

Background: The molecular mechanism of coronavirus PLPs suppressing the innate immune response remains unclear.
Results: PLP2 induces the degradation of p53 through stabilizing MDM2, and IRF7 is a novel target gene of p53.
Conclusion: PLP2 inhibits the p53-mediated production of type I IFN and apoptosis to ensure viral growth.
Significance: We identify the mechanism with which coronavirus induces the low dosage IFN production.
Keywords: Innate Immunity, Interferon, Mouse Double Minute 2 Homolog (MDM2), p53, Virus, PLP2, Coronavirus
Abstract
Infection by human coronaviruses is usually characterized by rampant viral replication and severe immunopathology in host cells. Recently, the coronavirus papain-like proteases (PLPs) have been identified as suppressors of the innate immune response. However, the molecular mechanism of this inhibition remains unclear. Here, we provide evidence that PLP2, a catalytic domain of the nonstructural protein 3 of human coronavirus NL63 (HCoV-NL63), deubiquitinates and stabilizes the cellular oncoprotein MDM2 and induces the proteasomal degradation of p53. Meanwhile, we identify IRF7 (interferon regulatory factor 7) as a bona fide target gene of p53 to mediate the p53-directed production of type I interferon and the innate immune response. By promoting p53 degradation, PLP2 inhibits the p53-mediated antiviral response and apoptosis to ensure viral growth in infected cells. Thus, our study reveals that coronavirus engages PLPs to escape from the innate antiviral response of the host by inhibiting p53-IRF7-IFNβ signaling.
Introduction
The tumor suppressor protein p53 is widely known as “the guardian of the genome” because of its ability to prevent the emergence of transformed cells by inducing cell cycle arrest and apoptosis (1). Recent studies indicate that P53 is also a direct target gene of the type I interferon (IFNα/β) pathway, and thus, it is activated by certain cytokines upon viral infection (2). This provides new insight into the function of p53 in antiviral innate immunity. Because virus infection activates p53, this tumor suppressor has been recently introduced as a new component of the cellular antiviral defense mechanism (2). In fact, p53 is a key player in antiviral innate immunity by both inducing apoptosis in infected cells and enforcing type I IFN production. Both actions coordinated by this tumor suppressor help thwart the replication of a wide variety of viruses both in vitro and in vivo (3–6). The finding that p53 is involved in antiviral immunity may help explain why this protein is conserved in invertebrate organisms, which do not suffer from cancer-related diseases, and why it is so frequently targeted by viral proteins.
Coronaviruses are mainly associated with respiratory, enteric, hepatic, and central nervous system diseases. However, until the late 1960s, coronaviruses were not recognized as pathogens that are responsible for human diseases, and it was only in 2003 when human coronaviruses (HCoVs)2 received worldwide attention after the emergence of severe and acute respiratory syndrome (SARS), which is caused by the coronavirus SARS-CoV. SARS-CoV has infected more than 8,000 people in 32 countries, with a mortality rate of up to 10%. The increasing amounts of research on coronaviruses soon led to the discovery of another human coronavirus, HCoV-NL63. Infection by the NL63 virus is prevalent in 7% of hospitalized patients and is associated with both upper and lower respiratory tract diseases, bronchiolitis, and possibly conjunctivitis in children and adults (7). Currently, no antiviral drugs are available to treat coronavirus infections; thus, potential drug targets need to be identified and characterized.
Coronaviruses are enveloped viruses with large RNA genomes (28–32 kb) (8, 9). Upon entry, coronavirus genomic RNA is translated to produce two large polyproteins, pp1a and pp1ab. These polyproteins are processed by viral cysteine proteases, both papain-like (PLPs/PLpro) and picornavirus 3C-like (3CLpro), to generate mature nonstructural proteins that assemble with host cell membranes to form double membrane vesicles (10–12). We previously identified HCoV-NL63 replicase gene products and characterized two viral PLPs, PLP1 and PLP2, that process the viral replicase polyprotein (7). HCoV-NL63 replicase can be detected at 24 h postinfection. These proteins accumulate in the perinuclear region, consistent with the function of membrane-associated replication complexes (7). Furthermore, NL63-PLP2 was found to exhibit deubiquitinase (DUB) activity and inhibit the expression of type I IFN (7, 9).
Previous studies have shown that type I interferons can inhibit the replication of coronaviruses and that IFNβ is more effective than IFNα (10, 11). However, clinical studies have revealed that coronavirus infections only induce very low levels of type I IFNs, which most likely contributes to rampant viral replication and a weakened immune response (12–14). The low level IFN response to this vigorously replicating RNA virus suggests that coronaviruses might either evade or inactivate the innate immune response. However, the molecular mechanism of the low dosage IFN production remains unclear. Here, we show that PLP2 decreases the stability and transcriptional activity of p53 by increasing the MDM2-mediated ubiquitination and nuclear export of p53. PLP2 inhibits antiviral responses by attenuating the p53-mediated production of type I IFN and apoptosis and, as a result, enhances viral replication. Intriguingly, we found that p53 directly transactivates IRF7 to regulate the transcription of type I IFN genes, which provides strong evidence for the role of p53 in the innate immune system.
EXPERIMENTAL PROCEDURES
Plasmid Constructs
Plasmids for the expression of human MDM2, p53, and their point mutants were previously described (15). HA-tagged ubiquitin plasmids were previously described (16). DNA constructs containing wild type and mutants of V5-tagged PLP2 and plasmids of IFN-β luciferase were previously described (9, 17).
Cell Culture, Transfection, and Luciferase Reporter Assay
Human lung adenocarcinoma H1299 cells (p53-deficient) and H1975 cells (p53-wild type) were maintained in RPMI 1640 medium containing 10% FBS (Hyclone) and 1% penicillin-streptomycin (Mediatech, Manassas, VA) at 37 °C and 5% CO2. Human p53+/+ HCT116 and p53−/− HCT116 colon cancer cells and p53−/− MEF and p53+/+ MEF cells were cultured in DMEM supplemented with 10% FBS (Hyclone) and 1% penicillin-streptomycin (Mediatech) at 37 °C and 5% CO2. Cell culture transfection was performed using Lipofectamine 2000 (Invitrogen) reagent according to the manufacturer's instructions. Forty-eight hours after transfection, cells were lysed in 90 μl of a passive lysis buffer, and luciferase activity was measured with the dual luciferase assay system (Promega) in accordance with the manufacturer's protocol.
