

Abstract
Burkholderia pseudomallei (Bp) is the causative agent of the infectious disease melioidosis. To investigate population diversity, recombination, and horizontal gene transfer in closely related Bp isolates, we performed whole-genome sequencing (WGS) on 106 clinical, animal, and environmental strains from a restricted Asian locale. Whole-genome phylogenies resolved multiple genomic clades of Bp, largely congruent with multilocus sequence typing (MLST). We discovered widespread recombination in the Bp core genome, involving hundreds of regions associated with multiple haplotypes. Highly recombinant regions exhibited functional enrichments that may contribute to virulence. We observed clade-specific patterns of recombination and accessory gene exchange, and provide evidence that this is likely due to ongoing recombination between clade members. Reciprocally, interclade exchanges were rarely observed, suggesting mechanisms restricting gene flow between clades. Interrogation of accessory elements revealed that each clade harbored a distinct complement of restriction-modification (RM) systems, predicted to cause clade-specific patterns of DNA methylation. Using methylome sequencing, we confirmed that representative strains from separate clades indeed exhibit distinct methylation profiles. Finally, using an E. coli system, we demonstrate that Bp RM systems can inhibit uptake of non-self DNA. Our data suggest that RM systems borne on mobile elements, besides preventing foreign DNA invasion, may also contribute to limiting exchanges of genetic material between individuals of the same species. Genomic clades may thus represent functional units of genetic isolation in Bp, modulating intraspecies genetic diversity.
Burkholderia pseudomallei (Bp) is the causative agent of melioidosis, a serious infectious disease of humans and animals and a leading cause of community-acquired sepsis and pneumonia in endemic regions (Currie et al. 2010). Initially thought to be confined to Southeast Asia and Northern Australia, the prevalence of Bp appears to be spreading (Wiersinga et al. 2012), and Bp has been designated a biothreat select agent in the United States. Bp can persist in extreme environmental conditions and can infect several plant and animal hosts, including birds, dolphins, and humans (Wuthiekanun et al. 1995; Howard and Inglis 2003; Sprague and Neubauer 2004; Larsen et al. 2013). Treatment of clinical melioidosis is challenging because the bacterium is inherently resistant to many antibiotics, and Bp infections can persist in humans for more than a decade (Hayden et al. 2012; Wiersinga et al. 2012).
The Bp genome comprises one of the largest and most complex bacterial genomes sequenced to date. Consisting of two large circular replicons (chromosomes) with a combined 7.2-Mb genome size (Holden et al. 2004), it contains a rich arsenal of genes related to virulence (e.g., Type III and Type VI secretion systems, polysaccharide biosynthesis clusters), metabolic pathways, and environmental adaptation (Wiersinga et al. 2012). Besides conserved regions, accessory genes on mobile elements and genomic islands may also contribute to phenotypic and clinical differences in microbial behavior (Currie et al. 2000; Sim et al. 2008). Analysis of the Bp genome has revealed previously unknown toxins and mechanisms of antibiotic resistance (Chantratita et al. 2011; Cruz-Migoni et al. 2011).
Most large-scale studies of Bp genetic diversity to date have analyzed strains using multilocus sequence typing (MLST). These studies have suggested a high degree of genetic variability between Bp strains and related Burkholderia species (Cheng et al. 2008), and have shown that Bp strains belonging to different sequence types (STs) can often coexist in the same locale and sometimes even within the same sample (Pitt et al. 2007; Wuthiekanun et al. 2009). However, due to the limited number of genes analyzed by MLST, these studies cannot comment on the global proportion of genetic material shared between strains of different STs nor on the relative contribution of recombination, mutation, and horizontal gene transfer on intraspecies genetic diversity. Moreover, although previous studies have applied whole-genome sequencing (WGS) to study global patterns of Bp genetic heterogeneity and evolution, earlier Bp WGS reports have been confined to a limited number of isolates (10–12) derived from diverse geographical regions (Nandi et al. 2010), where geophysical barriers likely limit the propensity of the analyzed strains to exchange genetic material. To achieve a comprehensive understanding of genetic variation among closely related Bp strains, WGS analysis of much larger strain panels, ideally performed on strains isolated from a common region and belonging to the same (or closely related) ST groups, is required.
In this study, we attempted to fill this important knowledge gap by performing WGS on 106 Bp strains drawn from a restricted Asian locale (Singapore and Malaysia). The WGS data, exceeding previous Bp WGS studies by 10-fold, enabled us to identify specific genomic clades of Bp, molecular features of Bp recombination at the whole-genome level, and accessory genome features contributing to recombination and horizontal gene transfer. We found a consistent pattern of genetic separation correlating with MLST, recombination haplotypes, shared accessory genes, and restriction modification (RM) systems. We provide evidence that restriction modification, beyond its role as defense against foreign DNA invasion, may have also partitioned the Bp species by restricting gene flow, resulting in the other observed correlations. Because RM systems are widely dispersed through the bacterial kingdom, it is possible that similar principles may apply to other bacterial species, implicating a potential role for epigenetic barriers as a driver of early incipient speciation.
Results
Bp genome sequencing
We analyzed 106 Bp strains, including 97 strains from Singapore and Malaysia (87/10) and nine strains from Thailand (Supplemental Table S1). The Singaporean and Malaysian strains were isolated from various clinical, animal, and environmental sources over a 10 yr period (1996–2005) (see Methods). MLST classified the strains into 22 sequence types (ST). Supporting their close phylogenetic relationships, 20 STs belonged to clonal complex CC48 (Supplemental Fig. S1A,B). The majority of strains were of ST51 (43 strains) and ST423 (16 strains).
Due to the high GC-bias of the Bp genome, we initially found that conventional Illumina sequencing protocols resulted in uneven genome coverage and suboptimal assemblies (median N50: 2907 bp) (Supplemental Table S2). We overcame this problem by applying a PCR amplification-free strategy (Kozarewa et al. 2009), resulting in markedly improved genome coverage and assemblies (median depth 100×; median N50: 102,577 bp). In total, we predicted 84,846 high quality SNPs in the WGS panel compared to the K96243 reference (Chr I: 43,829 and Chr II: 41,017). We validated the technical accuracy of the WGS data by Sanger sequencing of 50 randomly selected SNPs. Of the predicted SNPs, all 50 were confirmed by Sanger sequencing.
