is enriched in discrete chromatin foci where it recruits p300/CBP, and possibly other HATs, through the NUT moiety to stimulate regional histone hyperacetylation. This leads to enrichment of BRD4 and associated transcription factors like P-TEFb and MED1 to specifically activate gene expression within the hyperacetylated chromatin foci (Fig. 11). These highly acetylated chromatin foci may sequester transcription factors away from other genes, such as the differentiation regulator c-FOS, outside of the hyperacetylated chromatin regions to cause transcription repression (22). BRD4-NUT enriched at SOX2 and c-MYC stimulates their transcription (24, 25), providing strong support for this model. In line with this model, NUT fragment 346–593, which encodes the p300-binding site in NUT, functions as dominant-negative inhibitor to block p300 recruitment and to inhibit in situ transgene expression (Figs. 8 and 11). In addition, treatment with (+)-JQ1, which specifically dissociates BRD4 from chromatin without affecting the NUT protein tethered to the LacO transgene locus, inhibits transgene activation, demonstrating the supporting role of BRD4 in NUT-induced in situ transcriptional activation (Figs. 9 and 11). Together, these studies provide a comprehensive understanding of the mechanistic details underlying the transcription regulation activity of BRD4-NUT fusion oncogene, providing new insights to determine how BRD4-NUT fusion oncogene perturbs normal cellular gene expression to trigger oncogenic events in the highly lethal NMC carcinomas.
FIGURE 11.
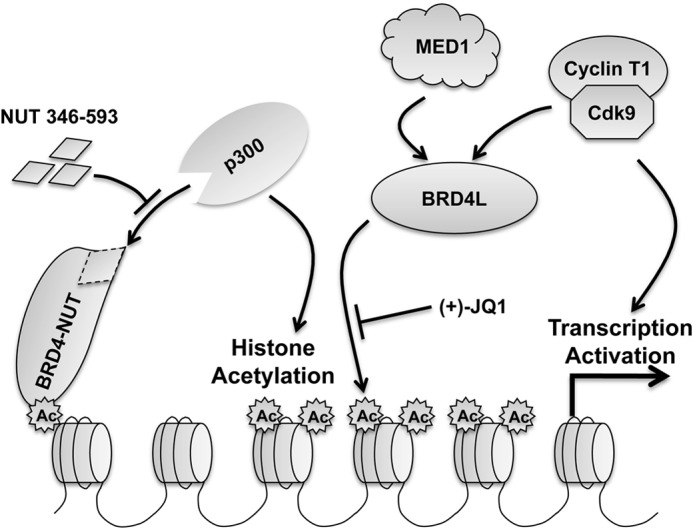
Model for the NUT-induced transcriptional activation. In NMC cells, NUT protein tethered to chromatin by BRD4, BRD3, or NSD3 recruits and activates p300, which acetylates nearby histones to induce the formation of hyperacetylated chromatin foci. The highly acetylated histones result in tighter chromatin binding of BRD4, which sequesters additional transcriptional factors, such as P-TEFb and MED1, to stimulate gene expression in the hyperacetylated chromatin domain while repressing genes present outside of these regions. NUT fragment 346–593 functions as a dominant-negative inhibitor to block p300 recruitment and to inhibit downstream transcription activation. In addition, (+)-JQ1 dissociates BRD4 and its interacting factors from the hyperacetylated chromatin region to repress downstream transcription activation.
Building on our observation, we hypothesized that BRD4-NUT-mediated HAT recruitment causes a dramatic alteration in the chromatin epigenetic landscape, perturbing normal BRD4 transcription function and downstream target gene expression, which cumulatively contribute to the highly aggressive tumorigenic activity in NMCs. It appears that BRD4-NUT may activate the genes it binds but represses the genes that localize outside of the BRD4-NUT hyperacetylated chromatin domain. However, this hypothesis needs to be formally tested in future studies using a parallel analysis of genome-wide BRD4-NUT target genes and BRD4-NUT chromatin binding sites to determine whether BRD4-NUT is specifically recruited to the activated genes but sequestered away from the repressed genes in NMC. Specific enrichment of BRD4-NUT at the activated genes will support our model, in which BRD4-NUT stimulates these genes in NMC by recruiting HATs, BRD4, and other transcription factors.
It has been reported previously that the mobility of GFP-BRD4-NUT was ∼2-fold less than that of GFP-BRD4, as determined by fluorescence recovery after photobleaching technology (4, 23). Both BRD4 and BRD4-NUT use the same double bromodomains to associate with chromatin. Therefore, it was unclear how the NUT moiety contributes to the relatively higher chromatin affinity of BRD4-NUT. In this study, our biochemical analysis showed that BRD4-NUT causes BRD4 to bind more tightly to chromatin because the NUT moiety of the fusion protein interacts with and stimulates HATs, inducing histone hyperacetylation. It is likely that NUT-stimulated histone hyperacetylation similarly contributes to the tight BRD4-NUT binding to chromatin.
