

Background: Cyclic di-AMP is an essential second messenger in eubacteria.
Results: The c-di-AMP receptor DarA was identified in B. subtilis. The crystal structure and ITC data revealed the nucleotide specificity of DarA.
Conclusion: DarA is a PII-like protein that undergoes conformational changes upon c-di-AMP binding.
Significance: A novel PII-like protein is involved in c-di-AMP signaling.
Keywords: Bacterial Signal Transduction, Crystal Structure, Cyclic Di-GMP (c-di-GMP), Cyclic Diadenosine Monophosphate (c-di-AMP), Isothermal Titration Calorimetry (ITC)
Abstract
The cyclic dimeric AMP nucleotide c-di-AMP is an essential second messenger in Bacillus subtilis. We have identified the protein DarA as one of the prominent c-di-AMP receptors in B. subtilis. Crystal structure analysis shows that DarA is highly homologous to PII signal transducer proteins. In contrast to PII proteins, the functionally important B- and T-loops are swapped with respect to their size. DarA is a homotrimer that binds three molecules of c-di-AMP, each in a pocket located between two subunits. We demonstrate that DarA is capable to bind c-di-AMP and with lower affinity cyclic GMP-AMP (3′3′-cGAMP) but not c-di-GMP or 2′3′-cGAMP. Consistently the crystal structure shows that within the ligand-binding pocket only one adenine is highly specifically recognized, whereas the pocket for the other adenine appears to be promiscuous. Comparison with a homologous ligand-free DarA structure reveals that c-di-AMP binding is accompanied by conformational changes of both the fold and the position of the B-loop in DarA.
Introduction
All living cells have to monitor their environment to sense any relevant changes, to transduce this information, and to respond by appropriate adaptation. Inability to survey the environment would inevitably result in loss of fitness, and other cells or species would rapidly take over. Therefore, interaction with the environment through adaptive responses is one of the hallmarks of life, and it contributes to the ability of organisms to live and survive in highly competitive ecosystems.
In bacteria, alterations in gene expression programs are a major factor in response to changing conditions. This is also reflected in the short half-lives of bacterial mRNAs that persist only in the minute range. In addition to adaptation at the level of gene regulation, pre-existing enzymes and proteins may receive signals to alter their activity. Both types of response often involve common principles of signal transduction such as the formation of second messengers or the use of dedicated signal transduction proteins.
We are interested in signaling pathways triggered by second messengers. Typically, specific nucleotides that are not incorporated into nucleic acids serve as second messengers, which are formed in response to extra- or intracellular signals. The paradigmatic prototype of this class of molecules, cyclic AMP (cAMP), is found in both pro- and eukaryotic cells. In Escherichia coli and other enteric bacteria, its presence is a signal for the lack of preferred carbon sources. The molecule binds to its receptor protein Crp, and this complex activates the expression of genes for the utilization of secondary carbon sources. In addition, cAMP is involved in a variety of functions including the control of virulence in many pathogenic bacteria (1, 2). In nearly all bacteria, the stringent response factor (p)ppGpp serves to switch off cellular household activities in response to nutrient limitation (3). In the past few years, cyclic dinucleotides have attracted much attention. Cyclic di-GMP is widespread in bacteria, and this second messenger is implicated in switching between two mutually exclusive lifestyles: motility and biofilm formation (4). Cyclic di-AMP, which was discovered in 2008 (5), is thought to be common in Gram-positive bacteria including many pathogens but seems to occur only in few other bacteria (6). Importantly, low-GC Gram-positive bacteria are unable to live in the absence of c-di-AMP as demonstrated by the essentiality of the genes for the diadenylate cyclases (7–10). Thus, c-di-AMP is so far the only known second messenger that is essential for the bacteria that produce it. Only recently, the composite cyclic dinucleotide cyclic GMP-AMP (3′3′-cGAMP)4 has been discovered in Vibrio cholerae. This second messenger is implicated in intestinal colonization of the pathogen (11).
The Gram-positive model organism Bacillus subtilis produces ppGpp, c-di-GMP, and c-di-AMP (9, 12–15). Cyclic di-AMP can be synthesized by three different enzymes in this organism. The DNA integrity-scanning protein DisA produces c-di-AMP in response to the integrity of the DNA. DisA interacts with several DNA repair and recombination proteins and is required to guarantee that outgrowing spores are free of DNA damage (5, 16–18). The second vegetative diadenylate cyclase CdaA seems to be the most important one as it is the sole enzyme present in all low-GC Gram-positive pathogens including Staphylococcus aureus and Listeria monocytogenes. In those organisms, the corresponding gene is essential, suggesting that c-di-AMP formed by CdaA is decisive for the essentiality of this second messenger (7, 8). CdaA-derived c-di-AMP has been implicated in cell wall metabolism and cell division as well as in the control of central metabolism and potassium homeostasis (9, 10, 19–21). Finally, the sporulation-specific diadenylate cyclase CdaS is required for efficient spore germination (22).
The various cellular functions of c-di-AMP as well as the essentiality of this second messenger ignited a strong interest in the identification of binding partners of the nucleotide. The first identified target was the DarR transcription factor from Mycobacterium smegmatis (17). In S. aureus, the potassium transporter KtrA, the putative transporter CpaA, the KdpD histidine kinase, and the PII-like protein PstA were identified (20). For KtrA, inhibition of potassium uptake upon binding of c-di-AMP was reported (20). This observation was recently confirmed for the corresponding protein CabP from Streptococcus pneumoniae (23). Further studies aimed at the identification of the controlling metabolite of the B. subtilis ydaO riboswitch identified the binding of c-di-AMP to this RNA structure (24, 25). It is interesting to note that the ydaO riboswitch controls two transcription units in B. subtilis: the ydaO gene and the ktrAB operon that encodes the c-di-AMP-responsive potassium transporter (26). Thus, c-di-AMP is the only second messenger that cannot only bind to two different classes of receptors, proteins and RNA, but that also controls one biological process by binding to a protein and by controlling its expression via RNA binding! Very recently, a proteome-wide study with L. monocytogenes confirmed c-di-AMP binding of PstA and led to the identification of the pyruvate carboxylase, an enzyme that replenishes the citric acid cycle as a direct target of c-di-AMP-mediated inhibition (21).
In this work, we aimed to identify c-di-AMP targets in the Gram-positive model organism B. subtilis. Our results suggest that the PII-like signal transduction protein DarA is a major target of c-di-AMP in this bacterium. This finding suggests the integration of two signal transduction principles, second messengers and small signal transduction proteins, in a common mechanism. The determination of the structure of DarA in complex with c-di-AMP defines specificity determinants for the interaction with c-di-AMP and reveals conformational changes upon c-di-AMP binding.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
All B. subtilis strains are derivatives of the wild type strain 168. E. coli DH5α (27) was used for cloning experiments and for the expression of recombinant protein. B. subtilis was grown in sporulation medium (28), in CSE defined minimal medium (28), or in Luria-Bertani broth (LB) (27). E. coli was grown in LB medium, and transformants were selected on plates containing ampicillin (100 μg/ml). LB and sporulation plates were prepared by the addition of 17 g of Bacto agar/liter (Difco) to LB or sporulation medium, respectively.