Whole-genome phylogenetic analysis resolves genomic clades
We excluded SNPs associated with regions of recombination as previously described (Croucher et al. 2011), resulting in a set of 10,314 SNPs representing mutations inherited by vertical descent along different lineages (“lineage SNPs” [L-SNPs]). Maximum likelihood phylogenies using the L-SNPs identified three major clades (“genomic clades”) containing all the Singapore and Malaysian strains, clustering apart from Thailand strains (Fig. 1A). Strains of the same ST grouped together within the same genomic clade, indicating strong similarities between phylogenies based on WGS and MLST. However, compared to MLST, the WGS phylogenies provided increased resolving power. For example, although MLST indicated a high degree of relatedness between ST50 and ST414 strains, WGS revealed that ST50 is more related to ST46 (Genomic Clade C), with ST414 being a more distant group (Fig. 1A). WGS also subdivided the ST51 strains into two distinct subclades—ST51a (39 strains) and ST51b (four strains) (Fig. 1B)—distinguished by ∼342 distinct L-SNPs (Supplemental Table S3). Notably, all three clades comprised a heterogeneous intermingling of strains from different isolation sources (e.g., clinical, animal, and environmental), arguing against the existence of a genetically distinct Bp subpopulation preferentially associated with human disease. The three clades also contained strains isolated during similar time periods (1996–2005), suggesting that genetically distinct Bp strains from different clades can coexist in the same region over many years.
Figure 1.

Whole-genome phylogeny and sequence variation of Bp strains. (A) Global phylogeny of Bp strains. The maximum likelihood tree was constructed using SNPs not associated with recombination events (see Results). Tip labels are colored according to the geographic locations of isolation ([red] Singapore; [green] Malaysia; [blue] Thailand; [black] unknown; [pink] imported to UK). Inset bars at right indicate the MLST scheme ([blue] ST51; [cyan] ST422; [red] ST423; [dark green] ST84; [pink] ST289; [light green] ST46). Three major genomic clades are identified: Clade A (ST51, ST422, ST414, ST169, nST4); Clade B (ST423, ST84, ST289, nST5); and Clade C (ST46, ST50). (B) Intra-ST subgroups resolved by WGS. ST51 strains cluster into two groups: ST51a and ST51b. Genomic locations of 342 L-SNPs (including both intra- and intergenic SNPs) distinguishing ST51a and ST51b are shown. The top and bottom panels with four rows show SNPs exclusively present in the two groups ST51a+ST51b− and ST51a−ST51b+, respectively. (C) Mutation spectra of ST51a: relative rates of six possible mutation categories. The most common mutations are C/G → T/A transitions. (D) Fraction of the three classes of cytosine mutations occurring at CG dinucleotides in the Bp genome compared with the expected fraction based on the average of 100 simulated genomes of the same size and composition (gray).
L-SNPs occurred at a ∼1.2-fold higher frequency on Chr II compared to Chr I (6.1 × 10−3 SNPs per site for Chr I versus 7.5 × 10−3 for Chr II, P = 3.08 × 10−14, χ2 = 57.68, χ2 test) (Supplemental Table S4a), suggesting a preferential accumulation of genetic mutations on Chr II during evolution. The majority of L-SNPs corresponded to C/G → T/A transitions (Fig. 1C; for Clade ST51a), in the context of CG dinucleotides (P = 3 × 10−10, binomial test, Fig. 1D), likely reflecting the tendency of methylated cytosines to form thymines (Kahramanoglou et al. 2012). For both chromosomes, L-SNPs preferentially localized to intergenic regions (Chr I: P < 2.2 × 10−16, χ2 = 101.42; Chr II: P = 4.196 × 10−14, χ2 = 57.04, χ2 test), and one-third of L-SNPs occurring within genes were nonsynonymous (Supplemental Table S4b). The dN/dS ratio (proportion of rate of nonsynonymous substitutions per site to rate of synonymous substitutions per site) for the major STs (e.g., ST51a, ST84) ranged between 0.17 and 0.64 per genome. Similar results were obtained when we analyzed a more restricted subset of 8035 L-SNPs associated with the Bp “core” genome (regions common to all Bp strains, estimated size 5.64 Mb) (Supplemental Table S5).
Widespread recombination among Bp isolates
Bp strains that are genetically distinct may interact within environmental reservoirs such as soil or water (Chantratita et al. 2008; Mayo et al. 2011) or in co-infected animals and human hosts (Pitt et al. 2007), thereby providing opportunities for recombination. Hence, it is vital to understand pathways and processes that facilitate or constrain gene flow between strains. To identify genomic characteristics of Bp recombination, we proceeded to analyze SNPs associated with recombination (R-SNPs). From 74,532 R-SNPs, we identified 2373 recombination events across the three genomic clades, with recombination tract lengths ranging from 3 bp to 71 kb (median ∼5 kb). We computed recombination/mutation (r/m) values, corresponding to the ratio of rates at which substitutions are introduced by recombination and mutation, across the entire population. The overall per site r/m ratio was 7.2. Based upon these data, we estimate that at least 78% of the BpK96243 reference genome (∼5.67 Mb) has undergone recombination, a level comparable to S. pneumoniae, a highly recombinogenic species (74K/85K R-SNPs for Bp; 50K/57K R-SNPs for S. pneumoniae) (Croucher et al. 2011). Similar to L-SNPs, higher recombination levels were observed for Chr II than Chr I (P < 2.2 × 10−16, Mann-Whitney U test),
Besides estimating whole-population metrics, we also computed clade-specific recombination and mutation rates for the three major Bp genomic clades, using a previously described Bayesian approach (ClonalFrame) (Didelot and Falush 2007). To minimize mapping artifacts, we excluded mobile genetic elements (e.g., phages, transposons, and genomic islands) and based our analysis on a reduced core genome of 5.6 Mb (see Methods). For all three clades, the ratio of mutation rate (theta) to recombination rate (rho) was close to one, suggesting that recombination and mutation both happen at approximately the same rates. Recombination was also found to introduce more substitutions than mutation (r/m = 4.5 in Clade A, r/m = 8.5 in Clade B, and r/m = 6 in Clade C) with the highest impact observed in Clade B (Supplemental Table S6). These values are in general agreement with the values obtained from the total population.
The high levels of recombination in the Bp clades motivated us to also analyze potential sources of recombination imports. We used previously established methods to assess intra- and interclade recombination flux (Didelot et al. 2009, 2011). Briefly, recombined fragments were compared with homologous sequences from other Bp genomes across the three clades, and a “match” was found if the sequence was identical or contained a single nucleotide difference. If a match was found to members of a single clade, the origin of the recombination event was attributed to this clade (matches to strains from multiple clades were categorized as ambiguous). If no matches were found, the origin was categorized as unknown. To estimate their relative impact on genomic diversification, the flux of genomic content between clades was summarized as the proportion of each genome originating from different origins. Of 2481, 821, and 334 recombination events detected within genomic Clades A, B, and C, respectively, we could assign sources (“matches”) for ∼60% of recombination events (1112 matches to single clades and 1059 matches to multiple clades). On average, ∼5% of each genome from a given clade was found to have originated from another clade and approximately another 7% from a source not present in our data set (Supplemental Table S7). Several of the interclade recombination events were found on recent branches of the clonal genealogy, suggesting that the isolation is not complete between the clades.