Immunoprecipitation and Immunoblotting
Forty-eight hours post-transfection, cell lysates were prepared in HEPES lysis buffer (20 mm HEPES, pH 7.2, 50 mm NaCl, 0.5% Triton X-100, 1 mm NaF, and 1 mm DTT) supplemented with protease inhibitor mixture (Roche Applied Science). Immunoprecipitations were performed using the indicated primary antibody and protein A/G-agarose beads (Santa Cruz) at 4 °C. Lysates and immunoprecipitates were examined using the indicated primary antibodies followed by detection with the related secondary antibody and the SuperSignal chemiluminescence kit (Pierce).
Protein Half-life Assay
For MDM2 and p53 half-life assay, Lipofectamine 2000 transfection was performed when HCT116 cells in 2-cm plates reached ∼60% confluence. Plasmids encoding for V5-PLP2 were used in transfection as indicated in individual experiments. Twenty-four hours later, cells were treated with the protein synthesis inhibitor cycloheximide (10 μg/ml) for the indicated durations before harvest.
In Vivo MDM2 Ubiquitination Assay
Lipofectamine 2000 transfection was performed when HCT116 cells in 10-cm plates reached ∼60% confluence. Plasmids encoding for MDM2, PLP2, and HA-tagged ubiquitin were used in transfection as indicated in individual experiments. Twenty-four hours after transfection, cells were treated with 20 μm proteasome inhibitor MG132 (Calbiochem) for 8 h. The cells were washed with PBS, pelleted, and lysed in 0.4 ml of HEPES buffer (20 mm HEPES, pH 7.2, 50 mm NaCl, 1 mm NaF, 0.5% Triton X-100) plus 0.1% SDS, 20 μm MG132, and protease inhibitor mixture. The lysates were centrifuged to obtain cytosolic proteins. Briefly, individual samples were incubated with anti-MDM2 antibody (Santa Cruz) for 3 h and protein A/G-agarose beads (Santa Cruz) for a further 8 h at 4 °C. Then the beads were washed three times with HEPES buffer. The proteins were released from the beads by boiling in 40 ml of 2× SDS-PAGE sample buffer for 10 min. Ten microliters of the samples were subjected to immunoblot against anti-HA monoclonal antibody (MBL) in individual experiments.
Fluorescence Microscopy
For detection of subcellular localization by immunofluorescence, after being fixed with 4% paraformaldehyde and permeabilized in 0.2% Triton X-100 (PBS), cells were incubated with the indicated antibodies (dilution 1:50; Abcam) for 8 h at 4 °C, followed by incubation with TRITC-conjugated or FITC-conjugated secondary antibody (dilution 1:200; Cwbio) for 1 h at 25 °C. The nuclei were stained with DAPI (Sigma), and images were visualized with a Zeiss LSM 510 Meta inverted confocal microscope.
Apoptosis Analysis
HCT116 cells were transfected with or without V5-PLP2. Transfected cells were treated with poly(I·C) (10 μg/ml, 24 h; Sigma) 24 h later. The apoptotic cells were then washed with PBS and stained with fluorescein isothiocyanate-annexin V and propidium iodide according to the manufacturer's protocol (Beijing Biosea Biotechnology annexin V kit). Apoptotic cells (annexin V-positive, propidium iodide negative) were then determined by flow cytometry.
RNA Interference
IRF9 siRNA-1 (5′-GCAUGAACCCCUUGUGCUG-3′), siRNA-2 (5′-UUCUCCGAACGUGUCACGU-3′), and nontargeting siRNA (5′-UUCUCCGAACGUGUCACGU-3′) were synthesized by Shanghai GenePharm. All siRNAs were transfected into the cells according to the manufacturer's protocol.
Sendai Virus (SeV) Infection and Poly(I:C) Treatment
Where indicated, cells were infected with 100 pfu/ml SeV for indicated hours prior to qPCR analysis as described previously (18). For poly(I·C) treatment, poly(I·C) (Sigma) was complexed with Lipofectamine 2000 (at 1:1 ratio) and loaded onto the cells for the indicated time period.
Quantitative Real Time RT-PCR
Total RNA was extracted from cells with TRIzol (Invitrogen). Reverse transcription was performed with 1 μg of RNA and the RNA PCR Kit (avian myeloblastosis virus) version 3.0 (TaKaRa), and quantitative PCR was performed with the IQ5 system (Bio-Rad). PCRs were performed in 25-μl reaction volumes with SYBR Green PCR master mix (Bio-Rad) and 0.2 μm specific primers. The primer sequences used for all qPCRs have been published (4, 17–19).
Electrophoretic Mobility Shift Assay
The double-stranded oligonucleotides used for EMSA were end-labeled with biotin. The labeled probes were incubated with the protein(s) for 30 min in binding buffer (10 mm Tris-HCl, pH 7.5, 5 mm KCl, 5 mm MgCl2, 10 μm ZnSO4, 50 μg/ml poly(dI-dC), 5 μg/μl BSA, 0.67 mm DTT, 0.67 mm PMSF, 2.5% glycerol) in the presence or absence of unlabeled probes. If an antibody was added to detect supershifts, the antibody and protein were preincubated for 20 min before the labeled probes were added. The protein/DNA samples were loaded onto a native 6–10% polyacrylamide gel in TBE buffer and then transferred to a Biodyne membrane. The membrane was blocked and incubated with HRP-conjugated streptavidin for 15 min. The membrane was washed three times, treated with SuperSignal detection reagents (Pierce Biotechnology), and exposed to Kodak Light films.
Chromatin Immunoprecipitation Assay
ChIP assays were performed as described previously (15). The primer sequences used for IRF7 were 5′-GGCATCTTGGCTGGTGGGGAATTGGG-3′ and 5′-GCAGCCTGAGGGCTGGCGACAGGTG-3′.
Statistical Analysis
Statistical evaluation was conducted using Student's t test.
RESULTS
PLP2 Deubiquitinates and Stabilizes MDM2 to Promote p53 Degradation
Previous studies have revealed that the structure of PLP2 is similar to PLpro (20) and HAUSP (21). Furthermore, PLP2 harbors similar DUB activity to HAUSP (22), which prefers to bind and deubiquitinate the oncoprotein MDM2 (23, 24). Thus, we hypothesized that PLP2 may regulate the stability of cellular MDM2. As expected, we found that MDM2 and PLP2 could be coimmunoprecipitated (Fig. 1, A and B); however, MDM2 could not be coimmunoprecipitated with another human DUB, USP13 (Fig. 1C). Moreover, MDM2 ubiquitination was significantly diminished by coexpression of PLP2 but not by USP13 (Fig. 1D). Meanwhile, PLP2 could not deubiquitinate Smurf1, which has been shown be a substrate of USP9x (25) and was confirmed in our experiment (Fig. 1E). These data suggest the specificity of PLP2 on MDM2.