DNA Manipulation
Transformation of E. coli and plasmid DNA extraction were performed using standard procedures (27). Restriction enzymes, T4 DNA ligase, and DNA polymerases were used as recommended by the manufacturers. DNA sequences were determined using the dideoxy chain termination method (27). Chromosomal DNA of B. subtilis was isolated as described (28).
Pulldown of Cytosolic Proteins via c-di-AMP-coupled Beads
To isolate c-di-AMP-binding proteins from B. subtilis, biotinylated c-di-AMP (2′-[biotin]-AHC-c-di-AMP; BIOLOG, Bremen, Germany) was coupled to Strep-Tactin-covered MagStrep “type 2HC” beads (IBA, Göttingen, Germany). Briefly, B. subtilis crude cell extracts were incubated in an assay mixture (1.2 mg of protein extract, 10% (w/v) glycerol, 4 mm EDTA, 50 μg/ml bovine serum albumin) in a total of 1 ml of binding buffer (1 mm MgCl2, 5 mm Tris-HCl, pH7.5, 230 mm NaCl, 0.5 mm DTT, 1× protease inhibitor (Roche Diagnostics)). To prepare the matrix, 40 μl of Strep-Tactin-covered beads were equilibrated twice with 800 μl of binding buffer containing glycerol (10%), bovine serum albumin (50 μg/ml), and EDTA (4 mm). After equilibration, 40 μl of biotinylated c-di-AMP was added, and the tubes were incubated with weak shaking for 15 min at room temperature. After two washes of the beads (800 μl of binding buffer containing glycerol (10%), bovine serum albumin (50 μg/ml), and EDTA (4 mm) to discard free c-di-AMP), the prepared protein extracts (1.2 mg) were added to the charged beads and incubated for 30 min with slight shaking at room temperature. After incubation, the samples were washed four times to eliminate all proteins that might have bound nonspecifically to c-di-AMP. The washing buffer contained Tris-HCl, pH 8.0 (100 mm), NaCl (150 mm), and EDTA (1 mm). Proteins bound to c-di-AMP were eluted by incubation with 50 μl of biotin buffer (28) that contained 5 mm biotin. Samples were analyzed by SDS-PAGE with subsequent silver staining. To confirm the specificity of binding, the experiment was repeated using 8′-[biotin]-AET-c-di-AMP (BIOLOG).
Proteins of interest were identified by mass spectrometry as described previously (29). Briefly, after destaining gel pieces, proteins underwent in-gel digestion with trypsin (10 ng/μl trypsin in 20 mm ammonium bicarbonate). Resulting peptides were extracted from gel pieces separated by a non-linear water-acetonitrile gradient in 0.1% acetic acid on a nanoAcquity UPLC reverse phase column (BEH130, C18, 100 μm × 100 mm; Waters Corp., Milford, MA) with a nano-UPLC system (Waters) coupled on line with an LTQ-Orbitrap Velos mass spectrometer (Thermo Electron, Bremen, Germany) operated in data-dependent MS/MS mode. Proteins were identified by searching all MS/MS spectra in.dta format against a B. subtilis protein database (4254 entries; extracted from SubtiList using SEQUEST version 2.7 revision 11 (Sorcerer build 4.04, Sage-N Research Inc., Milpitas, CA). Initial mass tolerances for peptide identification on MS and MS/MS peaks were 10 ppm and 1 Da, respectively. Up to two missed tryptic cleavages were allowed. Methionine oxidation (+15.99492 Da) and propionamide modification on cysteine (+71.037109 Da) were set as variable modifications. Protein identification results were evaluated by determination of probability for peptide and protein assignments provided by PeptideProphet and ProteinProphet (Institute of Systems Biology, Seattle, WA) incorporated in the Scaffold software package release 4.3.2 (Proteome Software, Portland, OR). Proteins were identified by at least two peptides with a peptide probability >90%, reflecting a protein probability of >99%. Annotation information was derived from the SubtiWiki web server (30).
Expression and Purification of DarA
To fuse the DarA protein (previously referred to as YaaQ; see SubtiWiki: DarA (30)) with a His tag at its N terminus, the darA coding sequence was amplified. The PCR product was digested with PstI and BamHI, and the resulting fragment was cloned into the expression vector pWH844 (31) to yield plasmid pGP2601. For the purification of His6-PtsH (SubtiWiki: PtsH (30)), we used plasmid pAG2 (32). Expression of the recombinant proteins was induced by the addition of isopropyl 1-thio-β-d-galactopyranoside (final concentration, 1 mm) to exponentially growing cultures (A600 of 0.8) of E. coli carrying the relevant plasmid. Cells were lysed by three passes at 18,000 p.s.i. through an HTU DIGI-F press (G. Heinemann, Germany). After lysis, the crude extract was centrifuged at 100,000 × g for 60 min and then passed over a Ni2+-nitrilotriacetic acid column (IBA). The protein was eluted with an imidazole gradient. After elution, the fractions were tested for the desired protein using 15% SDS-PAGE. The relevant fractions were combined and dialyzed overnight. The protein concentration was determined according to the method of Bradford (55) using the Bio-Rad dye binding assay and bovine serum albumin as the standard.
Assay for c-di-AMP Binding of Tagged Proteins Purified from E. coli
To investigate, whether His6-tagged DarA and PtsH bind to c-di-AMP, the proteins were expressed in E. coli (see above), and cytoplasmic cell extracts were incubated with 40 μl of biotinylated c-di-AMP (2′-[biotin]-AHC-c-di-AMP; 1 nm). For the negative control, cytoplasmic cell extracts were incubated without biotinylated c-di-AMP. After incubation, the samples were purified according to the standard protocol for purification of Strep-tagged proteins (28). The samples were analyzed by SDS-PAGE.
Construction of a darA Mutant Strain by Allelic Replacement
Deletion of the darA gene was achieved by transformation of B. subtilis 168 with a PCR product constructed using oligonucleotides to amplify DNA fragments flanking the darA gene and an intervening chloramphenicol resistance cassette (cat) as described previously (9). The integrity of the regions flanking the integrated resistance cassette was verified by sequencing PCR products of about 1,000 bp amplified from chromosomal DNA of the resulting mutant strain GP1712 (ΔdarA::cat).
Quantification of 2′3′-cGAMP and 3′3′-cGAMP in B. subtilis by LC-MS/MS
Cultures of B. subtilis 168 were grown in CSE minimal medium with glucose as the carbon source, pelleted, and lysed in a Micro Dismembrator S instrument (Sartorius). The resulting cell powder was used to extract the metabolites as described previously (14).