NMC tumors are unequivocally resistant to conventional chemotherapy (1). Currently there is no specific therapeutic regime available for curing this cancer. Our studies showed that the p300-binding NUT 346–593 domain functions as a dominant-negative inhibitor to significantly abolish p300 recruitment as well as NUT-induced gene transactivation (Fig. 8), demonstrating that sequestration of p300 by BRD4-NUT is critical for the downstream gene activation. This result provides a proof of principle example to support the hypothesis that breaking the BRD4-NUT and p300 interaction is an excellent strategy to abrogate the BRD4-NUT oncogenic activities. This study, therefore, establishes a platform for the future identification of small molecules that can specifically inhibit the abnormal BRD4-NUT and HAT interaction to prevent the growth of NMC tumors but have little effect on normal cells that do not express BRD4-NUT. These molecules will represent novel therapeutics to treat this highly malignant cancer that is resistant to conventional therapy (1).
Many epigenetic readers such as BRD4 recognize various histone modifications and translate the “histone codes” into gene transcription activities, providing a tight control for cell proliferation. Dysfunction of this process frequently leads to cancers and other human diseases, underscoring the biological significance of epigenetic signaling. BRD4 has been implicated in the pathogenesis of a variety of cancers (18–20, 33, 34). However, its role in the cancer development process remains largely undefined. The naturally occurring BRD4-NUT oncogenic mutation provides an exciting opportunity to determine how alteration of BRD4 function leads to cancer. By elucidating the BRD4-NUT function in modulating BRD4 chromatin association and transcription regulation in NMC tumors, our findings provide new insights for studying the oncogenic mechanisms of other BRD4-associated cancers.
Acknowledgments
We thank Susan M. Janicki for providing the U2OS 2-6-3 cell line and related reagent and members of our laboratory for helpful discussions and critical review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA148768 and R01CA142723.
- NMC
- NUT midline carcinoma
- NUT
- nuclear protein in testis
- BET
- bromodomain and extraterminal
- P-TEFb
- positive transcription elongation factor b
- HAT
- histone acetyltransferase
- CREB
- cAMP-response element-binding protein
- CBP
- CREB-binding protein
- TSA
- trichostatin A
- RGB
- red, green, and blue
- qPCR
- quantitative PCR.
REFERENCES
- 1. French C. A. (2012) Pathogenesis of NUT midline carcinoma. Annu. Rev. Pathol. 7, 247–265 [DOI] [PubMed] [Google Scholar]
- 2. French C. A., Miyoshi I., Kubonishi I., Grier H. E., Perez-Atayde A. R., Fletcher J. A. (2003) BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 63, 304–307 [PubMed] [Google Scholar]
- 3. Bauer D. E., Mitchell C. M., Strait K. M., Lathan C. S., Stelow E. B., Lüer S. C., Muhammed S., Evans A. G., Sholl L. M., Rosai J., Giraldi E., Oakley R. P., Rodriguez-Galindo C., London W. B., Sallan S. E., Bradner J. E., French C. A. (2012) Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin. Cancer Res. 18, 5773–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. French C. A., Ramirez C. L., Kolmakova J., Hickman T. T., Cameron M. J., Thyne M. E., Kutok J. L., Toretsky J. A., Tadavarthy A. K., Kees U. R., Fletcher J. A., Aster J. C. (2008) BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 27, 2237–2242 [DOI] [PubMed] [Google Scholar]
- 5. French C. A., Rahman S., Walsh E. M., Kühnle S., Grayson A. R., Lemieux M. E., Grunfeld N., Rubin B. P., Antonescu C. R., Zhang S., Venkatramani R., Dal Cin P., Howley P. M. (2014) NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discov. 4, 928–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. French C. A. (2010) NUT midline carcinoma. Cancer Genet. Cytogenet. 203, 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dey A., Chitsaz F., Abbasi A., Misteli T., Ozato K. (2003) The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U.S.A. 100, 8758–8763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maruyama T., Farina A., Dey A., Cheong J., Bermudez V. P., Tamura T., Sciortino S., Shuman J., Hurwitz J., Ozato K. (2002) A mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol. Cell Biol. 22, 6509–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Houzelstein D., Bullock S. L., Lynch D. E., Grigorieva E. F., Wilson V. A., Beddington R. S. (2002) Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell Biol. 22, 3794–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nishiyama A., Dey A., Miyazaki J., Ozato K. (2006) Brd4 is required for recovery from antimicrotubule drug-induced mitotic arrest: preservation of acetylated chromatin. Mol. Biol. Cell 17, 814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dey A., Ellenberg J., Farina A., Coleman A. E., Maruyama T., Sciortino S., Lippincott-Schwartz J., Ozato K. (2000) A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Mol. Cell Biol. 20, 6537–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mochizuki K., Nishiyama A., Jang M. K., Dey A., Ghosh A., Tamura T., Natsume H., Yao H., Ozato K. (2008) The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 283, 9040–9048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Z., He N., Zhou Q. (2008) Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell Biol. 28, 967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu S. Y., Chiang C. M. (2007) The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 282, 13141–13145 [DOI] [PubMed] [Google Scholar]
