

Abstract
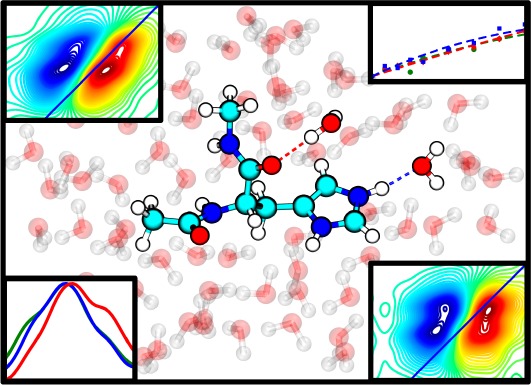
It is well known that histidine is involved in many biological functions due to the structural versatility of its side chain. However, probing the conformational transitions of histidine in proteins, especially those occurring on an ultrafast time scale, is difficult. Herein we show, using a histidine dipeptide as a model, that it is possible to probe the tautomer and protonation status of a histidine residue by measuring the two-dimensional infrared (2D IR) spectrum of its amide I vibrational transition. Specifically, for the histidine dipeptide studied, the amide unit of the histidine gives rise to three spectrally resolvable amide I features at approximately 1630, 1644, and 1656 cm–1, respectively, which, based on measurements at different pH values and frequency calculations, are assigned to a τ tautomer (1630 cm–1 component) and a π tautomer with a hydrated (1644 cm–1 component) or dehydrated (1656 cm–1 component) amide. Because of the intrinsic ultrafast time resolution of 2D IR spectroscopy, we believe that the current approach, when combined with the isotope editing techniques, will be useful in revealing the structural dynamics of key histidine residues in proteins that are important for function.
1. Introduction
The imidazole ring of histidine (His) can exist in two neutral tautomeric forms (π and τ) and also has a pKa of ∼6.5.1,2 Thus, these structural and ionization characteristics make His one of the most frequently used amino acids by nature to create protein binding sites, to coordinate metal ions, to carry out catalytic activities, and to facilitate signal transduction.3−11 For example, influenza A viruses cleverly exploit the ionization behavior of His at near-neutral pH and use it in the M2 transmembrane protein as a pH sensor and proton shuttle12−14 to initiate the process of viral replication in response to endocytosis-induced guest acidification.15 Other examples include the enzyme carbonic anhydrase II, where a histidine located in the active site plays a key role in the catalysis of CO2 hydration by acting as a proton shuttle.16 Because of the critical role that His plays in a wide range of biological activities and functions, many studies have been carried out to understand how the local protein environment affects the structural, dynamical, and ionization properties of its imidazole side chain. In this regard, NMR spectroscopy, due to the sensitivity of 15N and 13C chemical shifts to imidazole structures, has been the method of choice to characterize the structural distributions of histidines in proteins.13,17−22 For instance, Kay and coworkers have utilized NMR relaxation measurements and chemical shift titrations to characterize the protonation, rotamerization, and tautomerization of His61 in plastocyanin from Anabaena variabilis,18 and Cheng and coworkers have used solid-state NMR (ssNMR) to explore the tautomeric distributions of the imidazole side chain in a series of His-containing dipeptides.23 More recently, Hong and coworkers have shown, through ssNMR measurements, the existence of four distinct protonation states of histidine across a broad range of pH that not only associate differently with water, but also show pH-induced changes in side-chain rotameric distributions.1 Despite the excellent utility of NMR spectroscopy in characterizing the structure and dynamics of His residues in proteins, the technique lacks the ability to directly capture the dynamics of His side chains that occur on an ultrafast time scale, such as those associated with protonation and deprotonation of the imidazole ring, which is expected to occur on the picosecond time scale.24 Thus, this limitation of NMR spectroscopy underscores the need for methodologies that can visualize such ultrafast dynamics. Herein, we show that two-dimensional infrared (2D IR) spectroscopy is potentially a useful approach in this regard. The sensitivity of 2D IR spectroscopy to dynamical events that cause the vibrational frequency of the IR reporter to fluctuate or change stems from its ability to directly resolve the inhomogeneous contribution to the IR line shape in question as well as its time evolution through the frequency–frequency correlation function.25,26 Moreover, 2D IR signal strength scales as the square of the extinction coefficient of the vibrator, which significantly enhances the ratio of the desired signal over the solvent background.27 Thus, these unique capabilities of 2D IR spectroscopy suggest that it is ideally suited toward probing the structural and solvation dynamics of His residues in proteins.
The applicability of 2D IR spectroscopy to distinguish tautomers at equilibrium was recently demonstrated for the lactam–lactim system.28 Unlike such cyclic amides, the imidazole moiety lacks a strong vibrational mode,29 thus making the application of a side-chain vibration of His to study its tautomerization difficult. One potential approach to overcoming this challenge is to introduce a vibrational probe that has a higher extinction coefficient and is also spectrally isolated from other protein vibrations, such as a nitrile or azide moiety.30,31 However, this approach may alter the structural and ionization properties of the imidazole and thus is less attractive. Another approach is to use deuterated His residues.32,33 While this approach is nearly nonperturbative and Londergan and coworkers32,33 have shown that the C–D vibrational modes of a deuterated imidazole are indeed sensitive Raman probes of the structure and ionization state of His, the relatively low IR extinction coefficient of the C–D stretching vibration makes it a less appealing probe in linear and nonlinear IR measurements.