Genome-wide median recombination frequencies (RFs) were computed to identify genomic regions exhibiting elevated recombination rates and multiple recombination events (Fig. 2A). We identified 1630 protein-coding genes (Chr I: 897 genes; Chr II: 733) associated with regions of high recombination (RF > RFmedian + 3MAD, median absolute deviation). Genes experiencing high recombination frequencies were significantly enriched in intracellular trafficking and secretion pathways (corrected P = 0.0006, binomial test), whereas genes involved in protein translation were underrepresented (corrected P = 0.012, binomial test) (Supplemental Table S8). Examples of genomic regions exhibiting elevated recombination included a Type III secretion cluster (TTSS3; BPSS1520–BPSS1537) previously linked to mammalian virulence (Stevens et al. 2002), and a Type IVB pilus cluster (TFP8, Chr II: BPSS2185–BPSS2198) (Fig. 2B). Type IV pili (TFP), including those of the subtype IVB, encode surface-associated protein complexes involved in multiple cellular processes (Craig et al. 2004). To evaluate if TFP8 might modulate Bp virulence, we generated an isogenic Bp deletion strain lacking ∼12.9 kb of the TFP8 locus and assessed the virulence of the TFP8 mutant in a BALB/c mouse intranasal infection assay. The TFP8 deletion mutant exhibited significantly reduced virulence compared to parental Bp K96243 wild-type controls (P = 0.026, Mantel-Haenszel log-rank test, Fig. 2C). These results support a role for Type IVB pili in Bp murine virulence, and more generally that a subset of recombination hotspots in Bp may influence mammalian virulence.
Figure 2.

Recombination landscape of Bp. (A) Recombination hotspots in Bp. Circles: (outside) genome coordinates; (middle) compositionally biased regions identified by Alien hunter (Vernikos and Parkhill 2006) (green) and Bp core genome (violet); (innermost) regions of elevated recombination (height of red bars). Note that recombination levels are higher on Chr II than Chr I. Location of the TFP8 and TTSS3 clusters are indicated. (B) Local recombination events in the Type III secretion system and Type IVB pilus cluster. (Top) Genomic coordinates and location of protein-coding genes; (dark blue) predicted recombination events (R1 to Rn, n = number of recombination events) observed in Bp strains belonging to genomic clades (ST group 46, 51, 84, 289, 422, and 423). The recombination boundaries are indicated by the dark blue circles and the boundaries that fall beyond the depicted locus are shown as open ended. (C) Relative virulence of TFP8 deletion mutant. Graphs show survival curves of BALB/c mice following intranasal challenge with varying dosages of Bp (left: K96243 wild-type; right: TFP8 deletion mutant, units are colony forming units, CFU). See Methods for infection assay details. The TFP8 deletion mutant is significantly less virulent compared to Bp K96243 parental controls (P = 0.026, Mantel-Haenszel log rank test). (D) Distinct haplotypes at the TFP8 genomic locus. Each row represents an individual Bp strain arranged according to genomic clade/ST (shown on left with color bars indicating ST51 [blue]; ST422 [cyan]; ST423 [red]; ST84 [dark green]; and ST46 [light green]). Across each row (strain), SNP positions are ordered by genomic coordinate (top numbers, Bp Chr II, genomic locus 2,935,860–2,976,718), and color-coded according to nucleotide identity (A→ green; T→ blue; C→ orange; and G→ red). The right y-axis “Haplotypes” refers to the specific linear combination of SNPs exhibited by individual strains. In some cases, haplotypes can be composed of a specific combination of smaller recombination regions (R). For example, Haplotype H1 is composed of recombination regions R2, R7, and R8. Haplotype alignments were generated using Clustal X (Larkin et al. 2007).
Genome-wide recombination haplotype map of Bp
Extended genomic stretches with high recombination rates often displayed specific combinations of local independent recombination events in individual strains, resulting in the creation of distinct haplotypes. Using the TFP8 gene cluster as an example, some strains displayed recombination events R2, R7, and R8 (Haplotype 1 [H1]), whereas other strains displayed events R3, R7, and R8 (Haplotype 2 [H2]). In total, we identified five haplotypes (H1–H5) in the TFP8 gene cluster (Supplemental Table S9). We found that these five TFP8 haplotypes were tightly associated with specific clades; for example, haplotype H1 was associated with ST51 strains, whereas haplotype H4 (corresponding to recombination event R4) was associated with ST84 strains (Fig. 2D). To evaluate this association at the whole-genome level, we generated a whole-genome haplotype map of Bp, identifying 85 genomic regions exhibiting multiple (five or more) haplotypes (Supplemental Table S10). Similar to TFP8, the vast majority of haplotypes occurred in a genomic-clade specific pattern (Supplemental Fig. S2). Many of the multi-haplotype genomic regions were involved in specialized functions such as iron and cofactor metabolism, detoxification, and virulence (Supplemental Table S10). Almost half (48%) of the multiple-haplotype genomic regions exhibited at least one haplotype with an excess of nonsynonymous to synonymous SNPs, consistent with these regions having altered phenotypic properties. For instance, one haplotype overrepresented in nonsynonymous SNPs occurred in the virulence-associated TFP1 locus (BPSL0782-BPSL0783), within the pilA gene in ST51a strains. Notably, pilA plays a role in virulence yet its role in adherence and microcolony formation varies considerably in different Bp strains (Essex-Lopresti et al. 2005; Boddey et al. 2006). These findings suggest that haplotype variation may contribute to differences in Bp pathogenicity or survival in different strains.
Bp accessory genome elements exhibit clade restriction
The availability of WGS data for a large Bp panel also provided the opportunity to quantitatively assess the Bp accessory genome. Using the Velvet and NUCmer algorithms, we generated de novo sequence assemblies of genomic sequences not found in the K96243 reference genome. On average, ∼183 kb of novel accessory regions (NAE) were identified for each Bp strain (minimum region length 1 kb). We found that the Bp genome is “open,” with at least 2897 new non-K96243 genes associated with the accessory regions (Fig. 3A). The Bp pan genome (Bp core + Bp accessory) is thus at least 8802 genes, which is 2× the size of the Bp core genome. Accessory genes were characterized by a lower percentage GC content (median value: ∼59% ± 5.6) than the core genome (∼68%), consistent with their horizontally acquired nature. Accessory genes were also significantly enriched in pathways related to defense mechanisms (corrected P < 0.0005 relative to Bp core genes) (Fig. 3B).
Figure 3.
Accessory genome landscape of Bp. (A) Accumulation curves for Bp novel accessory genes (blue). Vertical bars represent standard deviation values based upon 100 randomized input orders of different Bp STs. The total number of accessory genes is indicated by the red dotted line. (B) Functional enrichment of Bp accessory genes. COG functional categories are indicated on the y-axis, and the percentage of genes in each COG category is shown on the x-axis. Dark blue columns represent novel accessory genes, and light blue columns indicate all Bp core genes with COG annotations. COG categories exhibiting a significant enrichment among the Bp accessory genes are highlighted by asterisks ([*] P < 0.0005, binomial test; after Bonferroni correction). The COG category “DNA replication, recombination and repair” was excluded as it was represented mainly by mobility genes, particularly transposases and integrases. (C) Distribution of accessory elements across Bp clades. The heatmap represents an all-pairwise strain comparison showing the degree of accessory element overlap between pairs of strains. Strains are arranged on the x- and y-axis according to their genomic clades and sequence types (ST51 [blue]; ST289 [pink]; ST422 [cyan]; ST423 [red]; ST84 [dark green]; and ST46 [light green]). The color scale bar at the bottom indicates the degree of accessory element sharing (more blue equates to increased sharing). The right-hand chart depicts the different types of restriction-modification (RM) systems associated with different clades. In each column, the RM systems are color-coded based on their encoded protein-coding sequences. In the first column, the bars in green and blue refer to two distinct sets of RM genes that belong to Type IC RM systems. Strain-specific RM systems are in black.