Reversed phase chromatographic separation of 2′3′-cGAMP and 3′3′-cGAMP was performed with an HPLC system (Shimadzu) using a Zorbax eclipse XDB-C18 1.8-μm column (50 × 4.6 mm) from Agilent connected to a C18 SecurityGuard (Phenomenex) and a 2-μm column saver (Supelco), all kept at 25 °C. The mobile phases were 3:97 methanol/water (v/v) (A) and 97:3 methanol/water (v/v) (B), each containing 50 mm ammonium acetate and 0.1% acetic acid. The following gradient was applied: 0–5 min, 0–50% B and 5–8 min, 0% B. The flow rate was 400 μl/min. Detection and quantification of 2′3′-cGAMP and 3′3′-cGAMP (authentic standards from BIOLOG) were carried out by a tandem mass spectrometer, 5500QTRAP (AB Sciex), equipped with an electrospray ionization source operating in positive ionization mode. For selected reaction monitoring detection, the following mass transitions were applied for 2′3′-cGAMP and 3′3′-cGAMP: m/z 328.0 [M + 2H]2+/136.2 [M + H]+ (quantifier), m/z 328.0 [M + 2H]2+/152.1 [M + H]+ (identifier). Tenofovir (obtained through the National Institutes of Health AIDS Research and Reference Reagent Program) was applied as an internal standard (m/z 288.0 [M + H]+/176.0 [M + H]+ (quantifier), m/z 288.0 [M + H]+/159.1 [M + H]+ (identifier)).
Assay of Nucleotide Binding of DarA by Isothermal Calorimetry
All ITC experiments were carried out with an ITC 200 microcalorimeter (MicroCal Inc., Northampton, MA; reaction cell volume, 210 μl). The protein solution containing His-tagged DarA was exhaustively dialyzed against 50 mm Tris-HCl, pH 7.5, 200 mm NaCl prior to ITC measurements, and tested ligands (c-di-AMP, c-di-GMP, 3′3′-cGAMP, 2′3′-cGAMP, AMP, ADP, and ATP) were individually dissolved in the same buffer. In a typical setup, DarA (82 μm in 50 mm Tris-HCl, pH 7.5, 200 mm NaCl) was placed in the sample cell, and the ligand (1 mm in the same buffer) was placed in the titration syringe. All experiments were carried out at 20 °C with a reference power of 7 and a stirring speed of 500 rpm. The parameters used for the titration series are given in Table 1. Data analysis was carried out using Microcal Origin software.
TABLE 1.
Titration parameters
| Injection no. | Injection volume | Injection duration | Spacing | Filter period | cligand in cell |
|---|---|---|---|---|---|
| μl | s | s | s | μm | |
| 1 | 0.2 | 0.2 | 150 | 2 | |
| 2–20 | 2 | 4 | 150 | 2 | 6.2–106.1 |
Crystallization and Data Collection
DarA was subjected to initial crystallization trials at a concentration of 7.5 mg/ml with a 2-fold molar excess of c-di-AMP (BIOLOG). The best diffracting crystals grew in 10% poly-γ-glutamic acid polymer LM (200–400-kDa low molecular mass polymer), 0.4 m sodium formate, and 0.1 m imidazole HCl, pH 6.5. The oscillation images were collected at the European Synchrotron Radiation Facility (ID23-1) and processed with XDS (33, 34). The scaling process revealed a primitive hexagonal lattice with unit cell parameters of a = b = 56.5, c = 58.2 Å, α = β = 90.00°, and γ = 120.00°. Systematic absences along the b axis indicated the space group to be P6(3). The Matthews coefficient (Vm = 2.3 Å3/Da) suggested one molecule in the asymmetric unit corresponding to a solvent content of 41%. The data collection statistics are summarized below (also see Table 2).
TABLE 2.
Data collection and refinement statistics
Values in parentheses refer to the highest resolution shell. r.m.s., root mean square; CC1/2, Pearson correlation coefficient calculated between intensities from random half-datasets.
| Wavelength (Å) | 0.91000 |
| Resolution range (Å) | 37.46–1.30 (1.35–1.30) |
| Space group | P6 (3) |
| Unit cell (Å; °) | 56.52, 56.52, 58.20; 90.0, 90.0, 120.0 |
| Unique reflections | 26,017 (2,789) |
| Multiplicity | 6.6 (6.3) |
| Completeness (%) | 99.96 (99.98) |
| Mean I/σ(I) | 38.86 (10.49) |
| Wilson B-factor (Å2) | 13.75 |
| Rmerge (%) | 2.5 (17.6) |
| CC1/2 | 100 (98) |
| Rwork | 0.1296 (0.2468) |
| Rfree | 0.1545 (0.2572) |
| Number of non-hydrogen atoms | 1,204 |
| Macromolecules | 1,012 |
| Ligands | 45 |
| Water | 145 |
| Protein residues | 116 |
| r.m.s. bonds (Å) | 0.009 |
| r.m.s. angles (°) | 1.47 |
| Ramachandran favored (%) | 95.0 |
| Ramachandran outliers (%) | 2.3 |
| Clash score | 8.15 |
| Average B-factor (Å2) | 35.30 |
| Macromolecules | 35.80 |
| Ligands | 15.20 |
| Solvent | 38.10 |
Structure Determination and Refinement
Initial phases were obtained by a molecular replacement method with PHASER using the structure of the protein of unknown function PEPE_1480 from Pediococcus pentosaceus ATCC 25745 (Protein Data Bank code 3M05) as search model. This search model was identified based on an HHpred (35) search and trimmed to the last common Cγ atoms using phenix.sculptor prior to the molecular replacement search. The model was manually rebuilt using Coot (36) and refined with PHENIX (37) using standard parameters including automatic building of solvent molecules. A random set of 3.5% of reflections was excluded from refinement to monitor Rfree (38). The final model has been refined at a resolution of 1.3 Å to R and Rfree factors of 12.96 and 15.45%, respectively. The model consists of one protein molecule encompassing residues −6 to 109, 145 water molecules, one c-di-AMP molecule, and one Ni2+ ion. Alternate side chain conformations were built for 14 residues (Ser-0, Met-1, Ser-13, Arg-26, Lys-29, Thr-33, Lys-38, Ser-39, Phe-44, Met-45, Val-48, Glu-49, Ser-58, and Met-70). The refinement statistics are summarized in Table 2. Surface complementarity coefficients and solvent-accessible surface areas were calculated with SC using a 1.7-Å-radius probe (39). Possible hydrogen bonds, salt bridges, and van der Waals contacts were detected with HBPLUS and CONTACSYM (40) using default parameters. The quality of the model was assessed using MolProbity as implemented in PHENIX. Secondary structure predictions were performed using DSSP (41). Coordinates were superimposed with LSQKAB (42) from the CCP4 program suite (39) or as implemented in PyMOL (Schrödinger, LLC, New York, NY).