Herein we aim to use the amide I vibrational mode of His as an IR reporter of its side-chain structure and dynamics. The amide I band of proteins mainly arises from the stretching vibrations of the backbone carbonyl groups and is an established structural reporter.34 For this reason and also because the amide I vibrational transition has a large extinction coefficient, many studies have utilized it to probe a wide range of structural and dynamical events of various biological systems.26,27,29,31,35,36 An additional advantage of using the amide I mode is that it offers site-specific resolution through isotopically labeling the backbone C=O unit of interest (i.e., replacing 12C=16O with 13C=16O or 13C=18O).34 For example, in conjunction with 2D IR spectroscopy, this isotopic labeling strategy has been used to study many dynamical processes occurring on the picosecond time scale,26,34,34,35,37−41 such as the dynamics of water inside the M2 proton channel26 and also in an amyloid fibril.42 On the basis of the established sensitivity of the amide I vibration to its local environment, we expect that the His amide I mode will reflect, at least to a certain degree, the protonation/tautomerization status and structural configuration of the imidazole ring, through its vibrational frequency, spectral line shape, and signal evolution dynamics. In other words, we propose that monitoring changes of the amide I spectral properties of a target His residue in a protein will allow us to detect the structural or environmental changes of its side chain. While this is an indirect approach as compared with NMR, where atoms on the imidazole ring can be probed directly, if proven feasible it will be quite useful in the study of various biological problems, such as understanding the mechanism of proton conduction by the M2 channel, the study of which currently lacks microscopic details due to intrinsic ultrafast time scale of the dynamics. To explore the feasibility of the proposed approach, here we report linear and 2D IR experiments on a model His dipeptide (i.e., Ac-His-CONHMe) that highlights the possibility of using the backbone amide mode of His for probing its conformational distributions and dynamics in proteins. In principle, the isolation of the amide I mode of a specific His residue could be achieved through isotopic labeling,43 although we note that such a strategy, to the best of our knowledge, has not yet been realized for His.
2. Experimental Section
Peptide Synthesis
A histidine amino acid acetylated (Ac) at the N-terminus and methyl amidated (NHMe) at the C-terminus, subsequently referred to as the “histidine dipeptide”, was used for the experiments reported in this article. This dipeptide (Ac-His-CONHMe) was synthesized using standard Fmoc-protocols employing 3-(methyl-Fmoc-amino)-methyl)-indol-1-yl) acetyl AM resin (EMD Chemicals). After the addition of the histidine residue, the peptide was acetylated using isotopically labeled (13C) acetic anhydride (Sigma-Aldrich). The peptide was purified to homogeneity by reverse-phase chromatography and characterized by mass spectrometry. Following purification, the residual trifluoroacetic acid from peptide synthesis, which has a sharp mid-IR band centered at 1673 cm–1, was mostly removed by multiple lyophilizations against a 0.1 M DCl solution. The peptide was additionally exchanged in D2O and lyophilized. The peptide sample was prepared in D2O and titrated to the desired pH with a final peptide concentration of ∼0.1 M.
Linear and 2D IR Spectroscopy
The peptide sample was placed between two calcium fluoride windows, separated by a 25 μm Teflon spacer, and used for both linear and 2D IR measurements. The optical density of the sample in the amide I region was <0.2. Linear IR spectra were collected on a Thermo Nicolet 6700 FTIR spectrometer. The experimental apparatus for collection of 2D IR spectra has been described elsewhere in detail.34 In short, a sequence of three ∼85 fs infrared pulses (center frequency of ∼1645 cm–1) was used to excite the samples, and signal in the phase-matched direction –k1 + k2 + k3 was heterodyned with a reference pulse (local oscillator). The combined signal and local oscillator pulses were dispersed on a monochromator having a 64-element mercury–cadmium–telluride (MCT) array detector (InfraRed Associates). The final frequency domain 2D spectrum was obtained by Fourier transforming the data along the time interval τ between the first and second pulses and along t, which denotes the evolution time of the signal. 2D spectra at different waiting times were collected by scanning the delay T between the second and third pulses.
Gaussian Calculations
Ab initio calculations were performed on neutral histidine peptides by means of the Gaussian03 package and B3LYP density functional theory (DFT) method with a basis of 6-31+G**. The structures were optimized, and an anharmonic frequency calculation was performed to determine assignments of specific vibrational transitions. No correction factor was applied to the frequencies calculated.
3. Results and Discussion
To test the utility of the amide I vibrational mode of His as a structural and dynamical IR reporter of His tautomerization and protonation status in proteins, we performed linear and nonlinear IR measurements on an N-terminally acetylated and C-terminally methylamidated His amino acid (Figure 1). Because this model compound contains two backbone carbonyl groups, it is referred to here as His dipeptide. In addition, to isolate the amide I band of His, 13C-acetic anhydride was used to cap the N-terminus. In the following text, only results pertinent to the unlabeled His amide I band are discussed.
Figure 1.