Using pairwise similarity metrics, we evaluated the extent to which accessory elements found in one Bp strain might be shared with other strains. Similar to the recombination haplotypes, strains belonging to the same genomic clade had a tendency to share many of the same accessory elements (Fig. 3C). For example, strains in genomic Clade A shared a 15-kb gene cluster of metabolic genes, including biotin carboxylase, NAD-dependent malic enzymes, mandelate racemase, and 5-enolpyruvylshikimate-3-phosphate synthase (Priestman et al. 2005; Tang et al. 2005; Li et al. 2009). Similarly, strains from genomic Clade B (ST423/ST84/ST289) shared accessory genes such as filamentous hemagglutinin, fhaC, which plays a crucial role in mediating adherence to eukaryotic cells (Relman et al. 1989).
Evidence of ongoing recombination and gene exchange within clades
The analyses described above revealed a strong correlation between Bp clades, core genome recombination haplotypes, and complements of accessory elements. We hypothesized that these correlations could be explained by two alternative models—“ongoing recombination” or “vertical descent” (Fig. 4A). In the first model, active recombination is ongoing in Bp, but preferentially restricted to exchange of DNA within a clade. To test for ongoing recombination, we computed within-clade nucleotide divergence levels in DNA sequences predicted to have undergone recombination and compared these to divergence levels within regions of nonrecombined DNA in strains exhibiting the recombination event. If recombination were ongoing among strains within a clade, then this would serve to homogenize the recombining sequences across strains, whereas nonrecombining regions would accumulate mutations independently in different strains (Fig. 4A, “Ongoing Recombination”). Thus, the within-clade nucleotide divergence of recombined regions would be predicted to be lower relative to nonrecombined regions. In the alternative model, recombination is not commonly taking place. Here, recombined regions would have entered a clade in its founder, and then would be found throughout the clade due mostly to strict vertical descent (Fig. 4A, “Vertical Descent”). In this case, recombined regions would be predicted to accumulate mutations at the same rate as nonrecombined regions.
Figure 4.

Distinguishing between ongoing recombination and vertical descent and in Bp. (A) Alternative models for clade-specific recombination haplotypes. (Left) In the “Ongoing Recombination” model, an imported fragment sweeps through the population via recombination, resulting in homogenization of the recombining fragment across strains. The recombining fragment should show lower levels of sequence diversity compared to nonimported regions. (Right) In the “Vertical Descent” model, an ancestral strain acquires a genomic fragment (yellow) from an external strain and subsequently transmits that fragment to all daughter strains in a clonal fashion. In this model, the imported fragment should accumulate new point mutations (green bars) at a similar rate to nonimported regions. (B) Within-clade sequence diversity of recombined regions compared to nonrecombined regions. Scatter plots comparing within-clade sequence diversity values of individual recombined regions (x-axis) to nonrecombined regions (y-axis) for the same strains in a given clade. Sequence diversity decreases in the direction of the red arrows (to right and upward). (*) Data points highlighted by the red bar correspond to recombined regions exhibiting 100% sequence identity. To visualize these points in a manner that captures both their density and extremely low sequence diversity, these were plotted within the x-axis range of 6.9–7.6 on a negative log scale. Sequence diversity is defined as the number of SNPs per kb. (C) Sequence features of nonrecombined regions (NR), recombined regions (R), and accessory elements (AE). (Top) Bp K96243 genomic tracks of Chr I and Chr II. Row 1: Genomic locations of recombined regions (red). Row 2: Genomic locations of 16 known Bp genomic islands (gray). (Bottom) Sequence feature comparison of genes in nonrecombined (white; NR), recombined (red; R), and accessory elements (gray; AE): (i) GC content (Puigbò et al. 2008); (ii) effective codon number (Puigbò et al. 2008); and (iii) sequence complexity (Pietrokovski et al. 1990). Each hourglass plot spans the 25th to 75th percentile (interquartile range [IQR]) of all genes in that category, with the bottleneck at the median. Horizontal tick marks show data ranges within 1.5 × IQR of the 25th and 75th percentiles. Open circles represent outliers outside this range. The width of the bottleneck (i.e., the length of the V-shaped notch) depicts the 95% confidence interval for the median. (D) Within-clade sequence diversity of accessory elements compared to nonaccessory elements. Accessory elements are defined as regions not present in the BpK96243 reference strain (see Methods). Scatter plots compare average sequence diversity values for individual accessory elements (x-axis) to corresponding nonaccessory elements (y-axis) for the same strain pairs in a given clade. Sequence diversity is defined as the number of SNPs per kb.
For each recombined region in a clade, we calculated the average sequence divergence level in that region, using strains exhibiting the recombination event (see Supplemental Fig. S3 and Supplemental Text for a detailed description of this analysis). To obtain a conservative set of nonrecombined sequences for comparison, we then took only those sections of the Bp genome predicted not to have undergone any recombination in any of the Bp strains; and for the same strains we calculated the sequence divergence levels in these nonrecombined sequences. We found that recombined regions in the core genome had uniformly lower sequence divergence than nonrecombined regions (Fig. 4B), suggesting that recombination is active and ongoing within clades. Extending this analysis to the gene level, we found that recombination has opposite effects on within- and between-clade divergence; genes in recombined regions had higher between-clade diversity compared with genes in nonrecombined regions, as expected, but much lower within-clade diversity (Supplemental Fig. S4). To rule out the possibility that the lower sequence divergence in recombined regions might be due to recombined regions possessing different sequence features or gene functions than nonrecombined regions (despite both regions being part of the same core genome), we also compared GC content, effective codon number, sequence complexity, and COG functions between the recombined regions, nonrecombined regions, and accessory elements. The latter was included because accessory elements are known to be distinct in gene function and sequence characteristics from the core genome (Kung et al. 2010). We found recombined and nonrecombined regions to be highly similar and distinct from accessory elements (Fig. 4C). For example, nucleotide frequencies between recombined and nonrecombined regions were similar (Chr I: χ2 = 0.0012, P-value = 1; Chr II: χ2 = 2 × 10−4, P-value = 1, χ2 test) (Supplemental Table S11), and COG analysis of all genes associated with recombination regions failed to reveal any significantly enriched biological pathways compared to the whole genome (Supplemental Table S12), indicating that general baseline recombination in Bp is not functionally selected.