RESULTS
Identification of the c-di-AMP Receptor DarA in B. subtilis
To isolate c-di-AMP-binding proteins from a crude extract of B. subtilis, we made use of a biotinylated derivative of the dinucleotide. Because of the strong interaction between biotin and Strep-Tactin, this 2′-[biotin]-AHC-c-di-AMP can be coupled to Strep-Tactin-coated beads. Proteins that interact with c-di-AMP should then bind to these beads and elute upon addition of biotin. To distinguish specific from nonspecific interaction candidates, we incubated Strep-Tactin-coated beads with B. subtilis protein extracts with or without prior saturation of the beads with biotinylated c-di-AMP. As shown in Fig. 1A, two major bands were observed when a crude extract was applied to the beads in the absence of c-di-AMP. These bands correspond to PycA and AccB, the two intrinsically biotinylated proteins of B. subtilis (29). This observation suggests that the experimental system is suited to bind and subsequently elute biotinylated samples. When the beads had been pretreated with biotinylated c-di-AMP, a different picture was obtained. The two biotinylated proteins were not able to bind, indicating that the beads had been saturated with the biotinylated c-di-AMP. In contrast, a single intensive band corresponding to a protein of about 13 kDa was observed after elution from the c-di-AMP-treated beads. To verify the binding of this protein to c-di-AMP, the experiment was repeated with a differently biotinylated dinucleotide, 8′-[biotin]-AET-c-di-AMP. Again, the same band of about 13 kDa was observed (data not shown). The protein was analyzed by mass spectrometry and identified to be the gene product YaaQ. As an additional control for the binding of YaaQ to c-di-AMP, we expressed His-tagged YaaQ in E. coli and performed affinity chromatography using a Strep-Tactin column that had been saturated with biotinylated c-di-AMP. In parallel, the same experiment was performed with His6-PtsH, a similarly small protein, from B. subtilis. As shown in Fig. 1B, YaaQ was specifically purified by this procedure, whereas no binding of PtsH was observed (data not shown). These findings confirm the specific interaction of YaaQ with the second messenger c-di-AMP.
FIGURE 1.

Identification of the c-di-AMP receptor DarA in B. subtilis. A, a pulldown assay was performed with B. subtilis crude extract to identify c-di-AMP-binding proteins. After incubation of Strep-Tactin-covered beads with 2′-AHC-biotinylated c-di-AMP and a B. subtilis crude extract, the samples were washed to eliminate nonspecifically bound proteins. Elution was performed with 5 mm biotin. Samples were analyzed by SDS-PAGE with subsequent silver staining. As the negative control, the crude extract was incubated with Strep-Tactin-covered beads alone (−). B, DarA was overexpressed in E. coli and subsequently incubated with 2′-AHC-biotinylated c-di-AMP. After incubation, the sample was purified using a Strep-Tactin column, and bound protein was eluted with 2.5 mm d-desthiobiotin. As a negative control, the overexpressed protein was purified without addition of 2′-AHC-biotinylated c-di-AMP (−). The last wash fraction (W7) and elution fractions (E1–E3) were analyzed by SDS-PAGE with subsequent silver staining.
Sequence homologs of YaaQ are the PstA proteins of S. aureus and L. monocytogenes that exhibit sequence similarities of 90.8 and 87.2% and identities of 65.1 and 66.1%, respectively, to YaaQ. Both PstA proteins have previously been found to bind c-di-AMP (20, 21). Because the designation PstA is already used in B. subtilis for a phosphate transporter, we renamed the YaaQ protein c-di-AMP receptor A, DarA, and the corresponding gene darA. The purification of DarA from c-di-AMP-coated beads in two different experimental setups as well as the absence of any other convincing candidate suggests that DarA is a major c-di-AMP-binding protein of B. subtilis.
DarA Is Not the Essential Target of c-di-AMP
Because DarA is a prominent c-di-AMP receptor in B. subtilis, it seemed reasonable to assume that it might be responsible for the essentiality of c-di-AMP in this organism. The essentiality of c-di-AMP might be caused either by the binding to an essential protein, triggering its function, or by binding to a toxic protein to counteract toxicity (43). None of the previously studied targets of c-di-AMP (KtrA, PycA, and the ydaO riboswitch) are essential, and c-di-AMP does not become dispensable upon their deletion, suggesting that c-di-AMP does not overcome the potential toxicity of any of these targets.5 To address the implication of darA in the essentiality of c-di-AMP, we first tested whether the gene can be deleted from the genome. This deletion could be obtained (strain GP1712), indicating that DarA is not an essential target of c-di-AMP. To test the alternative possibility that DarA might be toxic to the cell in the absence of c-di-AMP, we attempted to delete all genes encoding diadenylate cyclases (cdaA, disA, and cdaS) from the darA mutant strain. As observed for the wild type strain (9), at least one diadenylate cyclase had to be expressed during logarithmic growth phase for the viability of the darA mutant. Therefore, DarA is not implicated in the essentiality of c-di-AMP. This suggests that at least one other c-di-AMP-binding protein that is essential must exist.
The Crystal Structure of DarA
Crystallization of DarA in the presence of a 2-fold molar excess of c-di-AMP yielded hexagonal crystals diffracting up to 1.30-Å resolution. The crystallographic phase problem was solved by molecular replacement using the structure of a protein of unknown function (Protein Data Bank code 3M05), which was identified by sequence similarity as putative structural homolog, as the search model. The complete amino acid sequence of DarA could be built into the electron density as well as five histidines of the N-terminal His6 tag. Two of these histidines of each subunit are arranged around and coordinate a Ni2+ ion, which presumably has been stripped from the column during purification. The refinement of the crystal structure led to reasonable data statistics (see Table 2).
The overall secondary structure arrangement of DarA is β1-α1-β2-(T-loop)-β3/3′-α2-(B-loop)-β4 (Fig. 2A). This fold is reminiscent of that of PII signal transducer proteins like the PII proteins from E. coli (Protein Data Bank code 2PII) (44) and Synechococcus elongatus (Protein Data Bank code 4AFF) (45) as well as GlnK1 from Methanococcus jannaschii (Protein Data Bank code 2J9D) (46) and GlnK3 from Archaeoglobus fulgidus (47) (Fig. 2B). The most obvious differences between DarA and PII proteins are found in the T-loop (connecting β2 and β3/3′) and the B-loop (connecting α2 and β4). In contrast to the canonical PII proteins, the B- and T-loops of DarA are swapped with respect to their length. Although the B-loop of E. coli PII contains 9 residues, the B-loop of DarA is significantly elongated as it contains 32 residues. The T-loop of DarA is shortened to a small connecting loop of 7 residues, whereas it comprises 21 residues in E. coli PII. Further structural differences are found in the N-terminal region (Fig. 2B). The PII proteins carry an N-terminal extension forming a structurally defined motif with either α-helices or β-strands, which are lacking in DarA.
FIGURE 2.

Structure of DarA. A, the monomer structure of DarA presented as a schematic colored in rainbow mode from N terminus (blue) to C terminus (red). The numbering indicates the sequential arrangement of secondary structure elements (α-helices and β-strands). Functionally important B- and T-loops are indicated. B, the fold of DarA is highly similar to that of PII proteins as shown by the superposition of DarA (purple) and PII (Protein Data Bank code 2PII; colored gold). The most striking differences concern the length of the T- and B-loops and the N-terminal region, respectively. For clarity reasons, the N-terminal His6 tag of DarA is not shown. C, the unpublished structure of a protein of unknown function from P. pentosaceus (Protein Data Bank code 3M05; purple) clearly resembles DarA especially regarding the T- and B-loops and residues involved in c-di-AMP binding, indicating a homologous function. The structures of other homologous proteins (Protein Data Bank codes 2EG1, 2CE8, and 3L7P) belong to the family of canonical PII proteins. D, two perpendicular views of the DarA homotrimer. The three subunits are colored blue, green, and beige.