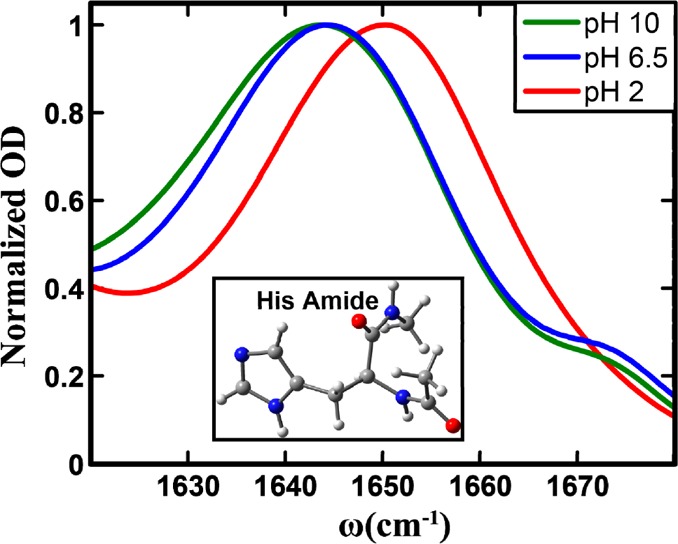
Normalized amide I′ bands of the histidine amide in the His dipeptide measured at different pH values, as indicated. Shown in the inset is the structure of the dipeptide, where the histidine side chain is in its π tautomer form.
As shown (Figure 1), FTIR spectra indicate that the amide I vibration of His gives rise to a single peak, centered approximately between 1620 and 1670 cm–1, as the weak band at ∼1674 cm–1 can be attributed to residual TFA ions from peptide synthesis. In addition, these spectra clearly show that the exact position and line shape of the amide I vibrational transition of His depend on pH. For example, when the pH is decreased from 10 to 2, which leads to protonation of the imidazole ring, the amide I band shifts from 1642 to 1650 cm–1. This result indicates that FTIR measurements alone can be used to qualitatively monitor the protonation/deprotonation of the His side chain. A closer inspection of the spectra indicates that while the spectra obtained at pH 10 and 6.5 show a similar peak frequency, the pH 10 spectrum is clearly broader. Perhaps more importantly, all three spectra show a certain degree of asymmetry, indicating the presence of underlying structural distributions that cannot be further resolved by linear spectroscopy. To further characterize the spectral and dynamical properties of these substructures, we subsequently carried out 2D IR measurements on this His dipeptide.
As shown (Figure 2), the 2D IR spectra of the His amide obtained at different pH values and two waiting times, 0 and 3 ps, all show distinct transitions that are not resolved in the corresponding FTIR spectra. Specifically, it is clear that besides the expected strong transition at ∼1644 cm–1, two weaker bands at ωτ of ∼1630 and 1656 cm–1 exist. The presence of these bands, which are denoted as A (1630 cm–1), B (1644 cm–1), and C (1656 cm–1), becomes even more evident in the diagonal traces of the 2D spectra at zero waiting time (Figure 3). The diagonal traces are obtained by taking a slice of the 2D IR spectra parallel to the diagonal line that runs through the positive peak. As expected, these bands, either their intensity, frequency, or both, show a measurable dependence on pH. The frequencies of all bands blue-shift by ∼2 cm–1 as the pH is lowered from 6.5 to 2, which indicates that the amide I vibrational frequency of His is sensitive to protonation/deprotonation of the imidazole ring. This result is consistent with recent work by Reppert et al.,44 which shows that the amide I vibration of a series of dipeptides depends on the protonation states of the N- and C-termini of the peptide. As shown (Figure 3), the diagonal traces also reveal, when the pH is decreased from 10 to 6.5, that the intensity of band A shows an appreciable decrease, whereas that of band C is virtually unchanged but shows a significant increase when the pH is further lowered to 2. Taken together, these results provide further evidence that the amide I vibration of His could be used to report on the structure and protonation state of its imidazole side chain, as explained in the following section.
Figure 2.

2D IR spectra of the histidine amide I′ vibrator in the His dipeptide measured at different pH values and waiting times, as indicated. The dashed line in each case indicates the peak position at zero waiting time.
Figure 3.
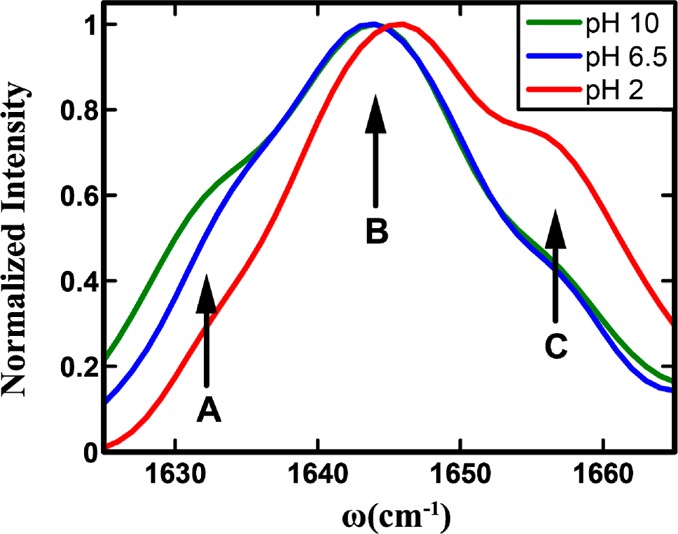
Normalized diagonal traces of the 2D IR spectra at zero waiting time in Figure 2. In each case, a linear baseline has been subtracted. The three conformers seen in the traces are shown with arrows and labeled A–C, respectively.