FIGURE 1.

PLP2 deubiquitinates and stabilizes MDM2. A, PLP2 interacts with MDM2 in vivo. Coimmunoprecipitation (IP) of PLP2 and exogenous MDM2 from p53+/+ HCT116 cells and whole cell lysates (Ly) were tested by HA and V5 antibodies. B, p53+/+ HCT116 cells transfected with the indicated plasmids were subject to immunoprecipitation with anti-V5 antibodies. The lysates and immunoprecipitates were analyzed. C, p53+/+ HCT116 cells transfected with the indicated plasmids were subjected to immunoprecipitation with anti-Myc antibodies. The lysates and immunoprecipitates were analyzed. D, MDM2 ubiquitination (Ub) is inhibited by coexpression of PLP2 in vivo. p53+/+ HCT116 cells were transfected with HA-ubiquitin, MDM2 and V5-PLP2, and ubiquitinated MDM2 was precipitated, followed by immunoblotting with anti-HA. E, p53+/+ HCT116 cells transfected with the indicated constructs were treated with MG132 for 8 h before harvest. Smurf1 was immunoprecipitated with anti-Flag and immunoblotted with anti-HA. F and G, PLP2 expression increases the steady-state level of endogenous (F) and exogenous (G) MDM2. HCT116 cells were transfected with increasing amounts of V5-PLP2. After 48 h, total lysates were immunoblotted to detect the expression of MDM2. H, half-life analysis of MDM2 in the presence of overexpressed PLP2. CHX, cycloheximide. I, subcellular localization of MDM2. MCF7 cells were fixed and stained with anti-MDM2 antibodies before visualization. J, regulation of the subcellular localization of MDM2 by PLP2. MCF7 cells transfected with the V5-PLP2 constructs were fixed and stained with anti-V5 and anti-MDM2 antibodies before visualization. K, deubiquitinase activity was required for PLP2 to increase MDM2 stability. Protein expression analysis was performed by Western blotting with the indicated antibodies for cells transfected with WT PLP2 (lane 2), the C1678A mutant (lane 3), the H1836A mutant (lane 4), and the D1849A mutant (lane 5).
The steady-state level of MDM2 was increased and the half-life of MDM2 was prolonged when PLP2 was overexpressed (Fig. 1, F–H). We also examined the subcellular localization of MDM2 and PLP2. As shown in Fig. 1I, MDM2 was predominantly localized in the nucleus but was also found in the cytoplasm. When PLP2 was overexpressed, MDM2 was restricted to the nucleus (Fig. 1J), suggesting that deubiquitinated MDM2 was exclusively localized in the nucleus. Previous studies found that the catalytic residues Cys-1678 and His-1836 of PLP2 are important for its DUB activity because mutation of these residues to alanine resulted in the partial reduction of DUB activity. In contrast, residue Asp-1849 is dispensable for DUB activity (9). We observed that the ability of PLP2 to up-regulate MDM2 levels was attenuated in the C1678A and H1836A mutants of PLP2 but not in the D1849A mutant (Fig. 1K), suggesting that PLP2 up-regulates MDM2 largely dependent of its DUB activity.
MDM2 is an important E3 ubiquitin ligase of p53, which down-regulates p53 levels by promoting ubiquitination-dependent protein degradation. Moreover, previous studies suggest that ubiquitination of p53 by MDM2 induces p53 translocation from the nucleus to the cytoplasm (26–29), which led us to hypothesize that PLP2 may promote the MDM2-mediated degradation and nuclear export of p53. PLP2 significantly decreased the steady-state level of p53 in a dose-dependent manner (Fig. 2A). PLP2-triggered p53 down-regulation was blocked by treatment with the proteasome inhibitor MG132 (Fig. 2B), indicating that PLP2 increases the degradation of p53 through the ubiquitin-proteasome pathway. Furthermore, analysis of the p53 half-life showed that the rate of p53 turnover was markedly increased when PLP2 was overexpressed (Fig. 2C). We then used a p53 mutant, 6KR, in which six ubiquitination sites at the COOH terminus were mutated to arginine residues (K370R/K372R/K373R/K381R/K382R/K386R) to evaluate the PLP2 activity on the ubiquitination-deficient p53. As expected, 6KR was less sensitive than p53-WT to the degradation mediated by PLP2 (Fig. 2D). This result supports that PLP2 regulates p53 through ubiquitination-dependent protein degradation. In addition, we performed immunofluorescence assays to detect the subcellular localization of p53. In control cells, p53 was readily detected in the nucleus (Fig. 2E). Strikingly, in PLP2-transfected cells, p53 was distributed in the cytoplasm (Fig. 2F). Moreover, using a luciferase assay to determine the effect of PLP2 on the transcriptional activity of p53, we found that PLP2 significantly inhibited the activity of both endogenous p53 in p53+/+HCT116 cells (Fig. 2G) and exogenous p53 transfected into p53−/−HCT116 cells (Fig. 2H). Moreover, we found that PLP2 could not regulate the protein levels of exogenous p53 in the absence of MDM2. When an exogenous MDM2 was reintroduced into the p53−/−MDM2−/− MEF cells, PLP2 restored the ability to down-regulate p53 (Fig. 2I), suggesting that PLP2 promotes p53 degradation dependent of the E3 ligase MDM2. Meanwhile, we observed that the ability of PLP2 to down-regulate p53 levels was attenuated in the C1678A and H1836A mutants of PLP2 but not in the D1849A mutant (Fig. 2J), suggesting that PLP2 down-regulates p53 largely dependent of its DUB activity. Importantly, the expression of p53 was enhanced in response to poly(I·C), a synthetic dsRNA analog, and nonstructural protein 3 (nsp3) of SARS-CoV significantly promotes p53 degradation by stabilizing MDM2 (Fig. 2K). Porcine epidemic diarrhea virus (PEDV) is an animal coronavirus that also codes papain-like protease, which is similar to human coronavirus papain-like protease with protease activity, deubiquitinase activity, and interferon antagonism activity. MDM2 was up-regulated, and p53 was down-regulated in Vero cells infected with PEDV (Fig. 2L). Collectively, these results suggest that PLP2 interacts with MDM2, deubiquitinates and stabilizes MDM2, and then promotes p53 degradation.
FIGURE 2.