Besides recombination in the core genome, the correlation between Bp clades and complements of accessory elements suggested a further test for whether recombination is ongoing in Bp. Similar to the logic for the core recombined sequences above, accessory elements that are pervasive throughout a given clade could be undergoing active, ongoing exchange (which would homogenize their sequence within the clade) or be inherited through vertical descent (and thus accumulate mutations similar to adjacent nonrecombining regions) (Fig. 4A). Both these possibilities are consistent with clade-specific complements of accessory elements (Fig. 3C). We found that within each clade, accessory elements also showed lower sequence divergence levels compared to nonaccessory elements in the same strains (Fig. 4D). Thus, these results suggest that in both the core and accessory genome, there is a strong signal for ongoing, active recombination within Bp clades.
Identification of clade-specific RM systems
The clade-specific pattern of haplotypes and accessory elements in Bp, coupled with evidence of ongoing gene flow within strains of the same clade, suggests that reciprocal barriers to gene exchange may exist between strains belonging to different clades. We hypothesized that these barriers might be due, at least in part, to the use of distinct restriction-modification (RM) systems in each clade. RM systems comprise different combinations of endonuclease, methylase, and DNA specificity domains that use specific methylation patterns to label endogenous “self” genomic DNA, whereas unmodified exogenous DNA is recognized as “non-self” and subsequently cleaved and destroyed (Ershova et al. 2012; Makarova et al. 2013). Studies have proposed that RM systems can act as barriers to horizontal gene transfer (Waldron and Lindsay 2006; Hoskisson and Smith 2007; Dwivedi et al. 2013). However, a role for RM systems in restricting intraspecies recombination is less well described (Waldron and Lindsay 2006).
By interrogating genes in the Bp accessory genome and mobile genetic elements, we identified four different Bp RM systems (I, II, III, and IV) (Roberts et al. 2007). Notably, specific sets of RM systems were found in association with genomic clades bearing distinct haplotypes and accessory genome features. For example, although clearly related by lineage (Fig. 1), clades ST51 and ST422 exhibit different recombination patterns and sets of accessory elements: We found that ST51 strains contained RM Type IIGC genes, whereas ST422 strains harbored both RM Type IIGC and RM Type IC systems. Similarly, strains from genomic Clade B (ST423/ST84/ST289) were largely dominated by RM Type IC and Type IBC systems, with type III RM genes additionally present in ST84 strains (Fig. 3C). The presence of these clade and ST-specific RM systems, which are predicted to result in clade-specific patterns of DNA methylation, may provide a molecular barrier to interclade gene sharing.
Methylome sequencing reveals clade-specific epigenetic profiles
To provide direct experimental data that Bp strains from different clades have distinct methylation profiles, we subjected one representative strain from Clade A (Bp35) and one strain from Clade B (Bp33) to whole-genome methylome analysis using SMRT sequencing technology (Murray et al. 2012). Because SMRT sequencing has the ability to measure DNA polymerase activity in real time, base modifications such as methylation can be detected as a change in the kinetics of base pair incorporation (Flusberg et al. 2010; Schadt et al. 2010).
SMRT sequencing followed by de novo assembly for Bp33 and Bp35 was performed to obtain two circular contigs of 4.0 and 3.1 Mb (average GC content is 68%) with 240× and 147× post-filter base coverage, 21× and 22× preassembled read coverage, respectively (Chin et al. 2013). We identified a 12.4-kb plasmid in Bp35 called pBp35 that to our knowledge represents the first plasmid described for Bp (Supplemental Fig. S5; Supplemental Data: plasmid sequence and annotation in GenBank format). We analyzed the local sequence contexts for all the methylated bases in both the strains and identified sequence motifs associated with these methylated bases. Both Bp strains showed methylation throughout their entire genomes. In total, six unique methylated motifs were identified in the two Bp strains (Table 1). Of these, one motif (5′-CACAG-3′) was shared by the two strains, whereas the other five were strain- or clade-specific. For example, the type II motif (5′-GTAWAC-3′) is unique to Bp35 (Clade A representative), whereas the type I motif (5′-GTCATN5TGG-3′) is present only in Bp33 (Clade B representative).
Table 1.
DNA methylation sequence motifs in Bp35 (Clade A) and Bp33 (Clade B)
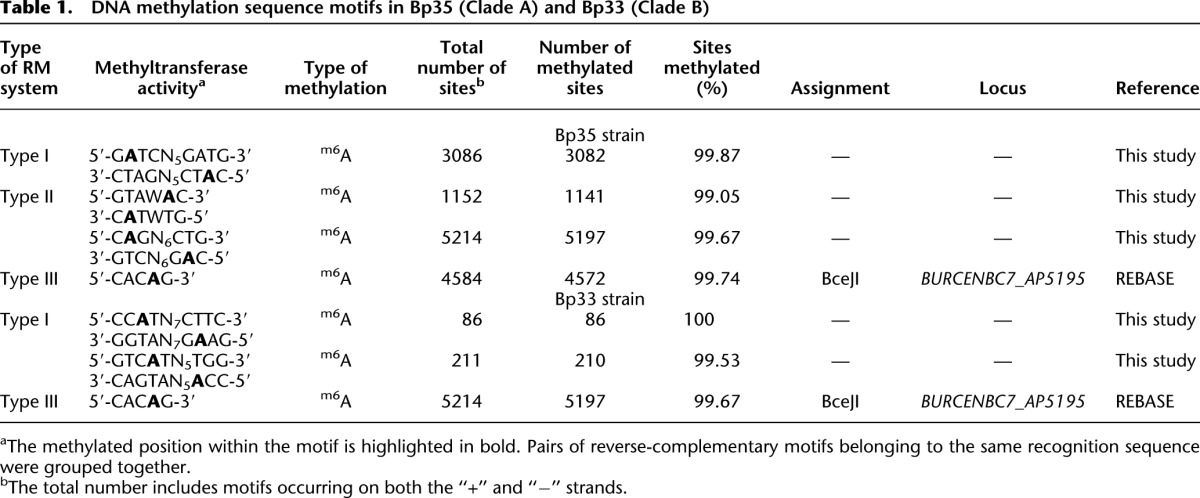
We proceeded to match the different motifs to specific RM systems found in the two genomes. Reassuringly, the shared CACAG motif was found to be associated with a conserved Type III RM system found in both strains. However, Bp 35 exhibited three strain-specific methylated motifs, and these could be associated with Type I and II systems found specifically in the Bp35 clade. Similarly, Bp 33 exhibited two strain-specific methylated motifs, and these could be associated with Type I RM systems found specifically in the Bp33 clade. For both strains, the fraction of strain-specific methylated motifs was close to 100%, consistent with their predicted methylation and restriction functions operating at high efficiency (Table 1). The demonstration that strains belonging to different Bp clades indeed have distinct methylation patterns is consistent with our hypothesis that clade-specific RM activity may represent a barrier to Bp interclade recombination and accessory element transfer.