For a given vibrational transition (e.g., amide I), the presence of multiple bands can often be attributed to the existence of different conformers in solution.38 In the current case, we can attribute the three resolvable bands observed within the profile of the amide I band of the His dipeptide to three states of the imidazole side chain, arising from tautomerization and protonation. This assignment is supported by the following observations: (1) a similar dipeptide that does not contain an imidazole side chain shows no splitting of its amide I band,45 (2) the possibility that the multiple bands observed arise from different molecular vibrations can be ruled out as the 2D IR spectra at zero waiting time do not show any significant intensity in the expected cross peak region,46 and (3) no significant backbone conformational changes of His-containing dipeptides have been reported between basic and acidic pHs.44 Hence it is logical to interpret the spectral changes observed in our experiments to be originating from protonation or conformational changes of the His side chain. Nevertheless, it should be noted that the measurements reported here do not directly probe the protonation state of the imidazole, and the subpopulations previously shown reflect the protonation/tautomerization state of the imidazole ring through its effects on the amide vibrational frequency and spectral dynamics, as shown later.
A distinctive advantage of 2D IR spectroscopy is the ability to probe ultrafast dynamic events that cause the initial vibrational excitation to scramble within the inhomogeneous spectral profile or transfer to a different state, through energy transfer, or to a different conformer, through chemical exchange.26,34,47,48 The latter is often manifested as a cross peak in the 2D IR spectrum that grows as a function of the waiting time.47,49 As shown (Figure 2), a comparison of the 2D IR spectra at waiting times of 0 and 3 ps reveals the presence of cross peaks that grow in with increasing waiting time, indicating ongoing exchange between two different states or conformers. However, significant buildup of cross-peak signal is observed only for the B–C pair and not for the A–B and A–C pairs. The time scale of these exchange dynamics was determined from fitting the corresponding cross-peak to diagonal-peak (i.e., SBB for state B and SBC for state C) ratio to a simple two-state exchange kinetic model,37,49 namely
| 1 |
where Keq is the equilibrium constant between the states B and C and ket represents the exchange rate constant. For simplicity, we have assumed that both states afford the same transition dipoles and vibrational lifetimes.37 The fitted curves to the experimental data are shown in Figure 4, and the fitted exchange time scales are tabulated in Table 1. As shown, the exchange kinetics between states B and C occur on a time scale of ∼3 ps at neutral and basic pH values and show no significant dependence on pH. However, at low pH, the exchange slows by a factor of ∼2. This suggests that the physical process underlying the development of the B–C cross peak is connected to the protonation–deprotonation process of the imidazole ring. Additionally, because imidazole tautomerization is expected to occur on a slower time scale than 3 ps, it is also unlikely that the B–C cross peak arises from an exchange process between two imidazole tautomers. The 3 ps exchange time is comparable to that observed for solvation dynamics of peptide backbone units.37 Thus, we attribute bandd B and C to a hydrated and dehydrated His amide, respectively, which are connected through a chemical equilibrium. This assignment is further supported by the fact that these two bands are separated by ∼10 cm–1, consistent with that observed in similar cases.26,37 Such solvated–unsolvated doublets have been seen for model amides in methanol.50 Similar observations have also been made for a tryptophan dipeptide in water, where the two states were shown to be related to a change in the torsional angle of the indole ring.37 Thus, attributing states B and C to different solvation states of the His amide is reasonable. In addition, for a His residue, where the imidazole ring offers a competing water binding site, these two states most likely represent two subpopulations where a water molecule is bound to the side chain and not the amide, and vice versa. Interestingly, as shown (Figure 3), the intensity of C remains virtually unchanged when the pH is decreased from 10 to 6.5 but shows a significant increase when the pH is further decreased to 2, indicating that this band is sensitive to the protonation status of the His side chain. This result is not entirely unexpected because the protonated form of the imidazole moiety is known to more strongly associate with water,1 thus making the amide unit more dehydrated, akin to the effect of trimethylamine N-oxide (TMAO).51 This is also consistent with the decrease in the B–C exchange rate at low pH; if water associates more strongly with the ring, the rate at which it switches between the amide and the imidazole is expected to be slower.
Figure 4.
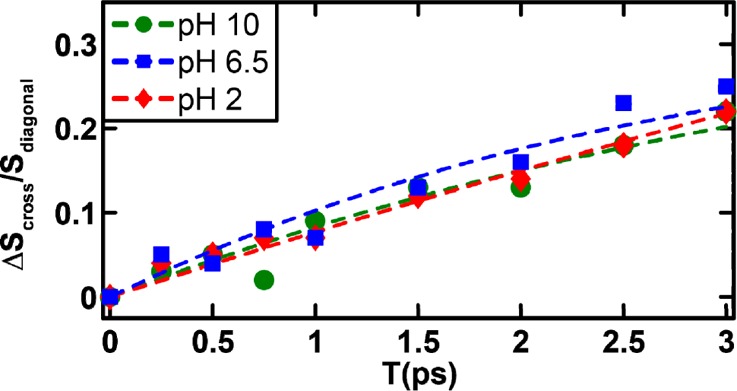
Evolution of the cross-peak to diagonal-peak ratio as a function of the waiting time for different pH values, as indicated. The dashed lines represent fits to a two-state kinetic model, as discussed in the text, and the resultant fitting parameters are listed in Table 1
Table 1. Parameters for Fitting the Evolution of the Cross to Diagonal Peak Ratios As Obtained from the 2D IR Spectra at Different pH Values.
