

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) is a major cause of nosocomial infection. Whole-genome sequencing of MRSA has been used to define phylogeny and transmission in well-resourced healthcare settings, yet the greatest burden of nosocomial infection occurs in resource-restricted settings where barriers to transmission are lower. Here, we study the flux and genetic diversity of MRSA on ward and individual patient levels in a hospital where transmission was common. We repeatedly screened all patients on two intensive care units for MRSA carriage over a 3-mo period. All MRSA belonged to multilocus sequence type 239 (ST 239). We defined the population structure and charted the spread of MRSA by sequencing 79 isolates from 46 patients and five members of staff, including the first MRSA-positive screen isolates and up to two repeat isolates where available. Phylogenetic analysis identified a flux of distinct ST 239 clades over time in each intensive care unit. In total, five main clades were identified, which varied in the carriage of plasmids encoding antiseptic and antimicrobial resistance determinants. Sequence data confirmed intra- and interwards transmission events and identified individual patients who were colonized by more than one clade. One patient on each unit was the source of numerous transmission events, and deep sampling of one of these cases demonstrated colonization with a “cloud” of related MRSA variants. The application of whole-genome sequencing and analysis provides novel insights into the transmission of MRSA in under-resourced healthcare settings and has relevance to wider global health.
Numerous organizations have recently expressed concern over the rising rates of antimicrobial resistance among human pathogens at a time when the development of new antimicrobial agents has stalled (UK Department of Health 2013; US Department of Health and Human Services, Centers for Disease Control and Prevention 2013; World Health Organization 2014). The problem has been recognized as a global one, since the localized emergence of a clinically significant resistance determinant is almost inevitably followed by a pattern of widespread dissemination (Johnson and Woodford 2013). Antimicrobial resistance in resource-poor settings is fueled by weak control of antibiotic prescribing and stewardship both in the community and hospitals, and by weak infection control infrastructure that fails to prevent the transmission of nosocomial pathogens—many of which are multidrug resistant.
Methicillin-resistant Staphylococcus aureus (MRSA) is a leading nosocomial pathogen worldwide (Nickerson et al. 2009a, b; Falagas et al. 2013). MRSA carriage in the community remains low in many countries, and healthcare-associated infection is often associated with acquisition within a healthcare setting. Rates of infection can be reduced dramatically by stringent infection control measures that prevent transmission of MRSA from one patient to another, as reflected by > 80% reduction in MRSA bacteremia rates observed in England since 2001. In high-income countries, an important component of MRSA control in hospitals is surveillance for suspected transmission events, followed by rapid interventions to prevent further transmission from occurring. Such events are investigated using epidemiological methods; low resolution bacterial genotyping has a very limited role because healthcare-associated MRSA (HA-MRSA) belong to a restricted number of successful lineages worldwide (Enright et al. 2002), so that in a given setting most HA-MRSA isolates will have the same genotype and appear identical (Ellington et al. 2010). In contrast, several studies have reported that whole-genome sequencing (WGS) provides sufficient discrimination between isolates of the same MRSA clone to be able to include or exclude cases in a suspected outbreak and to infer likely transmission pathways between staff, patients and their relatives (Eyre et al. 2012; Köser et al. 2012; Harris et al. 2013).
Previous studies have used WGS in settings with extensive infection control practices that restrict MRSA transmission and subsequent bloodstream infection. Yet the overwhelming burden of MRSA carriage and infection occurs in low- and middle-income countries, where infection control infrastructure and practice is frequently suboptimal. Almost nothing is known about S. aureus dynamics in developing country settings and to our knowledge, no large-scale prospective carriage studies have been carried out previously. The objective of this study was to use WGS to define phylogeny and transmission dynamics of MRSA in a hospital setting where MRSA transmission was common. Targeted research has the potential to provide accurate insights into transmission dynamics, which could be used to inform low-cost prevention planning and implementation.
Results
Study setting, patients, and bacterial isolates
We utilized a collection of MRSA isolated during a prospective carriage study conducted over a 3-mo period in 2008 on two intensive care units (ICUs; a general pediatric ICU and an adult surgical ICU) in a 1000-bed hospital in northeast Thailand. The dominant MRSA clone in this setting is ST 239, a highly successful global lineage for which the population structure and patterns of intercontinental spread are already described (Harris et al. 2010). Ethical approval was obtained from the Ethical and Scientific Review subcommittee of the Royal Thai Government Ministry of Public Health, and the Oxford Tropical Research Ethics Committee. All patients admitted to the two ICUs were eligible for inclusion and were enrolled after written informed consent was obtained. A multisite MRSA screen was performed on the day of admission and then twice weekly until discharge from the unit throughout the 3-mo period. Nasal swabs and fingertip cultures were taken from ICU nursing staff on three spaced time points. Further methodological details are provided in the Supplemental Text. We recruited 169 adults and 98 children on the adult and pediatric ICU, respectively. MRSA was isolated from one or more screens from 27 adults (16%), and 33 children (34%). MRSA acquisition occurred outside the ICU in approximately half the carriers on the basis that they were already MRSA positive on their first screen, and MRSA was assumed to have been acquired within the ICU in the remainder who had an initial negative MRSA screen followed at a later time point by a positive one. Staff screening demonstrated MRSA carriage by one of 22 nurses (5%) on the adult ICU and four of 15 nurses (27%) on the pediatric ICU. With one exception, each staff member was positive for MRSA on only one occasion.