Bp RM systems impede foreign DNA uptake in E. coli
To functionally test if clade-specific RM systems of Bp can impede the transfer of non-self DNA, we cloned and tested the Type I RM system associated with Genomic Clade A (Bp33) in E. coli (Janscak et al. 1999; Kasarjian et al. 2003). We engineered one plasmid to carry the Type I “restriction” endonuclease (R+), and a second separate plasmid to carry the “specificity” and “methylase” proteins (M+) (Fig. 5A). M+R+ and M+R− E. coli strains were then secondarily transformed with reporter plasmids carrying zero, one, and two copies of the RM recognition site predicted from SMRT sequencing (5′-GTCATN5TGG-3′; see Table 1), and efficiencies of transformation (EOT) were calculated (Fig. 5B). We found that when transformed into M+R+ strains, unmethylated reporter plasmids carrying one or two recognition sites exhibited a > 100-fold decrease in EOT compared to reporter plasmids with no recognition sites (P < 0.01; Student’s t-test) (Fig. 5C). Importantly, the Type I restriction endonuclease is required for this decrease, as no EOT differences were observed when the plasmids (zero, one, and two sites) were transformed into M+R− strains which only express the methylase (Fig. 5C). This result indicates that the restriction endonuclease of the Clade A-specific Type I RM system is indeed active and capable of impeding the uptake of non-self DNA harboring an unmethylated Type I recognition site.
Figure 5.

Restriction of non-self DNA by clade-specific Bp RM systems. (A) Molecular cloning of a Type I RM system specific to Bp genomic Clade A. The RM system comprises three genes: (R) “restriction,” (M) “methylase,” (S) “specificity.” Genes S (yellow) and M (blue) were cloned in plasmid pSLC-279 with kanamycin resistance (KmR) to give the M+ plasmid. Gene R (red) was cloned in plasmid pSLC-280 with ampicillin resistance (ApR) to give the R+ plasmid. Resistance genes are depicted in cyan. Green arrows represent the T5 promoter used to induce expression of the cloned genes. Plasmids are not drawn to scale. (B) Efficiency of transformation (EOT) assay. Reporter plasmids p0, p1, and p2 harbor zero, one, and two copies of the predicted Type I recognition site (5′-GTCATN5TGG-3′; indicated by green triangles). Plasmid p0 should not show any EOT changes because it does not contain Type I recognition sequences. Unmethylated plasmids p1 and p2, when transformed into M+R+ strains, should be recognized via their Type I sites and cleaved by the Type I restriction enzyme (registered as a drop in number of transformants). However, when transformed into M+R− strains that express the methyltransferase alone, no EOT differences should be observed. In contrast, methylated p1 and p2 plasmids (obtained by passage through M+R− strains; methylated sites indicated by red stars and superscript m), when transformed into M+R+ strains, should be recognized as “self” DNA by the Type I system and resist cleavage, resulting in minimal EOT changes. (C) EOT assay results using unmethylated plasmids. Host strains are MG1655 (M−R−, no RM system, cyan); SLC-623 (M+R+, complete RM system, red); and SLC-621 (M+R−, methyltransferase only, green). Reporter plasmids are pACYC184 (p0, control plasmid); pSLC-277 (p1, 1 recognition site); and pSLC-278 (p2, 2 recognition sites). Significant differences in EOT are observed between control plasmid p0 and plasmids p1 and p2 when transformed into M+R+ strains (P < 0.01) but not in host E. coli or M+R− strains. EOT in this study is the normalized number of CmR transformants obtained per unit amount of plasmid DNA. (D) EOT assay using methylated plasmids. Reporter plasmids were passaged through M+R− strains prior to transformation, which is predicted to cause recognition site methylation. No significant EOT differences are observed across the strains. All experiments were performed in triplicate, and data are presented as mean and standard deviations. Data are presented as log10 values of EOT. Student’s t-test was used to test for significant differences.
Next, we isolated plasmids with methylated recognition sites by passaging them through M+R− E.coli strains and transformed them into M+R+ strains. In contrast to the results using unmethylated plasmids, all three plasmids (zero, one, or two recognition sites) exhibited no significant EOT differences (Fig. 5D). This result indicates that, at least for one Bp clade-specific RM system, methylation of the recognition sites by RM methylases is sufficient to facilitate uptake of non-self DNA, even in the presence of its cognate restriction endonuclease.
Discussion
In this study, we performed WGS on a panel of Bp strains drawn from a restricted geographic locale to explore the contribution of gene mutation, recombination, and horizontal gene transfer to the molecular diversity of closely related Bp isolates. We found that Bp strains can be partitioned into distinct genomic clades and that a major proportion of the Bp core genome variation is strongly influenced by both mutation and recombination. Bp diversity is further enhanced by an accessory genome component that is at least double the Bp core genome. Moreover, using diverse approaches, including (1) sequence diversity comparisons in both recombination and accessory regions supporting active gene flow within but not across clades; (2) genome-wide methylome sequencing demonstrating clade-specific epigenetic profiles associated with distinct RM-systems; and (3) experimental demonstration in an E. coli system that Bp RM systems are functionally active and sufficient to mediate the methylation and restriction of non-self DNA, our results point toward a model in which Bp RM systems may function as a barrier to gene exchange between different Bp clades.
Phylogenetic analysis of the Bp clades revealed that they comprised mixtures of Bp isolates from animal, clinical, and environmental sources, arguing against the existence of a genetically distinct population of Bp capable of infecting humans. Supporting this model, in a separate analysis, we were unable to confidently identify a consistent set of signature genetic changes in strains associated with human disease (T Nandi and P Tan, unpubl.). The genetic similarity between clinical, animal, and environmental Bp strains raises the possibility that additional genetic changes may not be required for an environmental Bp strain to successfully cause human disease. This model is consistent with previous proposals that Bp is an “accidental pathogen,” in which adaptations incurred by Bp to survive in its natural reservoir (soil and potentially single-celled organisms located therein, e.g., amoebae) must have indirectly contributed to its ability to colonize a mammalian host (Casadevall and Pirofski 2007; Nandi et al. 2010). This “accidental virulence” hypothesis is further supported by epidemiological data in which patients with clinical melioidosis often possess pre-infection morbidities such as diabetes, which may contribute to a weakened host immune response (Currie et al. 2010).
Our data revealed several genome-wide features of the Bp recombination landscape. We found that recombination in Bp is pervasive, approaching levels previously reported for S. pneumonia (Croucher et al. 2011), and frequently involved defined sets of haplotypes. Importantly, analysis of genes in regions associated with high recombination suggests that haplotypes and recombination hotspots in Bp are not randomly distributed, but biased toward genomic regions associated with niche adaption, survival, and virulence. This included a TTSS cluster involved in intracellular survival of Bp and virulence (Stevens et al. 2002) and a type IVB pilus cluster (TFP8) that we have validated in this study as required for maximal virulence in murine infection assays. It is thus possible that the other regions of high recombination identified in this study may contain additional genes involved in Bp adaptation, survival, and virulence.