| pH | Keq | kex–1 (ps) |
|---|---|---|
| 10 | 0.45 | 3.7 ± 0.6 |
| 6.5 | 0.35 | 2.3 ± 0.4 |
| 2 | 1.2 | 7.4 ± 0.6 |
Unlike C, the intensity of A changes more gradually with pH (Figure 3). Thus, the possibility that A and C are connected through an equilibrium process, such as side chain protonation–deprotonation or tautomerization, can be ruled out. It has been shown from ssNMR experiments23 and theoretical modeling2 that in His-containing dipeptides the imidazole ring can sample different tautomeric states. Hence, it is most likely that A corresponds to one of the two tautomers of the His side chain. Consequently, this assignment would lead us to further attribute B to the other tautomer. To help better understand the structural nature of the conformations that give rise to bands A and B, we further performed vibrational frequency calculations on the isolated dipeptide using Gaussian 03. It should be noted that the structures obtained are the lowest energy conformations in the absence of water and thus may not reflect the exact pH-dependent conformational distribution. The calculations show that the amide I vibrational frequency of the π tautomer (1741.9 cm–1) is distinctively higher than that of the τ tautomer (1736.7 cm–1). On the basis of this result, we can assign band A as the τ tautomer and B as the π tautomer, as summarized in Figure 5. We note that the change of the A subpopulation with pH can also be attributed to a change in the orientation of the side chain with respect to the backbone. Such changes in the imidazole torsional angles have been observed by ssNMR measurements. Unfortunately, our results cannot discern between a tautomeric and a side-chain torsional equilibrium. However, for larger proteins and peptides, where the tautomeric distribution or side chain angles are known, the amide spectra can be more conveniently utilized to understand the His structural dynamics without ambiguity. For example, in the transmembrane M2 channels of the influenza virus, it is known from NMR investigations that the tautomeric equilibrium of His is heavily favored toward the τ tautomer in the neutral-acidic pH range. In such a case, the His amide vibrations can be used to track the pH-dependent protonation of the imidazole moiety and its interactions with the channel water.
Figure 5.
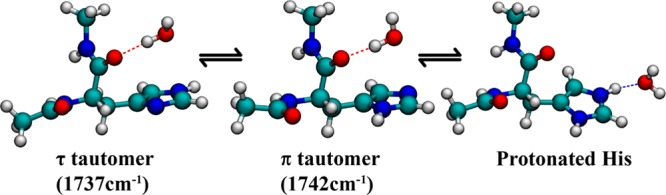
Summary of the experimental results depicting the equilibrium between the imidazole tautomeric and solvation states. Also listed are the calculated vibrational frequencies of the π and τ tautomers.
Furthermore, it should be noted that the sensitivity of the His amide to the protonation or conformation of the imidazole side chain arises primarily from the relative positions and values of the charges on the ring. Hence, the amide I vibrations of all amide units that are in close proximity to the imidazole are expected to exhibit a similar effect. This can be tested through inspection of the amide I band of the labeled acetyl end. As shown in the Supporting Information (Figures S1 and S2), the linear IR spectra and diagonal traces at zero waiting time of the acetyl group indeed exhibit a pH dependence that supports the presence of multiple states. As expected, the effect of the underlying structural transitions of the His side chain on the amide I mode of this nearby moiety is significantly weaker, which prevents a more quantitative assessment of the time evolutions of the subensembles, as indicated by the 2D traces.
In summary, the linear and 2D IR spectra presented in this study demonstrate the sensitivity of the His amide vibration not only to the protonation and tautomeric equilibria of its side chain but also to water dynamics near the residue, thus forwarding the applicability of infrared spectroscopy as a probe of protonation and hydration of His residues in larger proteins and biomolecular complexes, where this process often plays a key role in functional aspects of the protein involved. This sensitivity stems from the effects of the side-chain tautomerization/protonation on the corresponding amide I frequency and its spectral dynamics. More specifically, for the His dipeptide studied here we observed three spectrally resolvable features within the amide I vibrational band of His, which are positioned at approximately 1630 (A), 1644 (B), and 1656 cm–1 (C), respectively. On the basis of their dependence on pH and interactions, these three bands are assigned as follows: bands A and B represent two tautomeric states of the His side chain at neutral and basic pH values, whereas band C arises from a subpopulation of B whose amide is dehydrated. At pH values lower than the pKa of imidazole, band C corresponds to the desolvated amide in the protonated conformer of histidine, whereas B represents the solvated amide moiety.
Despite the aforementioned promising results, several hurdles need to be overcome to implement the proposed method to study real protein systems. First, site-specific isotope labeling of a His residue in peptides and proteins has not been reported. Second, even if the amide I band of the target His residue can be separated from the amide I transitions of the rest of the residues in a given peptide/protein, it could still overlap with vibrational bands from certain side chains, which would complicate data interpretation. Third, in a large peptide/protein system, other structural factors may also affect the spectral properties of the amide I mode of the His probe, making a quantitative assessment of the results difficult. Despite these challenges, it is our hope that the current study will inspire further efforts in the development of this method, as it does offer a unique approach to study the functional role of His residues in proteins.