- 15. Zhao R., Nakamura T., Fu Y., Lazar Z., Spector D. L. (2011) Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 13, 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rahman S., Sowa M. E., Ottinger M., Smith J. A., Shi Y., Harper J. W., Howley P. M. (2011) The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 31, 2641–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lovén J., Hoke H. A., Lin C. Y., Lau A., Orlando D. A., Vakoc C. R., Bradner J. E., Lee T. I., Young R. A. (2013) Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zuber J., Shi J., Wang E., Rappaport A. R., Herrmann H., Sison E. A., Magoon D., Qi J., Blatt K., Wunderlich M., Taylor M. J., Johns C., Chicas A., Mulloy J. C., Kogan S. C., Brown P., Valent P., Bradner J. E., Lowe S. W., Vakoc C. R. (2011) RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delmore J. E., Issa G. C., Lemieux M. E., Rahl P. B., Shi J., Jacobs H. M., Kastritis E., Gilpatrick T., Paranal R. M., Qi J., Chesi M., Schinzel A. C., McKeown M. R., Heffernan T. P., Vakoc C. R., Bergsagel P. L., Ghobrial I. M., Richardson P. G., Young R. A., Hahn W. C., Anderson K. C., Kung A. L., Bradner J. E., Mitsiades C. S. (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodriguez R. M., Huidobro C., Urdinguio R. G., Mangas C., Soldevilla B., Domínguez G., Bonilla F., Fernandez A. F., Fraga M. F. (2012) Aberrant epigenetic regulation of bromodomain BRD4 in human colon cancer. J. Mol. Med. 90, 587–595 [DOI] [PubMed] [Google Scholar]
- 21. Reynoird N., Schwartz B. E., Delvecchio M., Sadoul K., Meyers D., Mukherjee C., Caron C., Kimura H., Rousseaux S., Cole P. A., Panne D., French C. A., Khochbin S. (2010) Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J. 29, 2943–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yan J., Diaz J., Jiao J., Wang R., You J. (2011) Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. J. Biol. Chem. 286, 27663–27675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W. B., Fedorov O., Morse E. M., Keates T., Hickman T. T., Felletar I., Philpott M., Munro S., McKeown M. R., Wang Y., Christie A. L., West N., Cameron M. J., Schwartz B., Heightman T. D., La Thangue N., French C. A., Wiest O., Kung A. L., Knapp S., Bradner J. E. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grayson A. R., Walsh E. M., Cameron M. J., Godec J., Ashworth T., Ambrose J. M., Aserlind A. B., Wang H., Evan G. I., Kluk M. J., Bradner J. E., Aster J. C., French C. A. (2014) MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene 33, 1736–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang R., Liu W., Helfer C. M., Bradner J. E., Hornick J. L., Janicki S. M., French C. A., You J. (2014) Activation of SOX2 expression by BRD4-NUT oncogenic fusion drives neoplastic transformation in NUT midline carcinoma. Cancer Res. 74, 3332–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Janicki S. M., Tsukamoto T., Salghetti S. E., Tansey W. P., Sachidanandam R., Prasanth K. V., Ried T., Shav-Tal Y., Bertrand E., Singer R. H., Spector D. L. (2004) From Silencing to gene expression: real-time analysis in single cells. Cell 116, 683–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yan J., Li Q., Lievens S., Tavernier J., You J. (2010) Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 84, 76–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J., Li Q., Diaz J., You J. (2014) Brd4-mediated nuclear retention of the papillomavirus E2 protein contributes to its stabilization in host cells. Viruses 6, 319–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rafalska-Metcalf I. U., Powers S. L., Joo L. M., LeRoy G., Janicki S. M. (2010) Single cell analysis of transcriptional activation dynamics. PLoS ONE 5, e10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dang T. P., Gazdar A. F., Virmani A. K., Sepetavec T., Hande K. R., Minna J. D., Roberts J. R., Carbone D. P. (2000) Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J. Natl. Cancer Inst. 92, 1355–1357 [DOI] [PubMed] [Google Scholar]
- 31. McPhillips M. G., Ozato K., McBride A. A. (2005) Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 79, 8920–8932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang R., Li Q., Helfer C. M., Jiao J., You J. (2012) Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 287, 10738–10752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mertz J. A., Conery A. R., Bryant B. M., Sandy P., Balasubramanian S., Mele D. A., Bergeron L., Sims R. J., 3rd. (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U.S.A. 108, 16669–16674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crawford N. P., Alsarraj J., Lukes L., Walker R. C., Officewala J. S., Yang H. H., Lee M. P., Ozato K., Hunter K. W. (2008) Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. U.S.A. 105, 6380–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]