PLP2 increases p53 degradation through the ubiquitin-proteasome pathway. A, PLP2 expression decreases the steady-state level of p53. p53+/+ HCT116 cells were transfected with increasing amounts of V5-PLP2. After 48 h, total lysates were immunoblotted to detect the expression of p53. B, PLP2-triggered p53 down-regulation is blocked by treatment with the proteasome inhibitor MG132 (20 μm, 8 h). C, half-life analysis of p53 in the presence of overexpressed PLP2. Quantitative analysis was performed by measuring the integrated optical density. CHX, cycloheximide. D, p53−/− HCT116 cells were transfected with the indicated constructs. After 48 h, protein expression analysis was performed by Western blotting with the indicated antibodies, as shown. E, subcellular localization of p53. MCF7 cells were fixed and stained as indicated. F, MCF7 cells transfected with the V5-PLP2 constructs were treated with MG132 for 4 h to avoid the p53 degradation. Forty-eight hours later, the cells were fixed and stained with the indicated antibodies before visualization. G and H, PLP2 inhibits p53 activity. p53 activity in HCT116 cells was measured using a pG13L luciferase reporter gene assay, and the expression of PLP2 was determined by Western blot analysis. Representative results of three independent reporter assay experiments are shown. The data are shown as the means ± S.D. (n = 3). I, p53−/−MDM2−/− MEF cells transfected with the indicated constructs. The whole cell lysate was subjected to Western blot with indicated antibody. J, deubiquitinase activity was required for PLP2 to increase p53 degradation. Protein expression analysis was performed by Western blotting with the indicated antibodies for cells transfected with WT PLP2 (lane 2), the C1678A mutant (lane 3), the H1836A mutant (lane 4), and the D1849A mutant (lane 5). The differences are statistically significant (**, p value < 0.001). K, MEF cells were treated with poly(I·C) or transfected with V5-nsp3 (SARS-CoV), and the whole cell lysate was subjected to Western blot with indicated antibody. L, Vero cells were infected with PEDV, and the whole cell lysate was subjected to Western blot with indicated antibody. M protein is necessary for the assembly of virus, which is an indicator of viral successful infection.
PLP2 Inhibits the Innate Immune Response by Targeting the p53 Pathway
Studies showed that p53 plays a role in antiviral innate immunity by inducing apoptosis and enhancing the type I IFN response (3–6). The effect of PLP2 on p53 stabilization raised the possibility that PLP2 regulates p53-dependent transcriptional activity, the type I IFN response, and cell apoptosis. Therefore, we examined whether PLP2 could down-regulate the transcription of the IFN-β gene. As shown in Fig. 3A, the transactivation of the IFN-β luciferase reporter by poly(I·C) was higher in p53+/+ compared with p53−/− HCT116 cells. The expression of p53 enhanced the expression of the IFN-β gene in response to poly(I·C) treatment (Fig. 3, B and C). Interestingly, PLP2 significantly inhibited the p53-dependent transactivation of the IFN-β luciferase reporter (Fig. 3A) and the transcription of the IFN-β gene (Fig. 3, B and C). PLP2 WT and the PLP2 D1849A mutant more strongly inhibited the transcription of the IFN-β gene than the DUB-mutative C1678A or H1836A of PLP2 (Fig. 3D). This result suggests that this inhibitory effect of PLP2 is partially dependent on its DUB activity.
FIGURE 3.

PLP2 blocks type I interferon signaling by targeting the p53 pathway. A, PLP2 inhibits IFN-β luciferase activity. The transcription activity of IFN-β in HCT116 cells was measured by using an IFN-β luciferase reporter gene assay. Representative results of three independent reporter assay experiments are shown. The data are shown as the means ± S.D. (n = 3). B, PLP2 inhibits the transcription of the IFN-β gene. p53+/+ HCT116 cells and p53−/− HCT116 cells were transfected with V5-PLP2 or treated with poly(I·C), and IFN-β mRNA levels were analyzed by qPCR. The data are shown as the means ± S.D. (n = 3). C, p53-deficient H1299 cells were transfected with the indicated plasmids, and IFN-β mRNA levels were analyzed by qPCR. The data are shown as the means ± S.D. (n = 3). D, deubiquitinase activity is critical for PLP2 to block type I interferon signaling. IFN-β mRNA levels were detected by qPCR from cells transfected with WT PLP2 (lane 3), the C1678A mutant (lane 4), the H1836A mutant (lane 5), or the H1849A mutant (lane 6). E, PLP2 inhibits the transcription of the IRF9 gene in a p53-dependent manner. IRF9 mRNA levels in p53+/+ and p53−/− HCT116 cells transfected with or without V5-PLP2 were determined by qPCR. The data are shown as the means ± S.D. (n = 3). F, PLP2 inhibits the transcription of the IRF7 gene in a p53-dependent manner. IRF7 mRNA levels in p53+/+ and p53−/− HCT116 cells transfected with or without V5-PLP2 were determined by qPCR. The data are shown as the means ± S.D. (n = 3). G, PLP2 inhibits the transcription of the IFN-β gene promoted by LPS. p53+/+ HCT116 cells were transfected with V5-PLP2 or treated with LPS, and IFN-β mRNA levels were analyzed by qPCR. The data are shown as the means ± S.D. (n = 3). H, LPS does not affect the transcription of the P53 gene. p53+/+ HCT116 cells were transfected with V5-PLP2 or treated with LPS, and p53 mRNA levels were analyzed by qPCR. The data are shown as the means ± S.D. (n = 3). I, MEF cells were treated with poly(I·C) or transfected with V5-nsp3 (SARS-CoV), and IRF7 mRNA levels were analyzed by qPCR. The data are shown as the means ± S.D. (n = 3). J, Vero cells were infected with PEDV, and IRF7 mRNA levels were analyzed by qPCR. The data are shown as the means ± S.D. (n = 3). The differences are statistically significant (**, p value < 0.001). NS, not significant (p value > 0.05). Con, control.