4. Conclusions
The imidazole side chain of histidine can adopt two tautomeric forms and can ionize at near-neutral pH. Thus, histidine plays a key role in many enzymatic reactions and processes involving proton conduction. The effect of histidine protonation on the water structure near it or the interaction of histidine with water is therefore an important problem in biophysics. While the amide vibration of a histidine residue is an indirect probe of the side chain’s conformational distribution, it provides an approach to measure and understand these interactions, which occur on an ultrafast time scale, by observing the effects protonation has on the amide vibrational dynamics and frequency. To explore the potential utility of nonlinear IR spectroscopy in probing the structural and functional dynamics of the histidine side chain in proteins, we carried out 2D IR measurements on a model histidine dipeptide. Our results show that the amide I band of histidine consists of three spectrally resolvable components, at approximately 1630, 1644, and 1656 cm–1, which exhibit different dependences on pH. On the basis of results obtained at three pH values (i.e., 2, 6.5, and 10) and frequency calculations, we attribute the 1630 cm–1 component to the τ tautomer and the 1644 cm–1 component to a π tautomer. The 1656 cm–1 band is assigned to a desolvated amide mode that is present in both the protonated and the neutral π tautomeric states. Taken together, these results demonstrate that the amide I vibrational mode of histidine is sensitive to its side-chain conformation and protonation state and thus can be used as a viable IR reporter of biological processes that actively involve histidines.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM-012592). The 2D IR data were collected at the Ultrafast Optical Processes Laboratory under grant P41GM104605.
Supporting Information Available
Linear IR spectra of the histidine dipeptide obtained at different pH values, as indicated. Diagonal traces of the 13C=O labeled N-terminal amide obtained from 2D IR spectra at T = 0 and different pH values. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Li S.; Hong M. Protonation, Tautomerization, and Rotameric Structure of Histidine: A Comprehensive Study by Magic-Angle-Spinning Solid-State NMR. J. Am. Chem. Soc. 2011, 133, 1534–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila J. A.; Arnautova Y. A.; Vorobjev Y.; Scheraga H. A. Assessing the Fractions of Tautomeric Forms of the Imidazole Ring of Histidine in Proteins as a Function of pH. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5602–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin W. W.; Roberts J. D. N-15 Nuclear Magnetic-Resonance Spectroscopy - State of Histidine in Catalytic Triad of Alpha-Lytic Protease - Implications for Charge-Relay Mechanism of Peptide-Bond Cleavage by Serine Proteases. J. Am. Chem. Soc. 1978, 100, 8041–8047. [Google Scholar]
- Roberts V. A.; Iverson B. L.; Iverson S. A.; Benkovic S. J.; Lerner R. A.; Getzoff E. D.; Tainer J. A. Antibody Remodeling: A General Solution to the Design of a Metal-Coordination Site in an Antibody Binding Pocket. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 6654–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockel J.; Safar J.; Wallace A. C.; Cohen F. E.; Prusiner S. B. Prion Protein Selectively Binds Copper(II) Ions. Biochemistry 1998, 37, 7185–7193. [DOI] [PubMed] [Google Scholar]
- Wieland H. A.; Lüddens H.; Seeburg P. H. A Single Histidine in GABAA Receptors is Essential for Benzodiazepine Agonist Binding. J. Biol. Chem. 1992, 267, 1426–1429. [PubMed] [Google Scholar]
- Williams S.; Bledsoe R. K.; Collins J. L.; Boggs S.; Lambert M. H.; Miller A. B.; Moore J.; McKee D. D.; Moore L.; Nichols J.; Parks D.; Watson M.; Wisely B.; Willson T. M. X-ray Crystal Structure of the Liver X Receptor β Ligand Binding Domain: Regulation by a Histidine-Tryptophan Switch. J. Biol. Chem. 2003, 278, 27138–27143. [DOI] [PubMed] [Google Scholar]
- Tang X. S.; Diner B. A.; Larsen B. S.; Gilchrist M. L.; Lorigan G. A.; Britt R. D. Identification of Histidine at the Catalytic Site of the Photosynthetic Oxygen-Evolving Complex. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimahara H.; Yoshida T.; Shibata Y.; Shimizu M.; Kyogoku Y.; Sakiyama F.; Nakazawa T.; Tate S.; Ohki S.; Kato T.; Moriyama H.; Kishida K.; Tano Y.; Ohkubo T.; Kobayashi Y. Tautomerism of Histidine 64 Associated with Proton Transfer in Catalysis of Carbonic Anhydrase. J. Biol. Chem. 2007, 282, 9646–9656. [DOI] [PubMed] [Google Scholar]