Existence of numerous circulating clades of MRSA ST 239
We performed WGS on the first MRSA isolate and up to two repeat MRSA isolates for each case (a total of 76 isolates). The ST was defined using sequence data as ST 239 for all 76 isolates. We initially evaluated the sequence of the first MRSA isolate from 46 patients and five staff. Sequence reads were aligned to a reference ST 239 genome (TW20) to identify single-nucleotide polymorphisms (SNPs) in the core genome. This identified a total of 1535 SNPs for the 51 isolates, indicating considerable diversity within the lineage. We examined their recent evolution by constructing a phylogenetic tree based on core SNPs into which we included a further 20 MRSA isolates recovered from patients in the same hospital between October 2006 and April 2007 that had been sequenced previously during a study of global ST 239 and assigned to the Asian clade (Harris et al. 2010). This demonstrated that all 51 isolates belonged to the Asian clade (Supplemental Fig. S1). A striking feature of the tree was the presence of multiple distinct clusters, suggesting that endemic carriage in a single hospital was associated with the circulation of multiple clades of the same lineage.
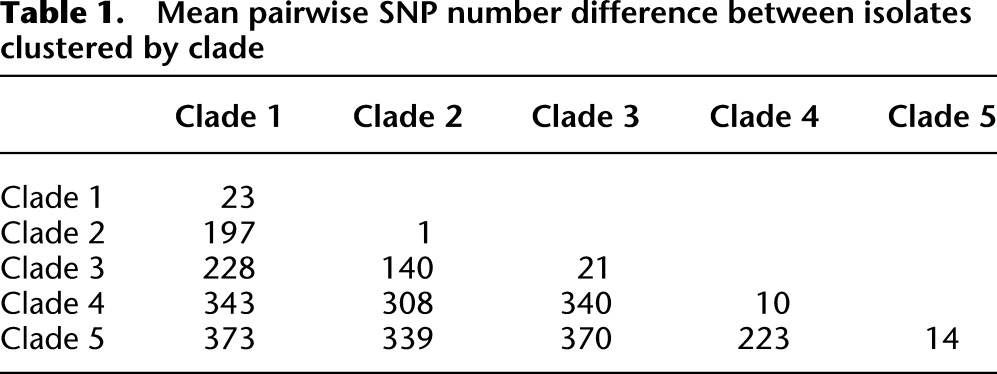
Next, we constructed a phylogenetic tree based on core genome SNPs using data for all 76 MRSA isolates from the 46 patients and five staff (Fig. 1). As before, several distinct clusters were observed. From the SNP alignments, pairwise SNP differences (Hamming distance) were calculated and plotted as a histogram (Fig. 2). This revealed a distribution with clustering for a cutoff of 60 core genome SNPs. We then defined clades using an iterative procedure that assigned each sequence to a particular clade if the Hamming distance to at least one other sequence within that clade was < 60 bp. This identified five main clades comprising 67/76 isolates (88%), which was consistent with the distinct clusters observed on the tree (labeled clades 1–5) (Fig. 1). The mean SNP number difference in pairwise comparisons ranged from 1–23 SNPs within each clade, and from 140–373 SNPs between clades (Table 1). These clades largely segregated by ward, with clades 2, 4, and 5 principally found on the adult ICU and clades 1 and 3 principally on the pediatric ICU.
Figure 1.

Existence of circulating clades of MRSA ST 239. Maximum likelihood phylogenetic tree based on core genome SNPs of the first and up to two repeat isolates from 46 patients and five healthcare workers. The first T number assigned to each represents the case study number. The second letter/number combination refers to the type of sample and the sample number from that particular site: (N) nasal swab; (T) throat swab; (A) axillae swab; (W) wound swab; (U) urine; (C) tracheal suction. For example, T178-N3 refers to the third nasal swab taken from case T178. Isolate labels are color-coded to signify isolation from a staff member (green), a patient on the adult ICU (red), or pediatric ICU (blue). Also shown (right-hand panel) is the presence (bold) or absence (pale) of four plasmids harboring antibiotic and antiseptic resistance genes. The colors used to denote clades 1 to 5 are cross-referenced in Figure 3.
Figure 2.

Histogram of pairwise SNP differences between 76 isolates from 46 patients and five healthcare workers. SNPs differences between isolates from the same clade are represented in red, and between isolates in different clades in blue.
Table 1.