Recent studies suggest that p53 induces the transactivation of IRF9 (interferon regulatory factor 9) (4), which contributes to the up-regulation of interferon-stimulated response element-dependent genes, such as IRF7 (30), and the enhancement of the IFN signaling pathway. Notably, poly(I·C) treatment also induced the expression of the IRF9 gene, whereas PLP2 inhibited poly(I·C)-triggered IRF9 transcription (Fig. 3E). By monitoring the IRF7 transcription levels, we found that poly(I·C) treatment also induced the expression of the IRF7 gene, and PLP2 inhibited its transcription (Fig. 3F). These results indicate that PLP2 inhibits the p53-mediated immune response. Additionally, the poly(I·C)-triggered p53-independent IFN response is also regulated by PLP2 (Fig. 3B). This result was verified by LPS treatment. LPS treatment promoted the production of IFN-β (Fig. 3G) but had no significant effects on p53 mRNA levels (Fig. 3H), suggesting that LPS-induced IFN-β production was independent of p53. However, PLP2 still inhibited the transcription of the IFN-β gene stimulated by LPS (Fig. 3G). Meanwhile, we found that nsp3 (SARS-CoV) and PEDV also significantly inhibited IRF7 gene transcription (Fig. 3, I and J).
Previous studies indicated that the IFN signaling-independent antiviral functions of p53 are critical in apoptosis. Therefore, we examined whether PLP2 could regulate p53-mediated apoptosis. As shown in Fig. 4A, overexpression of PLP2 decreased the expression of p53 target gene PUMA (p53 up-regulated modulator of apoptosis) in p53−/− MEF cells transfected with Flag-p53. When p53+/+ HCT116 cells were treated with poly(I·C), the levels of p53 expression (Fig. 4B) and cell apoptosis (Fig. 4C) were significantly increased. PLP2 overexpression decreased the apoptosis rate of p53+/+HCT116 cells (Fig. 4C). Poly(I:C)-induced apoptosis seemed to be dependent on p53 because poly(I·C) had no apparent effects on p53−/−HCT116 cells (Fig. 4D). These results suggest that PLP2 inhibits p53-dependent apoptosis.
FIGURE 4.
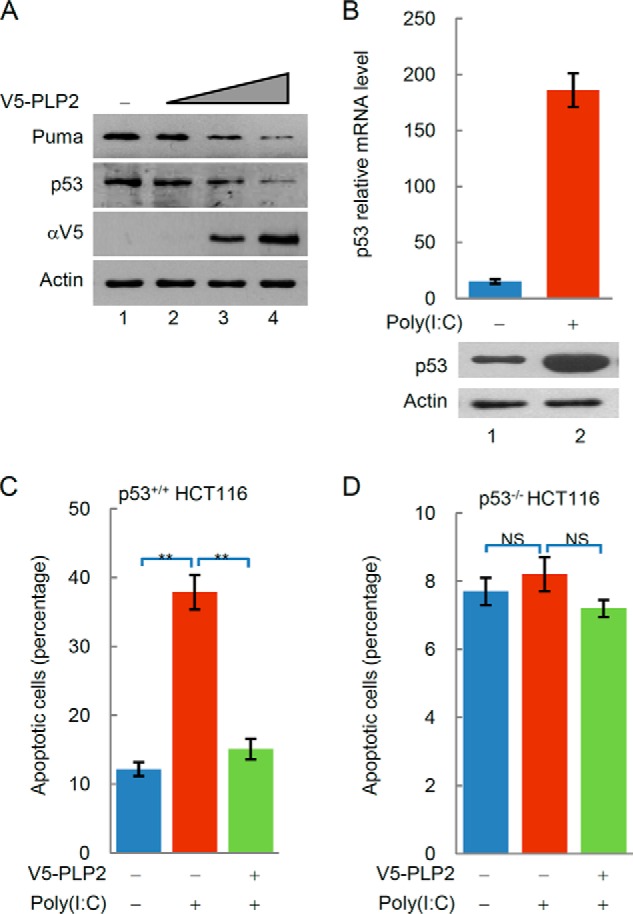
PLP2 inhibits the p53-dependent apoptosis. A, PLP2 expression decreases the steady-state level of Puma. p53−/− MEF cells were transfected with Flag-p53 and increasing amounts of V5-PLP2. After 48 h, total lysates were immunoblotted to detect the expression of Puma and p53. B, poly(I:C) increases the transcription of the P53 gene. p53 mRNA levels were analyzed by qPCR. The data are shown as the means ± S.D. (n = 3). C and D, overexpression of PLP2 decreases p53-dependent apoptosis. Apoptosis in p53+/+ and p53−/− HCT116 cells was determined by staining with annexin V. The data are shown as the means ± S.D. (n = 3). The differences are statistically significant (**, p value < 0.001). NS, not significant (p value > 0.05).
p53 Transactivates IRF7 to Control IFN-β Expression
It is worth noting that the poly(I·C)-stimulated transcription of the IRF7 gene was remarkably stronger than that of the IRF9 gene (Fig. 3, E and F). As previous studies suggest, p53 induces the transcriptional up-regulation of IRF9, leading to the up-regulation of the IRF7 gene, which encodes the IRF7 protein to further transactivate type I interferon genes. However, when the IRF9 gene was knocked down by siRNA in p53+/+HCT116 cells, the transcription of the IRF7 and IFN-β genes was not entirely inhibited (Fig. 5, A–C). These findings suggest that transcription of the IRF7 gene is regulated by factors other than IRF9. Comparing the mRNA levels of IRF7 in cells with or without p53, we found that the transcription of the IRF7 gene was reproducibly active in response to p53 induction (Fig. 3F). Moreover, our observation that p53 expression resulted in an increasing induction of IRF7 mRNA in p53 WT MEF cells compared with p53 knock-out MEF cells suggests that IRF7 is a potential transcriptional target of p53 (Fig. 5D). We identified a putative p53 binding site (p53BS) in the 3′-trailer region of the IRF7 gene (Fig. 5E), suggesting that IRF7 might be a putative target gene of p53. To evaluate the transcription-enhancing activity of the binding sequences, we performed a reporter assay with the heterologous luciferase gene fused to a p53BS upstream of the SV40 promoter (SV40-BS) of the pGL3 vector. As shown in Fig. 5F, expression of wild type p53 increased the luciferase activity significantly. ChIP analysis showed that specific p53 binding to the endogenous IRF7 3′-trailer region was enriched in chromatin immunoprecipitates using an anti-p53 antibody, especially after poly(I·C) treatment (Fig. 5G). To determine whether this sequence could directly bind to p53, we performed EMSAs to test whether p53 could bind to oligonucleotides corresponding to the p53BS sequence in the 3′-trailer region of the IRF7 gene. As shown in Fig. 5F, binding occurred in nuclear extracts of H1299 lung carcinoma cells transfected with p53, which was activated for DNA binding with the PAb421 antibody (31). When we added anti-p53 antibodies (DO-1) to the mixture, additional shifted bands were observed (Fig. 5H, lanes 5–7). Unlabeled self-oligonucleotides efficiently competed for the p53/IRF7-p53BS interaction (Fig. 5H, lanes 4 and 7). These results suggest that the p53 protein binds to IRF7-p53BS in vitro. Thus, the binding site that we identified serves as a p53 response element, indicating that IRF7 is a direct target of p53.