- Wang C.; Lamb R. A.; Pinto L. H. Activation of the M2 Ion Channel of Influenza Virus: A Role for the Transmembrane Domain Histidine Residue. Biophys. J. 1995, 69, 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu C. K.; Silverman D. N.; Forsman C.; Jonsson B. H.; Lindskog S. Role of Histidine-64 in the Catalytic Mechanism of Human Carbonic Anhydrase-Ii Studied with a Site-Specific Mutant. Biochemistry 1989, 28, 7913–7918. [DOI] [PubMed] [Google Scholar]
- Hong M.; DeGrado W. F. Structural Basis for Proton Conduction and Inhibition by the Influenza M2 Protein. Protein Sci. 2012, 21, 1620–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F. H.; Luo W. B.; Hong M. Mechanisms of Proton Conduction and Gating in Influenza M2 Proton Channels from Solid-State NMR. Science 2010, 330, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Fu R.; Nishimura K.; Zhang L.; Zhou H. X.; Busath D. D.; Vijayvergiya V.; Cross T. A. Histidines, Heart of the Hydrogen Ion Channel from Influenza A Virus: Toward an Understanding of Conductance and Proton Selectivity. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6865–6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L. H.; Lamb R. A. The M2 Proton Channels of Influenza A and B Viruses. J. Biol. Chem. 2006, 281, 8997–9000. [DOI] [PubMed] [Google Scholar]
- Tu C.; Silverman D. N.; Forsman C.; Jonsson B. H.; Lindskog S. Role of Histidine 64 in the Catalytic Mechanism of Human Carbonic Anhydrase II Studied with a Site-Specific Mutant. Biochemistry 1989, 28, 7913–7918. [DOI] [PubMed] [Google Scholar]
- Vila J. A.; Arnautova Y. A.; Vorobjev Y.; Scheraga H. A. Assessing the Fractions of Tautomeric Forms of the Imidazole Ring of Histidine in Proteins as a Function of pH. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5602–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass M. A. S.; Hansen D. F.; Christensen H. E. M.; Led J. J.; Kay L. E. Characterization of Conformational Exchange of a Histidine Side Chain: Protonation, Rotamerization, and Tautomerization of His61 in Plastocyanin from Anabaena Variabilis. J. Am. Chem. Soc. 2008, 130, 8460–8470. [DOI] [PubMed] [Google Scholar]
- Shimba N.; Takahashi H.; Sakakura M.; Fujii I.; Shimada I. Determination of Protonation and Deprotonation Forms and Tautomeric States of Histidine Residues in Large Proteins using Nitrogen-Carbon J Couplings in Imidazole Ring. J. Am. Chem. Soc. 1998, 120, 10988–10989. [Google Scholar]
- Hass M. A. S.; Yilmaz A.; Christensen H. E. M.; Led J. J. Histidine Side-Chain Dynamics and Protonation Monitored by C-13 CPMG NMR Relaxation Dispersion. J. Biomol. NMR 2009, 44, 225–233. [DOI] [PubMed] [Google Scholar]
- Harbison G.; Herzfeld J.; Griffin R. G. N-15 Chemical-Shift Tensors in L-Histidine Hydrochlorid Monohydrate. J. Am. Chem. Soc. 1981, 103, 4752–4754. [Google Scholar]
- Pelton J. G.; Torchia D. A.; Meadow N. D.; Roseman S. Tautomeric States of the Active-Site Histidines of Phosphorylated and Unphosphorylated III(GLC), a Signal-Transducing Protein from Escherichia-Coli, using Two-Dimentional Heteronuclear NMR Techniques. Protein Sci. 1993, 2, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F.; Sun H.; Zhang Y.; Mukkamala D.; Oldfield E. A Solid State 13C NMR, Crystallographic, and Quantum Chemical Investigation of Chemical Shifts and Hydrogen Bonding in Histidine Dipeptides. J. Am. Chem. Soc. 2005, 127, 12544–12554. [DOI] [PubMed] [Google Scholar]
- Nydegger M. W.; Dutta S.; Cheatum C. M. Two-Dimensional Infrared Study of 3-Azidopyridine as a Potential Spectroscopic Reporter of Protonation State. J. Chem. Phys. 2010, 133, 134506. [DOI] [PubMed] [Google Scholar]
- Roberts S. T.; Loparo J. J.; Tokmakoff A. Characterization of Spectral Diffusion from Two-Dimensional Line Shapes. J. Chem. Phys. 2006, 125, 084502. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Hochstrasser R. M. A Peptide’s Perspective of Water Dynamics. Chem. Phys. 2011, 390, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remorino A.; Korendovych I. V.; Wu Y. B.; DeGrado W. F.; Hochstrasser R. M. Residue-Specific Vibrational Echoes Yield 3D Structures of a Transmembrane Helix Dimer. Science 2011, 332, 1206–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C. S.; Tokmakoff A. Identification of Lactam-Lactim Tautomers of Aromatic Heterocycles in Aqueous Solution Using 2D IR Spectroscopy. J. Phys. Chem. Lett. 2012, 3, 3302–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth A.; Zscherp C. What Vibrations Tell Us About Proteins. Q. Rev. Biophys. 2002, 35, 369–430. [DOI] [PubMed] [Google Scholar]
- Tucker M. J.; Gai X. S.; Fenlon E. E.; Brewer S. H.; Hochstrasser R. M. 2D IR Photon Echo of Azido-Probes for Biomolecular Dynamics. Phys. Chem. Chem. Phys. 2011, 13, 2237–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M. J.; Getahun Z.; Nanda V.; DeGrado W. F.; Gai F. A New Method for Determining the Local Environment and Orientation of Individual Side Chains of Membrane-Binding Peptides. J. Am. Chem. Soc. 2004, 126, 5078–5079. [DOI] [PubMed] [Google Scholar]