Mean pairwise SNP number difference between isolates clustered by clade
WGS of MRSA isolated from nursing staff demonstrated that three of the four staff on the pediatric ICU (T40, T56, and T241) were colonized with isolates that clustered with one of the major lineages. This is consistent with transmission of MRSA between staff and patients on the pediatric ICU. In contrast, the MRSA isolated from the two remaining staff carriers (T59 and T353, one from each ICU) resided together in a discrete cluster (Fig. 1), suggesting that acquisition may have occurred elsewhere. This is supported by the observation that their isolates clustered with MRSA associated with healthcare-associated bacteremia drawn from the entire hospital population between 2006 and 2007 (Supplemental Fig. S1).
Genetic diversity of MRSA ST 239 involving clinically relevant determinants
Having identified that there were several circulating clades based on variation in the core genome, we also examined variation in the accessory genomes of the ST 239 isolates. Previous studies have shown that mobile genetic elements are one of the most plastic components of the S. aureus genome. Of note for its clinical relevance was variability in the presence of plasmids harboring antibiotic and antiseptic resistance genes (Fig. 1). Plasmid pTW20_1 (29,584 bp, harboring qacA encoding antiseptic resistance and often a mer operon encoding mercury resistance) was present in clades 1, 2, and 3, but absent in clades 4 and 5. The clade 1 variant of pTW20_1 lacked the mer operon, and clade 1 also had a deletion in the staphylococcal cassette chromosome mercury element (SCCmer), and hence these isolates lacked the mer operon. One or two copies of a pPR9-like plasmid harboring aadD (encoding aminoglycoside resistance) and ileS-2 (encoding high-level resistance to mupirocin) were present in a sublineage of clade 3. pKH4-like and pAY121857-like are small plasmids of ∼2400 bp that harbor a qacJ-like and qacC gene, respectively. pKH4-like was found in most clade 4 and 5 isolates, and pAY121857-like was in all clade 2 isolates. Ninety-two percent of patient isolates (defined for 50 isolates [46 first MRSA isolates and 4 new acquisition isolates]) carried at least one qac gene. The proportion of isolates with at least one qac gene was not significantly different for the isolates acquired after entering the ICU compared with those acquired beforehand (P = 0.43, Fisher’s exact test).
We also identified additional genetic variation in the accessory genome in the form of gene deletion, which was present both within and between the five clades (Supplemental Fig. S2). Compared to the reference TW20 genome, there were clade-specific large-scale deletions of up to 127 kb in prophage and the SCCmer regions, but no equivalent large-scale insertions were observed. Clade 5 contained deletion of prophage ϕSa3 that carries the staphylococcal complement inhibitor SCIN, staphylokinase, and enterotoxin A genes. Clade 2 had a partial deletion of the prophage ϕSPβ-like region, but retained the sasX gene that has been shown to be an important factor for colonization and pathogenicity (Li et al. 2012), as well as several genes related to antibiotic resistance. In contrast, the ϕSPβ-like region was entirely missing from three related isolates from healthcare worker T59.
Dynamic temporal flux of MRSA ST 239 clades
We then investigated whether WGS provided more insight into transmission dynamics than standard epidemiological investigation based on identifying outbreaks as temporal clusters. We define a transmission event to have occurred when a patient had one or more negative MRSA screening results following admission to the ICU followed by a first positive swab at least 48 h after admission, and acquired MRSA belonging to the same clade was defined as one carried by at least one other patient on that ICU during the 7 d prior to the date of the first positive swab. In the absence of WGS, the cases within wards did not appear to cluster in time (nonparametric ANOVA for explaining time of sample collection for each independent carriage episode by patient ward, P = 0.90, R2 = 0.02). In contrast, we found a significant clustering in time and within wards of the clades defined by WGS (nonparametric ANOVA for explaining time of sample collection by patient ward and sequence cluster, P = 0.006, R2 = 0.31). This result was not present when only first sequences from patients positive on admission were included (P = 0.34, R2 = 0.09), which demonstrates that the clustering was due to transmission in the wards. Thus, clades were not randomly represented over time.
The clade structure on each ICU was plotted for each day of the study. On the pediatric ICU, clades 1 (color-coded blue) and 3 (pink) cocirculated up to day 40, with clade 3 becoming predominant during days 41–94 (Fig. 3). On the adult ICU, clades 1 (blue), 2 (purple) and 4 (green) predominated up to study day 40, but were subsequently almost entirely replaced by clade 5 isolates (khaki) (Fig. 3). This indicates a dynamic temporal flux in both wards, with clustering evident in both time and space.
Figure 3.