A search for structurally homologous proteins by DALI (48) identified four unpublished structures with a high similarity to the structure of DarA (Protein Data Bank codes 2EG1, 3CE8, 3L7P, and 3M05). Three of these structures clearly belong to the family of canonical PII proteins as they share the typical arrangement of the N-terminal extension and the characteristic T-and B-loop length (Fig. 2C). The fourth structure (Protein Data Bank code 3M05) is of the functionally uncharacterized protein PEPE_1480 from P. pentosaceus ATCC 25745. Because of its high sequence similarity (51.8% identity and 82.4% similarity), it could be used successfully as a search model for the structure solution of DarA by means of molecular replacement. Like DarA, it lacks the N-terminal extension of the PII proteins, and the T- and B-loops are reminiscent of DarA with respect to length and arrangement within the secondary structure elements. The high overall structural similarity and the positioning of conserved residues involved in c-di-AMP binding (see below) strongly suggest that PEPE_1480 is a DarA ortholog.
Like PII proteins, the biologically functional unit of DarA is a homotrimer arranged in a way such that the β-sheets are located in the center of the trimer, forming an intertwined core stabilized by a β-sheet extension due to close interactions between strand β4 and the main chain atoms of the last 6 residues from the neighboring subunit (Fig. 2D). Furthermore, the side chains from these last residues of the neighboring subunit interact with helix α2, thus additionally stabilizing the trimer. The second helix is also located at the outside of the trimer, forming the periphery of the DarA trimer in concert with helix α1 and the C-terminally located 6 residues. Both the functionally relevant B- and T-loops are localized in close vicinity of the C-terminal end of helix α1 from the neighboring subunit. The long B-loop was modeled completely based on simulated annealing omit maps, albeit it reveals a high flexibility.
c-di-AMP-binding Pocket of DarA
c-di-AMP is bound by DarA in a deep pocket located in the interspace between two subunits as its location is unambiguously defined by the electron density (Fig. 3, A and B). Because of the homotrimeric architecture, three binding pockets exist with each containing one c-di-AMP molecule (Fig. 3C). The binding pocket is formed by residues of β-strands 2 and 3 and the C-terminally located residue Phe-99 from one subunit (su1) and by residues of the T-loop and residues located at the base of the B-loop and the succeeding β-strand 4 from the adjacent subunit (su2) (Fig. 3D). One adenine of c-di-AMP, here denoted as Ade1, is perfectly sandwiched by the side chains of Phe-36 and Arg-26, leading to π-π and cation-π interactions, respectively. Ade1 is also bound by hydrogen bonds with the side chain of Asn-24 and the carbonyl groups of Thr-21 and Phe-25 (Fig. 4A). The other adenine, Ade2, forms polar interactions with the side chain of Thr-28 and the carbonyl groups of Arg-26, Met-45, and Gly-47 (Fig. 4C). The above mentioned Phe-99 in its perpendicular orientation also interacts with Ade2. The cyclic arranged sugar and phosphate moieties are fixed predominantly by hydrogen bonds with the backbone nitrogen of Gly-35, Phe-36, Leu-37, Gly-97, and the carbonyl group of Glu-92 within su2. Moreover, the side chains of Asn-41 (su2) and Thr-28 of the other subunit (su1) also contribute to the binding of the ribose and phosphate moieties.
FIGURE 3.

c-di-AMP binding by DarA. A, |Fo − Fc| difference omit map of the bound c-di-AMP contoured at 3.0 σ. B, c-di-AMP binds in between two subunits with the adenine bases binding to one subunit and the sugar-phosphate backbone binding to the other. C, three molecules of c-di-AMP are bound to the DarA homotrimer. D, magnification of the binding site with hydrogen bonds to specific residues indicated. Only residues at a distance of 5 Å or less are shown, and interactions are indicated by red dashed lines. With regard to this figure, the upper adenine is denoted as Ade1, and the other adenine as Ade2.
FIGURE 4.

Discrimination of c-di-GMP by DarA. A, recognition of adenine Ade1 by DarA. B, binding of guanine to the Ade1 pocket appears to be possible as shown by the manually docked guanine. C, recognition of adenine Ade2 by DarA. D, binding of guanine to the Ade2 pocket appears to be incompatible as the guanine exocyclic NH2 group cannot be accommodated, and there are unfavorable contacts of the guanine carbonyl oxygen with the protein as shown by the manually docked guanine.
Specificity of DarA
Another important cyclic dinucleotide second messenger in bacteria is c-di-GMP, which is required for the switch between sessile and motile lifestyles (4). To address the question whether c-di-GMP could also be bound by DarA, we manually fitted it onto the c-di-AMP in the DarA structure (Fig. 4). The cyclic arrangement of the sugar and phosphate moieties would be fixed identically to those in the structure containing c-di-AMP, but c-di-GMP binding appears to be highly unlikely for several reasons. The exocyclic amino group of a guanine in place of Ade2 would be too close to the carbonyl group of Met-45, and the amide of the guanine would clash with Gly-47 (Fig. 4D). Moreover, the guanine carbonyl group would interfere with the carbonyl group of Gly-47. Hence, binding of guanine to the pocket of Ade2 appears to be incompatible. However, with a guanine in place of Ade1, only its carbonyl group would be in too close contact with the carbonyl group of Phe-25, and this could be circumvented by a slight rotation toward the periphery of the trimer (Fig. 4B). Therefore, binding of c-di-GMP to DarA appeared to be impossible, but binding of a mixed cGAMP dinucleotide could not be excluded.
Analysis of Nucleotide Binding and Specificity by Isothermal Titration Calorimetry
To analyze the binding of various nucleotides to DarA, we performed ITC experiments. The ITC data clearly confirm the binding of c-di-AMP to DarA (Fig. 5). The interaction is of an exergonic nature, and the obtained data suggest a binding stoichiometry of 1:1. A detailed analysis of the thermodynamic parameters is not straightforwardly possible because c-di-AMP itself is engaged in a multistate oligomeric equilibrium with the monomer, dimer, tetramer, and octamer species existing at the concentration used (49). The deoligomerization in the course of titration-dilution is an endothermic process as established in control experiments where c-di-AMP was titrated into buffer (Fig. 5). Because the endothermic deoligomerization of c-di-AMP and exothermic binding of its monomeric form to DarA are coupled equilibria, it is not possible to simply subtract the control experiment (c-di-AMP into buffer) from the ligand titration experiment (c-di-AMP to DarA). The steepness of the transition and the concentration profiles used suggest the KD to be in the lower μm range. Despite this caveat in quantitative data analysis, the binding and the 1:1 stoichiometry can be unambiguously demonstrated as proof of principle.
FIGURE 5.

Nucleotide binding studies. Binding of various mono- and dinucleotides was studied by means of ITC. Experimental results are shown for c-di-AMP, c-di-GMP, 3′3′-cGAMP, 2′3′-cGAMP, ADP, and ATP. At the concentrations of nucleotides used, clear binding was observed for c-di-AMP and 3′3′-cGAMP but not for any other mono- or dinucleotide. AMP might bind to DarA, but addition of AMP caused precipitation of the protein (data not shown).