Previous studies have shown that strains of different STs can be frequently co-isolated in the wild (Wuthiekanun et al. 2009). However, it is not known if such co-isolated strains are able to engage in an unrestricted exchange and transfer of genetic material. We found that strains associated with different Bp genomic clades tended to exhibit distinct sets of recombination haplotypes and accessory elements. These findings suggest that Bp clade/ST subgroups may represent a functional and potentially limiting unit of within-species genetic diversity. However, it is important to note that although our findings suggest a general scenario of recombination events being largely clade-specific, exceptions do exist. One example of a possible recombination across separate clades involved a heme/porphyrin locus (BPSS1245–BPSS1247), where a haplotype present in strain P171/04 from ST84 (but not other ST84 strains) was highly similar to the haplotypes of ST51 strains (Supplemental Fig. S6). One explanation for these exceptions is that although barriers to interclade gene exchange do exist, they may be incomplete or perhaps only recently established.
Two broad models of Bp evolution could explain this clade-restriction pattern. First, clade restriction might result from effective physical or niche separation, in which strains from different clades are adapted or restricted to distinct niches in the environment, and therefore do not share DNA. However, as mentioned above, strains of different STs can be co-isolated; and while this does not rule out the presence of microscale niche differences between strains, such “micro-niches” remain to be experimentally proven. Alternatively, the discovery of clade-specific RM systems provides an epigenetic explanation for Bp clade restriction. In this model, acquisition of a diversity of RM systems to combat invading DNA may thus have occurred as a primary event, and the resulting epigenetic differences may in turn have established barriers to intraclade DNA exchange. A synthesis of these two models is also possible, where early subspeciation is initially driven by epigenetic barriers and followed subsequently by traditional niche selection. Consistent with this model, we observed that among ST84 strains, the two Malaysian Bp strains, EY2 and EY5, clustered separately from the remaining seven Singapore strains, being separated by 13 L-SNPs mapping to 12 protein-coding genes. This scenario could be explained by initial RM-driven epigenetic isolation of the ST84 clade, followed by geographical separation between the Singapore and Malaysian ST84 strains. The presence of localized concentrations of nonsynonymous changes suggests the possible existence of specific selective pressures driving further divergence, and it is also possible that the driver for speciation in Bp is currently shifting from divergence due to genetic/epigenetic separation to divergence due to selection in different niches. As such, our results provide a potential snapshot of early incipient speciation in a microorganism associated with diverse genomes and habitats.
Methods
Ethics statement
This research was approved by the GIS Institutional Review Board. Animal studies were performed in accordance with the UK Scientific Procedures Act (Animals) (1986) and UK Codes of Practice for the Housing and Care of Animals Used in Scientific Procedures (1989).
Bacterial strains, plasmids, and primers
Strains used were obtained from DMERI, DSO National Laboratories. These include (1) 56 clinical isolates from melioidosis patients between 1996 and 2004; (2) 34 animal isolates from various species (e.g., monkeys, pigs, birds, and dogs) diagnosed with melioidosis between 1996 and 2005; and (3) 16 soil isolates from 1996 to 2000 (Supplemental Table S1). The isolates were sampled from a diversity of locations and not a single site (AL Tin, pers. comm.). For E. coli experiments, plasmids bearing predicted recognition sequences for the Clade A-specific Type I RM system (5′-GTCATN5TGG-3′) were generated by PCR-mediated insertion (see Supplemental Methods). Gene sequences encoding the Bp33 Type I restriction modification system proteins (“specificity,” “methylase,” and “restriction endonuclease”) were cloned into expression vectors driven by a T5 inducible promoter (DNA2.0, Singapore). All plasmids were propagated in E. coli K12 strain MG1655. Bacterial strains, plasmids, and primers are listed in Supplemental Table S13.
Genomic DNA extraction and multiplex sequencing
Live bacteria were grown in a BioSafety Level 3 facility in DSO National Laboratories. Genomic DNA was extracted using the Qiagen Genomic Tip 500/G kit (Qiagen). Unique index-tagged libraries for each sample were created, and up to 33 separate libraries were sequenced per lane on an Illumina HiSeq instrument with 100 base paired-end reads. Libraries were constructed using an amplification-free method (Kozarewa et al. 2009). Raw Illumina data were split to generate paired-end reads, and assembled using a de novo genome-assembly program, Velvet v0.7.03 (Zerbino and Birney 2008), to generate a multicontig draft genome for each Bp isolate.
Gene annotation, SNP, and phylogenetic analysis
Paired-end reads were mapped against the chromosomes of B. pseudomallei K96243 (accession numbers BX571965 and BX571965) (Holden et al. 2004). Bp genes were predicted using FGENESB (http://www.softberry.com). Gene orthologs were determined using OrthoMCL (Chen et al. 2006). RM systems were inferred based on the specificity sequences of homologs in REBASE (Roberts et al. 2007) and categorized into subtypes—IC, IBC, IIG, IIGC, IIP, and IIB—on the basis of their genetic organization, mode of action, recognition sites, and cleavage loci (Roberts et al. 2003). SNPs predicted to have arisen by homologous recombination were identified using Gubbins and excluded from phylogenetic reconstruction (Croucher et al. 2011). Indels were identified using Dindel (Albers et al. 2011). Maximum likelihood phylogenies were constructed using RAxML v0.7.4 (Stamatakis et al. 2005). SNPs ancestral and derived alleles (polarization) were determined according to the outgroup reference strain sequence.
Recombination analysis
The general time-reversible model with gamma correction was used for among-site rate variation for 10 initial random trees. To measure clade-specific recombination rates, ClonalFrame (Didelot and Falush 2007) was applied separately to each Bp clade. To reduce mapping artifacts, we focused on the 5.6-Mb portion of the core genome that excludes mobile genetic elements and other potentially biased regions such as surface polysaccharides, secretion systems, and tandem repeats. Recombination events were extracted from ClonalFrame as genomic fragments, where the probability of recombination for a given branch of the tree was consistently > 50% and reached 95% in at least one location. Potential origins of recombination imports were investigated as previously described (Didelot et al. 2009, 2011; Sheppard et al. 2013). To determine haplotypes, SNP alleles at the recombination loci were concatenated to give a single haplotype string for each strain. The aligned strings were then subjected to hierarchical clustering as implemented in R package “hclust.” The resulting dendrogram was used to assign strains to distinct haplotype groups using the “cutree” function in R. Within-clade sequence diversity comparisons between recombined and nonrecombined regions were performed as described in Supplemental Figure S3 and the Supplemental Text. Potential differences in sequence composition between recombined and nonrecombined regions were assessed using Artemis release 13.0 (Carver et al. 2012) or the K2 algorithm in CLC Main workbench 6.5 (http://www.clcbio.com) (Wootton and Federhen 1993).