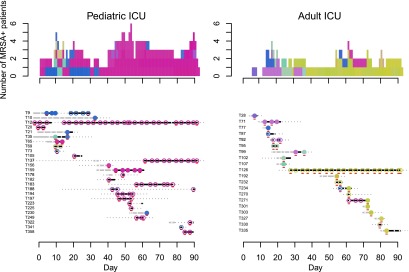
Dynamics of MRSA clades on the pediatric (left) and adult (right) ICUs. (Bottom) Each row provides details of a study patient (T number). Duration of admission to the hospital is shown in days, with a solid line for period(s) in the ICU ([gray] MRSA negative; [black] MRSA positive), and a dotted line for inpatient periods outside the study ICUs (MRSA status not tested). Superimposed on the lines are dots and circles ([white dot] MRSA negative; [colored circles] MRSA positive). An open colored circle signifies that the MRSA isolate was cultured from the first swab taken following ICU admission and was most likely acquired elsewhere in the hospital. A closed colored circle signifies that the MRSA isolate was cultured from a patient who had an initial negative MRSA screen in the ICU followed at a later time point by a positive one, which was interpreted as MRSA acquisition in the ICU. Colors were assigned based on the most recently sequenced isolate from the same patient and chosen to reflect sequence diversity. Each sequence was mapped to a unique color so that similar sequences have similar colors (see Methods). Representatives of the five clade colors used in Figure 1 are T35_N4 (clade 1, blue); T71_N3 (clade 2, purple); T106_N3 (clade 3, pink); T95_N1 (clade 4, green); and T126_N13 (clade 5, khaki). Isolates that were sequenced are underlined in red. (Top) The daily cumulative count of MRSA positive patients over the 3-mo study period. The colors used match the colored dots for MRSA isolates.
We sequenced more than one isolate from 21 patients, which showed that the MRSA clade changed in four patients, with a tendency for a switch to a predominant clade. For example, two patients (T234 and T271) on the adult ICU were initially colonized with nonclade 5 isolates but subsequently acquired a clade 5 isolate. Similarly, on the pediatric ICU, patient T35 was initially colonized with a clade 4 isolate but became colonized 7 d later with an isolate belonging to the circulating clade 1. Although this confirms acquisition of a new clade, this may also represent the development of carriage with more than one clade.
A notable feature of both ICUs was the presence of long-stay patients who were continuously MRSA positive and maintained the same clades (for example, clade 3 carried by patient T12 and clade 5 carried by patient T126). In both cases, these patients were the first detected carriers of these clades on these wards, and both of the clades became dominant over the period that these patients were on the ward. This suggests a process of introduction and sequential transmission events involving other patients following their admission.
Within-host genetic diversity of MRSA ST 239
Next, we undertook deep sequencing of ST 239 from a single patient to observe changes at a single time point, and over time. During the selection process of this case, we sought patients admitted for the longest period of time and with the longest period of MRSA carriage. The two possible choices were patient T12 on the pediatric ward and patient T126 on the adult ward, from which T126 was selected because more putative transmission events were noted to other patients on this ICU over time (based on new acquisition of ST 239). A total of 99 single colonies isolated over the 64 d of ICU admission were sequenced from T126, comprised of one primary plate colony taken from each of 19 screening samples taken at regular intervals throughout the 64 d stay, plus 20 primary plate colonies from each of four samples—the first and last sample and two equally spaced in time between these. We found that all 99 isolates clustered within clade 5, but that there was a cloud of diversity with 147 variable SNP sites and a mean pairwise SNP difference of 6.7 SNPs. This diversity was apparent at each of the four main sampling time points but with a subtle shift in where on the tree most variants resided at each time point (Supplemental Fig. S3). The patient was MRSA positive on the first screen, indicating that acquisition had occurred outside the ICU. Using linear regression of root-to-tip distances against the date of sampling, the most recent common ancestor of the T126 cloud was calculated to exist ∼55 d prior to the admission of patient T126 to the ICU (and 25 d prior to the commencement of the study), with a SNP accumulation rate of 9.1 × 10−6 (95% CI 5.8, 12.5 × 10−6) per site per year (Fig. 4).
Figure 4.

Linear regression plot of root-to-tip distances of the phylogenetic tree for MRSA isolates from patient T126 against the day of sampling. There were 99 isolates from patient T126 and seven isolates concurrently isolated from other patients on the adult ICU that belong to the same clade. The coefficient of determination was R2 = 0.22. Patient T126 was admitted to the ICU on day 29 of the study.
Patient T126 appeared to introduce clade 5 into the adult ICU where it predominated to the end of the study period. Comparison of variants from patient T126 with clade 5 isolates from other patients (all recovered from patients contemporaneously residing with patient T126 in the adult ICU) provided evidence that T126 acted as the donor for MRSA transmission to several other patients (Supplemental Fig. S4), who were colonized with isolates derived from variants within the T126 cloud. The tree did not support the hypothesis that newly colonized MRSA patients were the donors for new cases in a sequential transmission pathway. This is consistent with the observation that the inter-patient diversity was similar to the intra-patient diversity of patient T126, i.e., other patients were nested within the diversity cloud. The exception was clade 5 MRSA isolates from two patients (T232 and T270), which resided on a branch that was basal (ancestral) to the T126 cloud. These patients were positive on their first screen on admission to the ICU, consistent with MRSA acquisition outside of the ICU. The remaining patients carrying clade 5 isolates screened positive at least 72 h after ICU admission and had prior swabs, either negative for MRSA or positive with a different clade (patients T234 and T271), suggesting a new clade 5 acquisition. Deep sampling of patient T126 failed to demonstrate the presence of non-clade 5 isolates.