We further tested three naturally occurring dicyclic nucleotides that could potentially act as DarA effectors: c-di-GMP, 3′3′-cGAMP, and 2′3′-cGAMP. Whereas we could not detect any binding of c-di-GMP and 2′3′-cGAMP (at least not at the concentrations used), binding was clearly detectable in the case of 3′3′-cGAMP (Fig. 5). A direct comparison of the ITC data under identical conditions (DarA + c-di-AMP versus DarA + 3′3′-cGAMP) suggests that DarA binds c-di-AMP with higher affinity than it does the heterodinucleotide. This raises the questions whether or not two different dinucleotides regulate DarA activity in vivo and how these different regulatory modes would cross-talk in the cellular context. Our analysis of the metabolome of B. subtilis, however, indicates that 3′3′-cGAMP is not present in this organism, suggesting that c-di-AMP is the sole effector of DarA.
We further tested a potential binding of the mononucleotides AMP, ADP, and ATP (Fig. 5). Interestingly, neither ADP nor ATP bind to DarA under the conditions used. We noticed, however, that DarA reproducibly precipitated in the course of AMP titration, a phenomenon that was exclusively observed with AMP. Whether the apparent instability of DarA in the presence of AMP is a result of the experimental ITC setup (stirring and high concentration) or it reflects a physiologically relevant property remains unclear.
Conformational Changes of DarA upon c-di-AMP Binding
Because of their high sequence and structural similarity, the protein PEPE_1480 (Protein Data Bank code 3M05) from P. pentosaceus and DarA are most likely orthologs. Both proteins form a homotrimer and reveal high sequence conservation between residues involved in c-di-AMP binding, suggesting a similar function. Both the B-loop and T-loop of these two DarA proteins consist of the identical number of residues, respectively, but they differ in sequence. Loop residues that interact with the c-di-AMP by their side chain are conserved, e.g. Thr-97. Interestingly, the structure 3M05 represents the ligand-free or apo state of DarA. Comparison of the two structures reveals dramatic changes in the T-loop and B-loop obviously induced by c-di-AMP binding (Fig. 6). The orientation of Arg-26 (Arg-28 in the structure 3M05) changes from a position blocking the binding pocket to a position stacking with Ade1. In parallel, the T-loop of the neighboring subunit changes its orientation due to steric hindrance of the phosphate moiety, inducing a repositioning of Phe-36 (Phe-38 in the structure 3M05) from a perpendicular to a stacking conformation. In turn, the two ribose and two phosphate moieties attract the B-loop, which moves toward the c-di-AMP by 80°. This arrangement seems to reduce the flexibility of the B-loop as indicated by the missing residues in the crystal structure of the ligand-free form.
FIGURE 6.
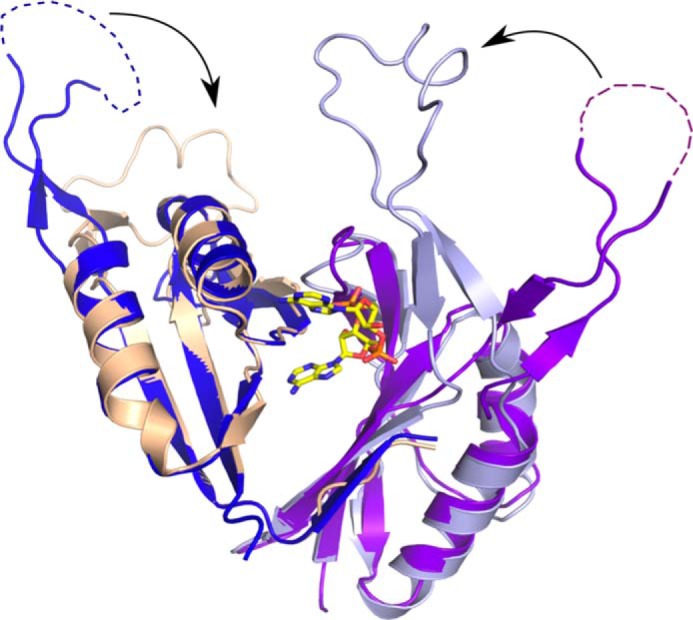
Conformational changes induced by c-di-AMP binding. Shown is the superposition of the structure of B. subtilis DarA (colored beige and light blue) and its putative ortholog from P. pentosaceus (Protein Data Bank code 3M05; colored purple and dark blue), which represents the ligand-free form. The B-loop of the ligand-free form is not fully defined in the crystal structure, and the missing part is indicated by dotted lines. Upon binding of c-di-AMP, the B-loop undergoes major conformational changes, thereby moving toward the core of the trimer and completing the binding pocket.
DISCUSSION
Our study identifies the PII-like signal transduction protein DarA as a target of the second messenger nucleotide c-di-AMP in B. subtilis. The interaction of c-di-AMP and DarA brings together two universally conserved principles of signal transduction: second messengers and small signal transduction proteins. PII signal transducer proteins play a central role in various signaling pathways in all kingdoms of life (50, 51). Our crystal structure analysis of the c-di-AMP-binding protein DarA from B. subtilis demonstrates that DarA is indeed a PII-like protein forming the canonical PII homotrimer. However, significant differences are found for the B-and T-loops, which are known to be major functional elements of PII proteins. Most importantly, in DarA, these loops are swapped with respect to their size. Although the B-loop is short in PII proteins, it is a rather large loop in DarA, and the large T-loop of PII proteins is a short loop in DarA. The function of PII proteins is triggered by the binding of the effector molecules ADP, ATP, and α-ketoglutarate. Superimposing the structure of the DarA·c-di-AMP complex and structures of PII proteins with bound ADP or ATP and α-ketoglutarate reveals that the Ade1 of the c-di-AMP is approximately located in the position of the ATP or ADP in PII proteins. Binding of AMP to DarA might in principal be possible, but it would be with significantly lower affinity as the amount of ligand-protein interactions is much smaller than for c-di-AMP. ADP and ATP binding, which is apparently hindered by the additional phosphate groups, could not be observed in the ITC experiments. Another important difference from PII proteins is the absence of the binding pocket for α-ketoglutarate in DarA as it is filled by the β-strands at the bottom of the B-loop.
The mode of binding c-di-AMP by a sandwich of an aromatic residue and an arginine side chain is reminiscent of the c-di-GMP binding of the PilZ domain (52). However, DarA is not capable to bind c-di-GMP as demonstrated by ITC. The structural basis for the discrimination is mainly found in the Ade2 pocket to which a guanine cannot bind (Fig. 4D). However, based on the DarA crystal structure, binding of guanine to the Ade1 pocket appeared feasible (Fig. 4B). Indeed, ITC experiments confirmed that the mixed 3′3′-cGAMP dinucleotide is bound by DarA. However, this property of DarA might have no physiological relevance as our analysis showed that cGAMP is not present in B. subtilis. Furthermore, there is no risk of mixing up signaling chains because of the ability of DarA to bind both c-di-AMP and cGAMP. In contrast, c-di-GMP is formed in B. subtilis, and therefore the signaling chains of these two second messengers must be kept separate.