FIGURE 5.

p53 transactivates IRF7 to regulate IFN-β. A, siRNA-mediated knockdown of endogenous IRF9. Protein expression analysis was performed by Western blotting with the indicated antibodies. B and C, the transcription of the IRF7 gene (B) and IFN-β gene (C) is not entirely inhibited in p53+/+HCT116 cells by siRNA-mediated knockdown of endogenous IRF9. D, p53 expression results in the increasing induction of IRF7 mRNA in p53 WT MEF cells compared with p53 knock-out MEF cells. E, putative p53 binding sites located in the 3′-trailer region of the IRF7 gene. F, this genomic region was cloned into the pGL3 firefly luciferase reporter vector (IRF7-p53BS luc). H1299 cells were cotransfected with IRF7-p53BS luc or p53 vectors and a Renilla luciferase construct to control for transfection efficiency and assessed for dual luciferase activity. G, ChIP assays were performed in U2OS cells that were transfected with or without poly(I·C) using control IgG or anti-p53 antibodies, and RT-PCR was performed for the indicated IRF7 3′-trailer regions. H, the binding activity of p53 to oligonucleotides containing p53-binding sites from the 3′-trailer region of IRF7 was determined by EMSA. The differences are statistically significant (**, p value<0.001). IP, immunoprecipitation.
PLP2 Positively Supports Viral Replication
Previous studies showed that the cells have rapid and strong IFN-β productions in response to the infection by SeV, which is an interferon-sensitive RNA virus (9, 32). To substantiate our hypothesis that PLP2 may be a general blocker of the type I IFN signaling pathway and positively support viral replication, we set to determine whether PLP2 inhibits IFN production induced by SeV and promotes the replication rate of SeV. We then compared the replication kinetics of the SeV in p53 WT and knock-out MEF cells that were transfected with PLP2 expression or control vectors with an input multiplicity of infection of 100 pfu/cell. SeV exhibited considerably faster kinetics in the p53 knock-out MEF cells than in the p53 WT MEF cells. However, when we examined p53 WT MEF cells transfected with PLP2, we found that p53 knock-out MEF cells transfected with the PLP2 expression or control vectors showed comparable virus titers and consistently showed much more severe viral replication than p53 WT MEF cells transfected with control vectors (Fig. 6A). To assess whether PLP2 positively supports viral replication through interferon antagonism via the p53 pathway, we measured the IFN-β mRNA levels in the corresponding cells. As expected, the IFN-β mRNA level was significantly higher in p53 WT MEF cells transfected with control vectors than in those cells transfected with PLP2 expression vectors, which had comparable IFN-β mRNA levels with p53 knock-out MEF cells transfected with either PLP2 expression or control vectors (Fig. 6B). SeV reproduction induces cell fusion and cytopathogenicity after infection. The p53 WT MEF cells transfected with PLP2 expression vectors consistently showed higher levels of cell fusion and cytopathogenicity rates than p53 WT MEF cells transfected with control vectors (Fig. 6C). The papain-like protease of SARS-CoV, PLpro, exhibits the identical effect to p53-mediated immune response with PLP2 (Fig. 6D). Ectopic expression of the PLP2/PLpro domain inhibits SeV-induced IFN response and facilitates viral replication (Fig. 6), suggesting that PLPs provides a general inactivation mechanism of the host anti-viral response by down-regulating p53-dependent type I IFN production.
FIGURE 6.

PLP2 positively supports viral replication. A and B, replication of SeV (A) and the change in the IFN-β mRNA level analyzed by qPCR (B) in p53 WT and knock-out MEF cells transfected with PLP2 expression or control vectors. Cells were infected at an multiplicity of infection of 100 pfu/cell and harvested at various hours, as indicated. C, moderation of cytopathogenicity of SeV for cells at 24 and 48 h p.i. under single cycle growth conditions. Infections were initiated with SeV at an multiplicity of infection of 100 pfu/cell. p.i., post-infection. Arrows, fused cells; arrowheads, cytopathogenicity. D, the change in the IFN-β mRNA level analyzed by qPCR in p53 WT and knock-out MEF cells transfected with PLpro expression or control vectors. Cells were infected at an multiplicity of infection of 100 pfu/cell and harvested at indicated hours.
DISCUSSION
In this study, we explored the mechanism of the CoV-induced degradation of p53 and subsequent inhibition of interferon signaling. PLP2 interacts with the cellular ubiquitin ligase MDM2, deubiquinates, and stabilizes MDM2. In this manner, PLP2 promotes p53 degradation and inhibits the p53-mediated antiviral response and apoptosis to ensure viral growth in infected cells. Our data established the new link between p53 and innate immunity, which contributes to explain why CoV infections tend to produce low levels of interferon.
Coronavirus Infection and p53-mediated Innate Immune Response
Upon viral infection, IFN and p53 are produced and employed by host cells as components of their antiviral defense. Therefore, viruses need to tightly oppose these host antiviral responses. Viruses have evolved elaborate mechanisms to subvert p53-mediated host innate immune responses. Both p53 and certain cytokines involved in antiviral responses are inactivated by various viral proteins by either silencing cellular immunity or protecting cells from apoptosis. The number of viruses that have been found to interfere with p53 activity has increased during recent years. Large T antigen of SV40, as well as BZLF1 and EBNA3C of the EBV, interact with p53 and inhibit its activity. The E1B-55K and E4-ORF6 proteins of adenovirus, E6 of human papillomavirus, EBNA-5 of EBV, and B1R kinase of vaccinia virus all induce the degradation of p53. The E1A of adenovirus and E7 of human papillomavirus inhibit its transcriptional activity, and the X protein of hepatitis B virus has been shown to interact with p53 and inhibit its function (1). However, whether coronavirus subverts the p53-mediated host immune response is not entirely clear. Our results indicate that PLP2 of the NL63 virus regulates p53-mediated apoptosis and the type I IFN response. As far as we know, the current findings provide the first evidence to the unfolding story that coronavirus encodes antagonistic proteins of p53 to inactivate the innate immune response.