The observation that variants from patient T126 were readily transmitted to other patients and replaced other lineages in donor patients suggests that the T126 variants may have had a fitness advantage. We considered the possible basis for this by comparing the genome sequence of the T126 cloud with the three ancestral isolates (from patients T232 and T270). The core genome of the ancestral isolates were a mean of only 15 SNPs different from any variant in the T126 cloud, but in their accessory (noncore) genomes all three lacked a qac gene encoding resistance to quaternary ammonium compound antiseptics. In contrast, all but one of the variants from the T126 cloud possessed qacJ carried on a 2.4-kb pKH4-like plasmid. The importance of resistance to quaternary ammonium compounds with regard to fitness and transmission in this ICU setting remains to be clarified, but our observation suggests this warrants further exploration.
Discussion
The population structure of MRSA in this single hospital is remarkably diverse. Although the isolates all belonged to a single ST, and therefore would be indistinguishable by a routinely used molecular typing technique, they exhibit considerable diversity at the genomic level. Previously, it has been noted that the ST 239 isolates from this hospital contain more diversity than an entire South America clade (Harris et al. 2010). It is likely therefore that the ST 239 lineage studied here in a single facility in northeast Thailand has been established in this region for a considerable length of time. Comparison of the ST 239 population collected during the prospective cohort study with that from the same hospital collected 12 mo previously shows there is very little overlap, which suggests that there is not a single stably maintained clone of ST 239 within this hospital. At any one time there were multiple clades circulating in the hospital, which are likely to have resulted from introductions from the wider ST 239 population and subsequent local transmission within the hospital.
We showed that the MRSA population of this hospital is under flux, with continuous cycles of introduction, transmission, and extinction. A corollary of this is that variants of ST 239 carrying different complements of mobile genetic elements are repeatedly introduced. Our analysis shows that plasmids are one of the most fluid components of ST 239 genomes. This is of particular clinical significance since these elements often encode resistance to antimicrobials. In particular, we found variation in genes encoding resistance to quaternary ammonium compounds. The high rate of qac gene carriage in ST 239 associated with an MRSA outbreak has been noted previously in an ICU in the United Kingdom, where the initiation of daily chlorhexidine bathing for all patients was associated with greatly reduced transmission of endemic UK clones but no reduction in the transmission of ST 239 (Batra et al. 2010; Cooper et al. 2012). Recent studies have determined that carriage of plasmid borne qac genes can confer at least tolerance, and possibly resistance, to widely used antiseptics such as chlorhexidine gluconate, and may provide a selective advantage in the nosocomial environment (Smith et al. 2008; Batra et al. 2010; Lee et al. 2011; Otter et al. 2013). Where there are no broadly available means of phenotypic identification of resistance, such as with quaternary ammonium compounds, WGS allows for the detection of genetic elements associated with resistance. In addition, molecular assays for qac genes only detect certain qac variants and lack sensitivity in detection compared with WGS. Variability was also noted in our study in the presence of genes encoding resistance to aminoglycosides and mupirocin. These features may reflect the capability of this nosocomial pathogen to undergo adaptation in response to environmental and therapeutic cues.
This study has demonstrated that WGS resolves transmission clusters in a way that is substantially more powerful than epidemiological information alone. We observed a dynamic flux of distinct MRSA ST 239 clades in two ICUs over time, each of which had its own distinctive clade pattern. In both ICUs, an individual was admitted after the study had commenced who was already carrying MRSA and who stayed until the study ceased. It was notable that the clade carried by each of these patients went on to become the predominant carriage clade within their respective ICU. Our findings indicate multiple new transmission events, a high risk of MRSA acquisition in these units, and the impact that even a single MRSA carrier can have on a hospital ward. The rate of transmission events is likely to be an underestimate, since we did not perform repeated sequencing of MRSA throughout the admission time course for each patient. Infection control in the study hospital does not mirror that in highly resourced health care settings. Side rooms are not available in either ICU, and gloves and aprons are not routinely used when nursing MRSA positive patients. Thus, the findings are in contrast to those of a study conducted in an ICU in the UK, where acquisitions of MRSA through patient-to-patient transmission were rare (Price et al. 2014).