Other bacterial c-di-AMP-binding proteins have recently been identified, and for one of them, pyruvate carboxylase, the crystal structure was determined (21). The binding of c-di-AMP by the pyruvate carboxylase is rather different from that of DarA because the sandwiching arginine is missing, and a c-di-AMP dimer in which one adenine stacks between the two adenines of the other c-di-AMP molecule is bound.
The physiological role of DarA is unknown, and we showed that the DarA protein is not essential for the viability of B. subtilis. It remains unclear which protein is the receptor of DarA-mediated c-di-AMP signaling. Trimeric PII proteins are known to bind a trimeric target protein as seen in the structure of the AmtB·GlnK complex (53); hence, it is tempting to speculate that the DarA target is also a homotrimeric protein. Alternatively, the structure of the PII·PipX complex showed that PII proteins might bind three molecules of a monomeric target protein (54). The DarA/PstA protein is present only in Gram-positive bacteria closely related to the genus Bacillus including Staphylococcus, Listeria, Lactobacillus, and Clostridium but not in Streptococcus and Lactococcus. All bacteria that encode a DarA/PstA protein also contain the diadenylate cyclase CdaA. This suggests that the functions of DarA and of this particular diadenylate cyclase are coupled to each other. The observation that the growth of the darA mutant is not impaired as compared with the wild type strain (data not shown) suggests that DarA might be involved in a subtle regulation. This is not unprecedented for PII proteins: uridylation of the E. coli PII proteins has only a minor impact on their interactions with other proteins (54).
Because c-di-AMP is the only essential second messenger, both the enzymes that synthesize it and the proteins involved in c-di-AMP-mediated signaling are excellent candidate targets for novel antimicrobial compounds. As DarA is conserved among the Gram-positive bacteria including many important pathogens such as multiresistant S. aureus strains, it remains an important future goal to identify the target of DarA and to understand the physiological relevance of the DarA·c-di-AMP complex.
Acknowledgments
We are grateful to the staff of the European Synchrotron Radiation Facility beam line ID23-1 for support during data collection. We thank Annette Garbe for excellent help in LC-MS/MS analyses and Felix Mehne for helpful discussions.
This work was supported in part by Deutsche Forschungsgemeinschaft Grants HI 291/13-1 and SFB860.
The atomic coordinates and structure factors (code 4RLE) have been deposited in the Protein Data Bank (http://wwpdb.org/).
J. Gundlach, A. Dickmanns, K. Schröder-Tittmann, P. Neumann, J. Kaesler, J. Kampf, C. Herzberg, E. Hammer, F. Schwede, V. Kaever, K. Tittmann, J. Stülke, and R. Ficner, unpublished results.
- cGAMP
- cyclic GMP-AMP
- ITC
- isothermal titration calorimetry
- 2′-[biotin]-AHC-c-di-AMP
- 2′-O-(6-[biotinyl]aminohexylcarbamoyl)-cyclic diadenosine monophosphate
- 8′-[biotin]-AET-c-di-AMP
- 8-(2-[biotinyl]aminoethylthio)-cyclic diadenosine monophosphate
- UPLC
- ultraperformance LC
- su
- subunit.
REFERENCES
- 1. Görke B., Stülke J. (2008) Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624 [DOI] [PubMed] [Google Scholar]
- 2. McDonough K. A., Rodriguez A. (2012) The myriad roles of cyclic AMP in microbial pathogens: from signal to sword. Nat. Rev. Microbiol. 10, 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boutte C. C., Crosson S. (2013) Bacterial lifestyle shapes stringent response activation. Trends Microbiol. 21, 174–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hengge R. (2009) Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7, 263–273 [DOI] [PubMed] [Google Scholar]
- 5. Witte G., Hartung S., Büttner K., Hopfner K. P. (2008) Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 30, 167–178 [DOI] [PubMed] [Google Scholar]
- 6. Corrigan R. M., Gründling A. (2013) Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 11, 513–524 [DOI] [PubMed] [Google Scholar]
- 7. Chaudhuri R. R., Allen A. G., Owen P. J., Shalom G., Stone K., Harrison M., Burgis T. A., Lockyer M., Garcia-Lara J., Foster S. J., Pleasance S. J., Peters S. E., Maskell D. J., Charles I. G. (2009) Comprehensive identification of essential Staphylococcus aureus genes using transposon-mediated differential hybridisation (TMDH). BMC Genomics 10, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woodward J. J., Iavarone A. T., Portnoy D. A. (2010) c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mehne F. M., Gunka K., Eilers H., Herzberg C., Kaever V., Stülke J. (2013) Cyclic di-AMP homeostasis in Bacillus subtilis: both lack and high level accumulation of the nucleotide are detrimental for cell growth. J. Biol. Chem. 288, 2004–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo Y., Helmann J. D. (2012) Analysis of the role of Bacillus subtilis σM in β-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83, 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davies B. W., Bogard R. W., Young T. S., Mekalanos J. J. (2012) Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kriel A., Bittner A. N., Kim S. H., Liu K., Tehranchi A. K., Zou W. Y., Rendon S., Chen R., Tu B. P., Wang J. D. (2012) Direct regulation of GTP homeostasis by (p)ppGpp: a critical component of viability and stress resistance. Mol. Cell 48, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao X., Mukherjee S., Matthews P. M., Hammad L. A., Kearns D. B., Dann C. E., 3rd (2013) Functional characterization of core components of the Bacillus subtilis cyclic-di-GMP signaling pathway. J. Bacteriol. 195, 4782–4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Diethmaier C., Newman J. A., Kovács A. T., Kaever V., Herzberg C., Rodrigues C., Boonstra M., Kuipers O. P., Lewis R. J., Stülke J. (2014) The YmdB phosphodiesterase is a global regulator of late adaptive responses in Bacillus subtilis. J. Bacteriol. 196, 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oppenheimer-Shaanan Y., Wexselblatt E., Katzhendler J., Yavin E., Ben-Yehuda S. (2011) c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep. 12, 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Campos S. S., Ibarra-Rodriguez J. R., Barajas-Ornelas R. C., Ramírez-Guadiana F. H., Obregón-Herrera A., Setlow P., Pedraza-Reyes M. (2014) Interaction of apurinic/apyrimidinic endonucleases Nfo and ExoA with the DNA integrity scanning protein DisA in the processing of oxidative DNA damage during Bacillus subtilis spore outgrowth. J. Bacteriol. 196, 568–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang L., He Z. G. (2013) Radiation-sensitive gene A (RadA) targets DisA, DNA integrity scanning protein A, to negatively affect cyclic di-AMP synthesis activity in Mycobacterium smegmatis. J. Biol. Chem. 288, 22426–22436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bejerano-Sagie M., Oppenheimer-Shaanan Y., Berlatzky I., Rouvinski A., Meyerovich M., Ben-Yehuda S. (2006) A checkpoint protein that scans the chromosome for damage at the start of sporulation in Bacillus subtilis. Cell 125, 679–690 [DOI] [PubMed] [Google Scholar]