- Hoffman K. W.; Romei M. G.; Londergan C. H. A New Raman Spectroscopic Probe of Both the Protonation State and Noncovalent Interactions of Histidine Residues. J. Phys. Chem. A 2013, 117, 5987–5996. [DOI] [PubMed] [Google Scholar]
- Romei M. G.; Hoffman K. W.; Londergan C. H. 2-Deuterated Histidine is a Raman Reporter of Histidine’s Protonation State, Hydrogen Bonding, and Metal Coordination. Biophys. J. 2013, 104, 685a–685a. [Google Scholar]
- Kim Y. S.; Hochstrasser R. M. Applications of 2D IR Spectroscopy to Peptides, Proteins, and Hydrogen-Bond Dynamics. J. Phys. Chem. B 2009, 113, 8231–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganim Z.; Chung H. S.; Smith A. W.; Deflores L. P.; Jones K. C.; Tokmakoff A. Amide I Two-Dimensional Infrared Spectroscopy of Proteins. Acc. Chem. Res. 2008, 41, 432–441. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Kwak K.; Fayer M. D. Ultrafast 2D IR Vibrational Echo Spectroscopy. Acc. Chem. Res. 2007, 40, 75–83. [DOI] [PubMed] [Google Scholar]
- Bagchi S.; Charnley A. K.; Smith A. B.; Hochstrasser R. M. Equilibrium Exchange Processes of the Aqueous Tryptophan Dipeptide. J. Phys. Chem. B 2009, 113, 8412–8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A.; Qiu J.; DeGrado W. F.; Hochstrasser R. M. Tidal Surge in the M2 Proton Channel, Sensed by 2D IR Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6115–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C.; Bauman J. D.; Das K.; Remorino A.; Arnold E.; Hochstrasser R. M. Two-Dimensional Infrared Spectra Reveal Relaxation of the Nonnucleoside Inhibitor TMC278 Complexed with HIV-1 Reverse Transcriptase. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Middleton C. T.; Singh S.; Reddy A. S.; Woys A. M.; Strasfeld D. B.; Marek P.; Raleigh D. P.; de Pablo J. J.; Zanni M. T.; Skinner J. L. 2DIR Spectroscopy of Human Amylin Fibrils Reflects Stable Beta-Sheet Structure. J. Am. Chem. Soc. 2011, 133, 16062–16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton C. T.; Marek P.; Cao P.; Chiu C. C.; Singh S.; Woys A. M.; de Pablo J. J.; Raleigh D. P.; Zanni M. T. Two-Dimensional Infrared Spectroscopy Reveals the Complex Behaviour of an Amyloid Fibril Inhibitor. Nat. Chem. 2012, 4, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. S.; Liu L.; Axelsen P. H.; Hochstrasser R. M. 2D IR Provides Evidence for Mobile Water Molecules in Beta-Amyloid Fibrils. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17751–17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marecek J.; Song B.; Brewer S.; Belyea J.; Dyer R. B.; Raleigh D. P. A Simple and Economical Method for the Production of 13C,18O-Labeled Fmoc-Amino Acids with High Levels of Enrichment: Applications to Isotope-Edited IR Studies of Proteins. Org. Lett. 2007, 9, 4935–4937. [DOI] [PubMed] [Google Scholar]
- Reppert M.; Tokmakoff A. Electrostatic Frequency Shifts in Amide I Vibrational Spectra: Direct Parameterization Against Experiment. J. Chem. Phys. 2013, 138, 134116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. S.; Hochstrasser R. M. Dynamics of Amide-I Modes of the Alanine Dipeptide in D2O. J. Phys. Chem. B 2005, 109, 6884–6891. [DOI] [PubMed] [Google Scholar]
- Falvo C.; Zhuang W.; Kim Y. S.; Axelsen P. H.; Hochstrasser R. M.; Mukamel S. Frequency Distribution of the Amide-I Vibration Sorted by Residues in Amyloid Fibrils Revealed by 2D-IR Measurements and Simulations. J. Phys. Chem. B 2012, 116, 3322–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J. R.; Kwak K.; Asbury J.; Chen X.; Piletic I. R.; Fayer M. D. Ultrafast Dynamics of Solute-Solvent Complexation Observed at Thermal Equilibrium in Real Time. Science 2005, 309, 1338–1343. [DOI] [PubMed] [Google Scholar]
- Kwak K.; Zheng J. R.; Cang H.; Fayer M. D. Ultrafast Two-Dimensional Infrared Vibrational Echo Chemical Exchange Experiments and Theory. J. Phys. Chem. B 2006, 110, 19998–20013. [DOI] [PubMed] [Google Scholar]
- Kim Y. S.; Hochstrasser R. M. Chemical Exchange 2D IR of Hydrogen-Bond Making and Breaking. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 11185–11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woutersen S.; Mu Y.; Stock G.; Hamm P. Hydrogen-Bond Lifetime Measured by Time-Resolved 2D-IR Spectroscopy: N-Methylacetamide in Methanol. Chem. Phys. 2001, 266, 137–147. [Google Scholar]
- Pazos I. M.; Gai F. Solute’s Perspective on How Trimethylamine Oxide, Urea, and Guanidine Hydrochloride Affect Water’s Hydrogen Bonding Ability. J. Phys. Chem. B 2012, 116, 12473–12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.