The deep sequencing of isolates from a single individual (T126) over time confirmed the presence of a genetic cloud of diversity. We note that the methodological approach taken only provides a limited snapshot of the bacterial diversity across multiple hosts; therefore, it does not capture the full diversity of the population. However, comparison of the extent of the cloud with other isolates from patients on the same ward at the same time revealed an overlap, suggesting a common origin for these isolates and that extensive transmission had occurred. This observation has important implications for the application of WGS as a tool to identify transmission events in a clinical setting. Previous studies have largely been based on sequencing of a single bacterial colony from each human host. This assumes that there is limited within-host diversity of the MRSA clone and that a single colony is sufficiently representative. However, as with viral infections, where a mixed population of a single lineage can be present within a host (Ojosnegros et al. 2011), there is growing evidence that the same is true for bacteria. Harris et al. (2013) sequenced 20 individual colonies of an MRSA clone isolated from the nose of a healthcare worker during a suspected outbreak, which demonstrated a cloud of genetic diversity. This phenomenon is not confined to MRSA; for example, Snitkin et al. (2012) reported the isolation of genetic variants of the same KPC (Klebsiella pneumoniae carbapenemase)-harboring K. pneumoniae clone at different time points from a single patient and used this information to infer three separate transmission events from this index patient. Within-host bacterial diversity could confound analyses of between-host transmission and indicates a need to investigate intra-host and inter-host diversity in the same patient population. An alternative means of investigating within host genetic heterogeneity of microorganisms is a metagenomic approach (Loman et al. 2013; Schloissnig et al. 2013). Future studies could incorporate a comparison of sequencing multiple isolates recovered from a sample with sequencing of all microbial DNA from a sample. Either way, as evidenced by this study and other simulated work (Worby et al. 2014), gaining an accurate understanding of transmission networks will require measures of within-host bacterial diversity as well as traditional “shoe-leather” epidemiological data.
In the case of the individual targeted in our study, T126, they were MRSA positive on admission to the ICU, and therefore had possibly been colonized with ST 239 for a period of time prior to this. From the genome data, the estimate for the most recent common ancestor of the cloud was ∼55 d prior to the admission of the patient to the ICU. Notably, the SNP accumulation rate of 9.1 × 10−6 per site per year calculated for the T126 cloud is slightly higher than that estimated previously (Harris et al. 2010) for the broader ST 239 population of 3.3 × 10−6 but would be consistent with a very recent clonal expansion, where purifying selection has not had a chance to act since diversification (Rocha et al. 2006). In light of the limitations of infection control, we predict that a combination of a diverse founder population and repeated transmission account for most of the diversity, rather than a single acquisition by patient T126. Furthermore, the phylogenetic clade structure described here provides a framework for ruling out a transmission event between two patients in the event that they are carrying an isolate belonging to a different clade. Ruling a direct transmission event in or out when two patients are carrying an isolate belonging to the same clade requires epidemiological information on other patients who are positive for that clade and their movement pattern and opportunity for contact.
Previous microbial sequence-based studies have been undertaken in settings where strenuous efforts have been made to optimize infection control (typically with hand hygiene in excess of 50%), although the importance of low resource countries for the emergence and global spread of multidrug-resistant bacteria is now being recognized. In addition, most of the previous studies using whole-genome sequencing to explore S. aureus dynamics have been in the context of known outbreaks; therefore, they do not provide a valid basis for generalizing about more typical transmission dynamics. In contrast, this study was a prospective study with a prespecified protocol designed to investigate the typical pattern of MRSA spread. A key strength of this study is that it delves into transmission patterns in a setting where infection control is very underdeveloped. What to expect was far from obvious given the very large differences in demographics, crowding, patterns of antibiotic use, and environmental factors compared to high income settings. In this hospital, despite attempts by the infection control team to promote hand hygiene, compliance has remained <10%.
As demonstrated here, WGS can be applied to understand the transmission dynamics of nosocomial pathogens, and illustrate how their dynamics differ compared to that seen in high resource settings. Because of resource constraints, it remains the case that no routine MRSA screening is performed at the study hospital; our findings are likely to reflect the ongoing real-world patterns of transmission both here and in other hospitals with similar infection control practices. The degree of transmission demonstrated has directly led to prioritization of improved hand hygiene practices at the hospital and to the development of an ongoing study to evaluate a hand hygiene intervention in this setting.
Methods
Sequencing and bioinformatics analyses
Isolates from 46 patients (20 adults, 26 children) and five staff members (51 individuals in total) remained available for use in this study. Unique index-tagged libraries were created for each isolate, and multiplexed libraries were sequenced on either the Illumina Genome Analyzer GAII with 108 basepair (bp) paired-end reads or Illumina HiSeq with 75 bp paired-end reads. To detect SNPs in the core genome, paired-end reads were mapped using SMALT v 0.5.8 (http://www.sanger.ac.uk/resources/software/smalt/) against the chromosome of the reference Staphylococcus aureus ST 239 strain TW20 (accession number FN433596) (Holden et al. 2010). Reads were only mapped if they matched the reference with an identity > 90%. Variation statistics at each base were calculated using SAMtools mpileup, followed by BCFtools view from the SAMtools package (Li et al. 2009). All bases were then filtered to remove those with uncertainty in the base call. The BCFtools variant quality score was required to be greater than 50 and mapping quality greater than 30. If not all reads gave the same base call, the allele frequency (as calculated by BCFtools) was required to be either zero for bases called the same as the reference or one for bases called as a SNP. The majority base call was required to be present in at least 80% of reads mapping at the base, and the minimum mapping depth allowed was four reads, at least two of which had to map to each strand. If any of these filters were not met, the base was called as uncertain. SNPs falling within MGEs regions were excluded from the core genome. Phylogenetic reconstruction was carried out using the maximum-likelihood program RAxML v 7.0.43 with a GTR model of evolution (Stamatakis 2006). To investigate the accessory genomes of isolates, detailed comparisons of sequences was conducted on the de novo assemblies using BLASTN (Altschul et al. 1990), facilitated by using the Artemis Comparison Tool (ACT) (Carver et al. 2005). Raw Illumina data were assembled using a de novo genome-assembly program, Velvet v0.7.03 (Zerbino and Birney 2008) to generate multicontig draft genomes for each isolate. For a summary of the de novo assemblies and mapping data, see Supplemental Table S1.