- 19. Corrigan R. M., Abbott J. C., Burhenne H., Kaever V., Gründling A. (2011) c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 7, e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Corrigan R. M., Campeotto I., Jeganathan T., Roelofs K. G., Lee V. T., Gründling A. (2013) Systematic identification of conserved bacterial c-di-AMP receptor proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 9084–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sureka K., Choi P. H., Precit M., Delince M., Pensinger D. A., Huynh T. N., Jurado A. R., Goo Y. A., Sadilek M., Iavarone A. T., Sauer J. D., Tong L., Woodward J. J. (2014) The cyclic dinucleotide c-di-AMP is an allosteric regulator of metabolic enzyme function. Cell 158, 1389–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mehne F. M., Schröder-Tittmann K., Eijlander R. T., Herzberg C., Hewitt L., Kaever V., Lewis R. J., Kuipers O. P., Tittmann K., Stülke J. (2014) Control of the diadenylate cyclase CdaS in Bacillus subtilis: an autoinhibitory domain limits cyclic di-AMP production. J. Biol. Chem. 289, 21098–21107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bai Y., Yang J., Zarrella T. M., Zhang Y., Metzger D. W., Bai G. (2014) Cyclic di-AMP impairs potassium uptake mediated by a cyclic di-AMP binding protein in Streptococcus pneumoniae. J. Bacteriol. 196, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nelson J. W., Sudarsan N., Furukawa K., Weinberg Z., Wang J. X., Breaker R. R. (2013) Riboswitches in eubacteria sense the second messenger c-di-AMP. Nat. Chem. Biol. 9, 834–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones C. P., Ferré-D'Amaré A. R. (2014) Crystal structure of a c-di-AMP riboswitch reveals an internally pseudo-dimeric RNA. EMBO J. 33, 2692–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Block K. F., Hammond M. C., Breaker R. R. (2010) Evidence for widespread gene control function by the ydaO riboswitch candidate. J. Bacteriol. 192, 3983–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sambrook J., Russell D. (2001) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 28. Commichau F. M., Herzberg C., Tripal P., Valerius O., Stülke J. (2007) A regulatory protein-protein interaction governs glutamate biosynthesis in Bacillus subtilis: the glutamate dehydrogenase RocG moonlights in controlling the transcription factor GltC. Mol. Microbiol. 65, 642–654 [DOI] [PubMed] [Google Scholar]
- 29. Meyer F. M., Gerwig J., Hammer E., Herzberg C., Commichau F. M., Völker U., Stülke J. (2011) Physical interactions between tricarboxylic acid cycle enzymes in Bacillus subtilis: evidence for a metabolon. Metab. Eng. 13, 18–27 [DOI] [PubMed] [Google Scholar]
- 30. Michna R. H., Commichau F. M., Tödter D., Zschiedrich C. P., Stülke J. (2014) SubtiWiki—a database for the model organism Bacillus subtilis that links pathway, interaction and expression information. Nucleic Acids Res. 42, D692–D698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schirmer F., Ehrt S., Hillen W. (1997) Expression, inducer spectrum, domain structure, and function of MopR, the regulator of phenol degradation in Acinetobacter calcoaceticus NCIB8250. J. Bacteriol. 179, 1329–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galinier A., Haiech J., Kilhoffer M. C., Jaquinod M., Stülke J., Deutscher J., Martin-Verstraete I. (1997) The Bacillus subtilis crh gene encodes a HPr-like protein involved in carbon catabolite repression. Proc. Natl. Acad. Sci. U.S.A. 94, 8439–8444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brünger A. T. (1993) Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr. D Biol. Crystallogr. 49, 24–36 [DOI] [PubMed] [Google Scholar]
- 39. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 40. Sheriff S., Hendrickson W. A., Smith J. L. (1987) Structure of myohemerythrin in the azidomet state at 1.7/1.3 Å resolution. J. Mol. Biol. 197, 273–296 [DOI] [PubMed] [Google Scholar]
- 41. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 42. Kabsch W., Kabsch H., Eisenberg D. (1976) Packing in a new crystalline form of glutamine synthetase from Escherichia coli. J. Mol. Biol. 100, 283–291 [DOI] [PubMed] [Google Scholar]
- 43. Commichau F. M., Pietack N., Stülke J. (2013) Essential genes in Bacillus subtilis: a re-evaluation after ten years. Mol. Biosyst. 9, 1068–1075 [DOI] [PubMed] [Google Scholar]
- 44. Carr P. D., Cheah E., Suffolk P. M., Vasudevan S. G., Dixon N. E., Ollis D. L. (1996) X-ray structure of the signal transduction protein from Escherichia coli at 1.9 Å. Acta Crystallogr. D Biol. Crystallogr. 52, 93–104 [DOI] [PubMed] [Google Scholar]
- 45. Zeth K., Fokina O., Forchhammer K. (2012) An engineered PII protein variant that senses a novel ligand: atomic resolution structure of the complex with citrate. Acta Crystallogr. D Biol. Crystallogr. 68, 901–908 [DOI] [PubMed] [Google Scholar]
- 46. Yildiz O., Kalthoff C., Raunser S., Kühlbrandt W. (2007) Structure of GlnK1 with bound effectors indicates regulatory mechanism for ammonia uptake. EMBO J. 26, 589–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maier S., Schleberger P., Lü W., Wacker T., Pflüger T., Litz C., Andrade S. L. (2011) Mechanism of disruption of the Amt-GlnK complex by PII-mediated sensing of 2-oxoglutarate. PLoS One 6, e26327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gentner M., Allan M. G., Zaehringer F., Schirmer T., Grzesiek S. (2012) Oligomer formation of the bacterial second messenger c-di-GMP: reaction rates and equilibrium constants indicate a monomeric state at physiological concentrations. J. Am. Chem. Soc. 134, 1019–1029 [DOI] [PubMed] [Google Scholar]
- 50. Forchhammer K. (2008) PII signal transducers: novel functional and structural insights. Trends Microbiol. 16, 65–72 [DOI] [PubMed] [Google Scholar]
- 51. Huergo L. F., Chandra G., Merrick M. (2013) PII signal transduction proteins: nitrogen regulation and beyond. FEMS Microbiol. Rev. 37, 251–283 [DOI] [PubMed] [Google Scholar]
- 52. Benach J., Swaminathan S. S., Tamayo R., Handelman S. K., Folta-Stogniew E., Ramos J. E., Forouhar F., Neely H., Seetharaman J., Camilli A., Hunt J. F. (2007) The structural basis of cyclic diguanylate signal transduction by PilZ domains. EMBO J. 26, 5153–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Conroy M. J., Durand A., Lupo D., Li X. D., Bullough P. A., Winkler F. K., Merrick M. (2007) The crystal structure of the Escherichia coli AmtB-GlnK complex reveals how GlnK regulates the ammonia channel. Proc. Natl. Acad. Sci. U.S.A. 104, 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao M. X., Jiang Y. L., Xu B. Y., Chen Y., Zhang C. C., Zhou C. Z. (2010) Crystal structure of the cyanobacterial signal transduction protein PII in complex with PipX. J. Mol. Biol. 402, 552–559 [DOI] [PubMed] [Google Scholar]
- 55. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]