Most mammalian cells, including both immune and nonimmune cells, can produce type I IFNs in response to various viral infections. However, clinical studies have revealed that coronavirus infection induces very low levels of type I IFNs, which likely contribute to the rampant viral replication rate and high risk of related diseases. Recently, the PLP proteins of coronavirus have been identified as modulators of interferon antagonism, but the molecular mechanism remains poorly understood. Our findings uncover PLP2 as an important negative regulator of p53-mediated apoptosis and the type I IFN response and indicate a molecular framework involving both p53 and PLP2 in the virus-infected cells (Fig. 7). At the early stage of infection, p53, as a virus-induced cellular stress sensor, acts as an important transcription factor to transactivate the IRF9 and IRF7 genes. Meanwhile, p53 activates the PUMA gene to induce apoptosis (Fig. 7, left). At the advanced stage of infection, viral nucleic acids in the cells encode viral proteins, including PLP2, which directly interacts with and deubiquitinates MDM2. The deubiquitinated MDM2 is accumulated in the nucleus, where it interacts with p53 and recruits p53 to the cytoplasm for proteasomal degradation. Thus, PLP2 strongly inhibits p53-mediated IFN-β production and apoptosis while promoting viral growth in cells (Fig. 7, right). Additionally, IFN-β production is not only mediated by p53. Previous studies indicate that IRF3 also functions as a transcription factor of the IFN-β gene, and IRF3 activation by dsRNA is sufficient to induce the transcription of IFN-β (33). Therefore, the inhibitory regulation of PLP2 on IFN-β maybe not only depend on p53 signaling manner, but on other cell signaling, such as IRF3 signaling. In a word, our work validates PLP2 as a candidate drug target for the development of therapeutics against coronavirus.
FIGURE 7.
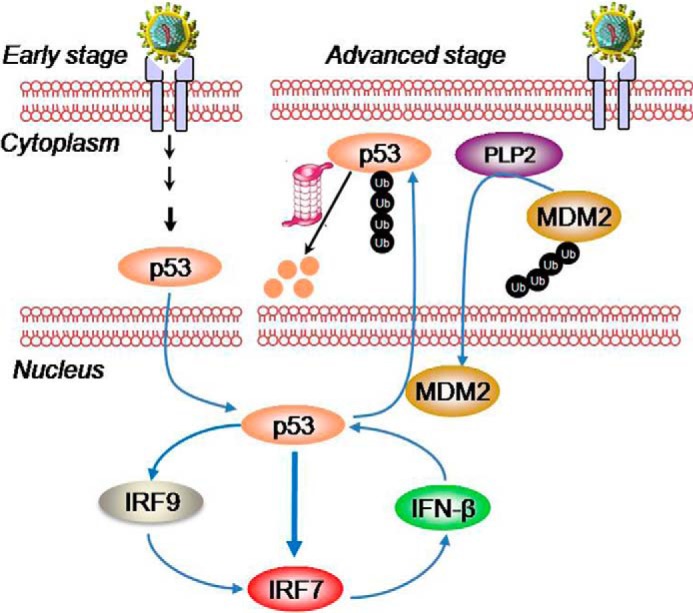
Proposed model for the regulation of p53-dependent apoptosis and the type I IFN response by PLP2. At the early stage of infection, p53 transactivates the IRF9 and IRF7 genes. At the advanced stage of infection, viral nucleic acids in the cells encode viral proteins, including PLP2, which decreases the stability and transcriptional activity of p53 by increasing the MDM2-mediated ubiquitination and nuclear export of p53 and, in turn, strongly inhibits p53-mediated IFN-β production and apoptosis while promoting viral growth in cells.
IRF7 Is a Direct Target of p53 in Immune Response
In addition to DNA damage and oncogene activation, p53 is also responsible for responses to hypoxia, nutrient deprivation, and viral infection. The recently identified functions of p53 in both virus-induced apoptosis and the up-regulation of the type I IFN response suggest that p53 may act as a virus-induced cellular stress sensor. However, how p53 exhibits its functions in immune response remains not fully understood. In this study, we found that p53 induces the expression of IRF7, which may later activate type I interferon genes, thereby contributing another page to the unfolding story of how p53 activates the innate immune response. At least four pieces of evidence support the true transcriptional regulation of p53 on IRF.: First, in vitro reporter assays showed that p53 transactivates IRF7-p53BS luciferase activity in cultured cells. Second, in vitro EMSA assays demonstrated the direct binding of p53 to the IRF7-p53BS sequence. Third, in vivo mRNA detection illustrated that the IRF7 gene is transactivated by p53. Fourth, in vivo ChIP assays showed the significant binding of p53 to the IRF7-p53BS sequence. Therefore, we propose that p53 functions as a bona fide transcriptional factor of the IRF7 gene. It is well known that LPS, viral infections, IFN, and certain chemical reagents, such as sodium butyrate, can induce the expression of IRF7. In the early stage of viral infection, IRF7 that is modified by carboxyl-terminal phosphorylation is translocated to the nucleus, where together with IRF3, it induces the expression of early interferon genes and ultimately triggers many biological pathways. Late in infection, IRF7 induces the high expression of IFNα and IFNβ, thus activating anti-viral gene expression. Gene targeting tests indicate that without IRF7, early and late IFN genes cannot be transcribed, which suggests that IRF7 participates in all stages of type I interferon production.
In summary, the results of our study indicate that inactivation of p53 by PLP2 may contribute to the weak IFN response in coronavirus infections. Our work validates PLP2 as a candidate drug target for the development of therapeutics against coronavirus. Ultimate understanding of the overall interactions between coronaviruses and the host innate immune system will provide further insight into the pathogenesis of viruses in this class and open a new avenue of therapeutic target exploration against coronavirus infections.
The work was supported by National Basic Research Programs 2012CB910702, 2011CB910802, and 2013CB910803 and National Natural Science Foundation Projects 81201271, 31125010, and 81221004.
- HCoV
- human coronavirus
- SARS
- severe and acute respiratory syndrome
- PLP
- papain-like protein
- DUB
- deubiquitinase
- TRITC
- tetramethylrhodamine isothiocyanate
- qPCR
- quantitative PCR
- PEDV
- porcine epidemic diarrhea virus
- SeV
- Sendai virus
- MEF
- murine embryonic fibroblast.