The presence of antibiotic resistance genes residing in MGEs was initially investigated by mapping sequence reads to a pseudomolecule consisting of concatenated antibiotic resistance genes, as previously described (Köser et al. 2012). We noted the presence of genes conferring resistance to antiseptic agents (qac) and mupirocin (ileS-2) and manually searched for these genes in the de novo assemblies. Comparative analysis using BLASTN identified contigs containing these genes, with best hits to four known S. aureus plasmids: pPR9 (accession no. GU237136), pTW20 (accession no. FN433597), qacC containing plasmid (accession no. AY121857), and pKH4 (accession no. U81980). BLASTN comparisons were visualized using ACT to compare to these known plasmid sequences, demonstrating that contigs for isolates in the current study had deletions in pTW20 (nucleotide positions 17853 to 20819, CDS p1180 to p1200) and pPR9 (nucleotide positions 6378 to 12922, CDS p06 to p11 [Tn552]). We then created a concatenated pseudomolecule consisting of mecA (as a control), each of the plasmids (trimmed of the above-mentioned deletions), and mapped reads back against this plasmid pseudomolecule. Large-scale deletions in the genome of each strain were visualized by plotting mapping depth of reads against the reference TW20 genome.
Determination of clades with pairwise SNP comparisons
Pairwise SNP differences (Hamming distance) were calculated using MEGA5 (Tamura et al. 2011) and plotted as a histogram, which revealed a distribution with clustering for a cutoff of 60 core genome SNPs. Clades were defined using an iterative procedure that assigned each sequence to a particular clade if the Hamming distance to at least one other sequence within that clade was < 60 bp. If a sequence was assigned to two or more clades, the clades were merged. If a sequence could not be assigned to any existing clade, it was assigned to a new clade. These SNP distance-based clades demonstrated excellent concordance with the clades seen “by eye” on the phylogenetic tree. The colors assigned to clades in Figures 1 and 2 were obtained by using nonmetric-multidimensional scaling to assign a location in two-dimensional space using the matrix of pairwise Hamming distances between sequences (Sammon 1969). Coordinates were then mapped to a color wheel in the CIELUV color space.
Data access
Genome data from this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/) under study accession number ERP000130.
Supplementary Material
Acknowledgments
We thank Drs. Pramot Srisamang, Weera Manavanakul, and Manas Kanoksil and the staff at Sappasithiprasong Hospital for their support in the bacterial isolate collection. The authors acknowledge financial support from the UKCRC Translational Infection Research (TIR) Initiative and the Medical Research Council (Grant number G1000803), with contributions to the grant from the Biotechnology and Biological Sciences Research Council, the National Institute for Health Research on behalf of the Department of Health, and the Chief Scientist Office of the Scottish Government Health Directorate (to Professor Peacock); from Wellcome Trust grant number 098051 awarded to the Wellcome Trust Sanger Institute; and the NIHR Cambridge Biomedical Research Centre (to Professor Peacock). S.Y.C.T. is an Australian National Health and Medical Research Council Career Development Fellow (1065736).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.174730.114.
Freely available online through the Genome Research Open Access option.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Batra R, Cooper BS, Whiteley C, Patel AK, Wyncoll D, Edgeworth JD. 2010. Efficacy and limitation of a chlorhexidine-based decolonization strategy in preventing transmission of methicillin-resistant Staphylococcus aureus in an intensive care unit. Clin Infect Dis 50: 210–217. [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. 2005. ACT: the Artemis comparison tool. Bioinformatics 21: 3422–3423. [DOI] [PubMed] [Google Scholar]
- Cooper BS, Kypraios T, Batra R, Wyncoll D, Tosas O, Edgeworth JD. 2012. Quantifying type-specific reproduction numbers for nosocomial pathogens: evidence for heightened transmission of an Asian sequence type 239 MRSA clone. PLoS Comput Biol 8: e1002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington MJ, Hope R, Livermore DM, Kearns AM, Henderson K, Cookson BD, Pearson A, Johnson AP. 2010. Decline of EMRSA-16 amongst methicillin-resistant Staphylococcus aureus causing bacteraemias in the UK between 2001 and 2007. J Antimicrob Chemother 65: 446–448. [DOI] [PubMed] [Google Scholar]
- Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. 2002. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci 99: 7687–7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre DW, Golubchik T, Gordon NC, Bowden R, Piazza P, Batty EM, Ip CL, Wilson DJ, Didelot X, O’Connor L, et al. 2012. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2: e001124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas ME, Karageorgopoulos DE, Leptidis J, Korbila IP. 2013. MRSA in Africa: filling the global map of antimicrobial resistance. PLoS ONE 8: e68024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, et al. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SR, Cartwright EJ, Török ME, Holden MT, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, et al. 2013. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 13: 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Lindsay JA, Corton C, Quail MA, Cockfield JD, Pathak S, Batra R, Parkhill J, Bentley SD, Edgeworth JD. 2010. Genome sequence of a recently emerged, highly transmissible, multi-antibiotic- and antiseptic-resistant variant of methicillin-resistant Staphylococcus aureus, sequence type 239 (TW). J Bacteriol 192: 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AP, Woodford N. 2013. Global spread of antibiotic resistance: the example of New Delhi metallo-β-lactamase (NDM)-mediated carbapenem resistance. J Med Microbiol 62: 499–513. [DOI] [PubMed] [Google Scholar]
- Köser CU, Holden MT, Ellington MJ, Cartwright EJ, Brown NM, Ogilvy-Stuart AL, Hsu LY, Chewapreecha C, Croucher NJ, Harris SR, et al. 2012. A neonatal MRSA outbreak investigation using rapid whole genome sequencing. N Engl J Med 363: 2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Macedo-Vinas M, François P, Renzi G, Schrenzel J, Vernaz N, Pittet D, Harbarth S. 2011. Impact of combined low-level mupirocin and genotypic chlorhexidine resistance on persistent methicillin-resistant Staphylococcus aureus carriage after decolonization therapy: a case-control study. Clin Infect Dis 52: 1422–1430. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Du X, Villaruz AE, Diep BA, Wang D, Song Y, Tian Y, Hu J, Yu F, Lu Y, et al. 2012. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat Med 18: 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ, Quick J, Weir JC, Quince C, Smith GP, Betley JR, et al. 2013. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309: 1502–1510. [DOI] [PubMed] [Google Scholar]
- Nickerson EK, West TE, Day NP, Peacock SJ. 2009a. Staphylococcus aureus disease and drug resistance in resource-limited countries in south and east Asia. Lancet Infect Dis 9: 130–135. [DOI] [PubMed] [Google Scholar]
- Nickerson EK, Hongsuwan M, Limmathurotsakul D, Wuthiekanun V, Shah KR, Srisomang P, Mahavanakul W, Wacharaprechasgul T, Fowler VG, West TE, et al. 2009b. Staphylococcus aureus bacteraemia in a tropical setting: patient outcome and impact of antibiotic resistance. PLoS ONE 4: e4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojosnegros S, Perales C, Mas A, Domingo E. 2011. Quasispecies as a matter of fact: viruses and beyond. Virus Res 162: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otter JA, Patel A, Cliff PR, Halligan EP, Tosas O, Edgeworth JD. 2013. Selection for qacA carriage in CC22, but not CC30, methicillin-resistant Staphylococcus aureus bloodstream infection isolates during a successful institutional infection control programme. J Antimicrob Chemother 68: 992–999. [DOI] [PubMed] [Google Scholar]
- Price JR, Golubchik T, Cole K, Wilson DJ, Crook DW, Thwaites GE, Bowden R, Walker AS, Peto TE, Paul J, et al. 2014. Whole-genome sequencing shows that patient-to-patient transmission rarely accounts for acquisition of Staphylococcus aureus in an intensive care unit. Clin Infect Dis 58: 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EP, Smith JM, Hurst LD, Holden MT, Cooper JE, Smith NH, Feil EJ. 2006. Comparisons of dN/dS are time dependent for closely related bacterial genomes. J Theor Biol 239: 226–235. [DOI] [PubMed] [Google Scholar]
- Sammon JW., Jr 1969. A nonlinear mapping for data structure analysis. IEEE Trans Comput C-18: 401–409. [Google Scholar]
- Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, et al. 2013. Genomic variation landscape of the human gut microbiome. Nature 493: 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, Gemmell CG, Hunter IS. 2008. The association between biocide tolerance and the presence or absence of qac genes among hospital-acquired and community-acquired MRSA isolates. J Antimicrob Chemother 61: 78–84. [DOI] [PubMed] [Google Scholar]
- Snitkin ES, Zelazny AM, Thomas PJ, Stock F, NISC Comparative Sequencing Program Group, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4: 148ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK Department of Health 2013. UK Five Year Antimicrobial Resistance Strategy 2013–2018. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/244058/20130902_UK_5_year_AMR_strategy.pdf.
- US Department of Health and Human Services, Centers for Disease Control and Prevention 2013. Antibiotic resistance threats in the United States, 2013. http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf.
- Worby CJ, Lipsitch M, Hanage WP. 2014. Within-host bacterial diversity hinders accurate reconstruction of transmission networks from genomic distance data. PLoS Comput Biol 10: e1003549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization 2014. Antimicrobial resistance: global report on surveillance 2014. http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf?ua=1.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.