REFERENCES
- 1. Rivas C., Aaronson S. A., Munoz-Fontela C. (2010) Dual role of p53 in innate antiviral immunity. Viruses 2, 298–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takaoka A., Hayakawa S., Yanai H., Stoiber D., Negishi H., Kikuchi H., Sasaki S., Imai K., Shibue T., Honda K., Taniguchi T. (2003) Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 424, 516–523 [DOI] [PubMed] [Google Scholar]
- 3. Munoz-Fontela C., Garcia M. A., Garcia-Cao I., Collado M., Arroyo J., Esteban M., Serrano M., Rivas C. (2005) Resistance to viral infection of super p53 mice. Oncogene 24, 3059–3062 [DOI] [PubMed] [Google Scholar]
- 4. Muñoz-Fontela C., Macip S., Martínez-Sobrido L., Brown L., Ashour J., García-Sastre A., Lee S. W., Aaronson S. A. (2008) Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 205, 1929–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pampin M., Simonin Y., Blondel B., Percherancier Y., Chelbi-Alix M. K. (2006) Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J. Virol. 80, 8582–8592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nguyen M. L., Kraft R. M., Aubert M., Goodwin E., DiMaio D., Blaho J. A. (2007) p53 and hTERT determine sensitivity to viral apoptosis. J. Virol. 81, 12985–12995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Z., Wang Y., Ratia K., Mesecar A. D., Wilkinson K. D., Baker S. C. (2007) Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol. 81, 6007–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perlman S., Netland J. (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7, 439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clementz M. A., Chen Z., Banach B. S., Wang Y., Sun L., Ratia K., Baez-Santos Y. M., Wang J., Takayama J., Ghosh A. K., Li K., Mesecar A. D., Baker S. C. (2010) Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84, 4619–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cinatl J., Morgenstern B., Bauer G. (2003) Treatment of SARS with human interferons. Lancet 362, 293–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng B., He M. L., Wong K. L., Lum CT, Poon L. L., Peng Y., Guan Y., Lin M. C., Kung H. F. (2004) Potent inhibition of SARSassociated coronavirus (SCOV) infection and replication by type I interferons (IFN-α/β) but not by type II interferon (IFN-gamma). J. Interferon Cytokine Res. 24, 388–390 [DOI] [PubMed] [Google Scholar]
- 12. Reghunathan R., Jayapal M., Hsu L. Y. (2005) Expression profile of immune response genes in patients with severe acute respiratory syndrome. BMC Immunol. 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J., Subbarao K. (2007) The immunobiology of SARS. Annu. Rev. Immunol. 25, 443–472 [DOI] [PubMed] [Google Scholar]
- 14. Frieman M., Heise M., Baric R. (2008) SARS coronavirus and innate immunity. Virus Res. 133, 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tian C., Xing G., Xie P., Lu K., Nie J., Wang J., Li L., Gao M., Zhang L., He F. (2009) KRAB-type zinc-finger protein Apak specifically regulates p53-dependent apoptosis. Nat. Cell Biol. 11, 580–591 [DOI] [PubMed] [Google Scholar]
- 16. Lu K., Yin X., Weng T., Xi S., Li L., Xing G., Cheng X., Yang X., Zhang L., He F. (2008) Targeting WW domains linker of HECT-type ubiquitin ligase Smurf1 for activation by CKIP-1. Nat. Cell Biol. 10, 994–1002 [DOI] [PubMed] [Google Scholar]
- 17. Devaraj S. G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C. J., Tseng C. T., Baker S. C., Li K. (2007) Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282, 32208–32221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kato A., Kiyotani K., Sakai Y., Yoshida T., Nagai Y. (1997) The paramyxovirus, Sendai virus, V protein encodes a luxury function required for viral pathogenesis. EMBO J. 16, 578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sato M., Suemori H., Hata N., Asagiri M., Ogasawara K., Nakao K., Nakaya T., Katsuki M., Noguchi S., Tanaka N., Taniguchi T. (2000) Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13, 539–548 [DOI] [PubMed] [Google Scholar]
- 20. Chaudhuri R., Tang S., Zhao G., Lu H., Case D. A., Johnson M. E. (2011) Comparison of SARS and NL63 papain-like protease binding sites and binding site dynamics: inhibitor design implications. J. Mol. Biol. 414, 272–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ratia K., Saikatendu K. S., Santarsiero B. D., Barretto N., Baker S. C., Stevens R. C., Mesecar A. D. (2006) Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U.S.A. 103, 5717–5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nicholson B., Leach C. A., Goldenberg S. J., Francis D. M., Kodrasov M. P., Tian X., Shanks J., Sterner D. E., Bernal A., Mattern M. R., Wilkinson K. D., Butt T. R. (2008) Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 17, 1035–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li M., Brooks C. L., Kon N., Gu W. (2004) A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13, 879–886 [DOI] [PubMed] [Google Scholar]
- 24. Cummins J. M., Rago C., Kohli M., Kinzler K. W., Lengauer C., Vogelstein B. (2004) Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 10.1038/nature02501 [DOI] [PubMed] [Google Scholar]
- 25. Xie Y., Avello M., Schirle M., McWhinnie E., Feng Y., Bric-Furlong E., Wilson C., Nathans R., Zhang J., Kirschner M. W., Huang S. M., Cong F. (2013) Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J. Biol. Chem. 288, 2976–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stommel J. M., Marchenko N. D., Jimenez G. S., Moll U. M., Hope T. J., Wahl G. M. (1999) A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J. 18, 1660–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boyd S. D., Tsai K. Y., Jacks T. (2000) An intact MDM2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol. 2, 563–568 [DOI] [PubMed] [Google Scholar]
- 28. Geyer R. K., Yu Z. K., Maki C. G. (2000) The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol. 2, 569–573 [DOI] [PubMed] [Google Scholar]
- 29. Li M., Brooks C. L., Wu-Baer F., Chen D., Baer R., Gu W. (2003) Mono-versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302, 1972–1975 [DOI] [PubMed] [Google Scholar]
- 30. Lu R., Au W. C., Yeow W. S., Hageman N., Pitha P. M. (2000) Regulation of the promoter activity of interferon regulatory factor-7 gene. J. Biol. Chem. 275, 31805–31812 [DOI] [PubMed] [Google Scholar]
- 31. Hupp T. R., Meek D. W., Midgley C. A., Lane D. P. (1992) Regulation of the specific DNA binding function of p53. Cell 71, 875–886 [DOI] [PubMed] [Google Scholar]
- 32. Zheng D., Chen G., Guo B., Cheng G., Tang H. (2008) PLP2, a potent deubiquitinase from murine hepatitis virus, strongly inhibits cellular type I interferon production. Cell Res. 18, 1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peters K. L., Smith H. L., Stark G. R., Sen G. C. (2002) IRF-3-dependent, NF-κB and JNK-independent activation of the 561 and IFN genes in response to double-stranded RNA. Proc. Natl. Acad. Sci. U.S.A. 99, 6322–6327 [DOI] [PMC free article] [PubMed] [Google Scholar]