Bp mutagenesis and mouse virulence studies
Isogenic Bp mutants carrying a 12.9-kb deletion TFP8 were generated in a two-step process as previously described (see Supplemental Methods; Essex-Lopresti et al. 2005; Boddey et al. 2006). Virulence of wild-type and mutant Bp strains was assessed using an intranasal BALB/c mouse model (Essex-Lopresti et al. 2005). Briefly, groups of six age-matched BALB/c female mice were anesthetized and infected intranasally with 10-fold dilutions (101–106) of either wild-type Bp K96243 or TFP8 deletion strains grown overnight at 37°C with shaking. Mice were recovered, and survival was recorded for up to 51 d. Analysis was performed using the Mantel-Haenszel log rank test in GraphPad Prism 4 or by Regression with Life Data in MiniTAB v13.0, using a significance threshold of P = 0.05.
Accessory genome analysis
Nucmer (Kurtz et al. 2004) was used to generate alignments of Velvet contigs against the reference strain Bp K96243 to identify novel accessory regions (NAE). NAE values for individual Bp strains were defined as blocks with a minimal 1000-bp length that was absent in Bp K96243 (median NAE per strain = 183,482 bp). Sequence diversity comparisons between accessory and nonaccessory regions utilized accessory regions > 1000 bp and performed using MUMmer 3.20 under DNAdiff default settings (Kurtz et al. 2004).
SMRT sequencing and data analysis
Twenty micrograms of gDNA was processed to create SMRTbell sequencing templates > 10 kb (average insert size 17 kb) and sequenced using a PacBio RS II System in which polymerase-MagBead-bound templates were loaded at an on-plate concentration of 150 pM. Templates were subsequently sequenced using DNA Sequencing Kit 2.0, with data collection of 180 mins (Pacific Biosciences). Genomes were assembled using HGAP (Chin et al. 2013) with default parameters in SMRT Analysis Suite version 2.1 (Pacific Biosciences). Additional manual assembly of contigs was carried out in cases of unique overlapping sequence. Consensus sequence polishing was done using the Quiver algorithm in Genomic Consensus version 0.7.0. Base modification analysis was performed by mapping SMRT sequencing reads to the respective assemblies using the BLASR mapper (Chaisson and Tesler 2012) and SMRT Analysis Suite version 2.1 using standard mapping protocols. Clustering of sequence motifs was performed using Motif Finder (https://github.com/PacificBiosciences/DevNet/wiki/Motiffinder). See Supplemental Methods for further details.
Restriction-modification assay
Plasmids containing methylated and unmethylated Type I restriction sites were transformed into E. coli strains engineered to express all three proteins of the Type I RM system or only the specificity and methylase units. Efficiency of transformation (EOT) values were computed by comparing bacterial titers (colony forming units per mL, cfu/mL) on antibiotic selection plates divided by the corresponding titers from LB plates. EOT values were log transformed and plotted for analysis of RM system restriction activity. EOT values from triplicate experiments were compared using a two-tailed Student’s t-test. See Supplemental Methods for further details.
Statistical analysis
All statistical analyses were performed using R-2.15.1 (Ihaka and Gentleman 1996).
Data access
The data from this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under accession number ERP000251. Methylation data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE55168.
Supplementary Material
Acknowledgments
This study was supported by a core grant to P.T. from the GIS, an A-STAR research institute. The sequencing of the Burkholderia pseudomallei strains was supported by Wellcome Trust grant 098051 to J.P.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.177543.114.
References
- Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R. 2011. Dindel: accurate indel calls from short-read data. Genome Res 21: 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddey JA, Flegg CP, Day CJ, Beacham IR, Peak IR. 2006. Temperature-regulated microcolony formation by Burkholderia pseudomallei requires pilA and enhances association with cultured human cells. Infect Immun 74: 5374–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. 2012. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadevall A, Pirofski LA. 2007. Accidental virulence, cryptic pathogenesis, martians, lost hosts, and the pathogenicity of environmental microbes. Eukaryot Cell 6: 2169–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantratita N, Wuthiekanun V, Limmathurotsakul D, Vesaratchavest M, Thanwisai A, Amornchai P, Tumapa S, Feil EJ, Day NP, Peacock SJ. 2008. Genetic diversity and microevolution of Burkholderia pseudomallei in the environment. PLoS Negl Trop Dis 2: e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantratita N, Rholl DA, Sim B, Wuthiekanun V, Limmathurotsakul D, Amornchai P, Thanwisai A, Chua HH, Ooi WF, Holden MT, et al. 2011. Antimicrobial resistance to ceftazidime involving loss of penicillin-binding protein 3 in Burkholderia pseudomallei. Proc Natl Acad Sci 108: 17165–17170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Mackey AJ, Stoeckert CJ Jr, Roos DS. 2006. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res 34: D363–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AC, Ward L, Godoy D, Norton R, Mayo M, Gal D, Spratt BG, Currie BJ. 2008. Genetic diversity of Burkholderia pseudomallei isolates in Australia. J Clin Microbiol 46: 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Craig L, Pique ME, Tainer JA. 2004. Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2: 363–378. [DOI] [PubMed] [Google Scholar]
- Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, et al. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331: 430–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Migoni A, Hautbergue GM, Artymiuk PJ, Baker PJ, Bokori-Brown M, Chang CT, Dickman MJ, Essex-Lopresti A, Harding SV, Mahadi NM, et al. 2011. A Burkholderia pseudomallei toxin inhibits helicase activity of translation factor eIF4A. Science 334: 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie BJ, Fisher DA, Howard DM, Burrow JN. 2000. Neurological melioidosis. Acta Trop 74: 145–151. [DOI] [PubMed] [Google Scholar]
- Currie BJ, Ward L, Cheng AC. 2010. The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis 4: e900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Falush D. 2007. Inference of bacterial microevolution using multilocus sequence data. Genetics 175: 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Barker M, Falush D, Priest FG. 2009. Evolution of pathogenicity in the Bacillus cereus group. Syst Appl Microbiol 32: 81–90. [DOI] [PubMed] [Google Scholar]
- Didelot X, Bowden R, Street T, Golubchik T, Spencer C, McVean G, Sangal V, Anjum MF, Achtman M, Falush D, et al. 2011. Recombination and population structure in Salmonella enterica. PLoS Genet 7: e1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi GR, Sharma E, Rao DN. 2013. Helicobacter pylori DprA alleviates restriction barrier for incoming DNA. Nucleic Acids Res 41: 3274–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ershova AS, Karyagina AS, Vasiliev MO, Lyashchuk AM, Lunin VG, Spirin SA, Alexeevski AV. 2012. Solitary restriction endonucleases in prokaryotic genomes. Nucleic Acids Res 40: 10107–10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essex-Lopresti AE, Boddey JA, Thomas R, Smith MP, Hartley MG, Atkins T, Brown NF, Tsang CH, Peak IR, Hill J, et al. 2005. A type IV pilin, PilA, contributes to adherence of Burkholderia pseudomallei and virulence in vivo. Infect Immun 73: 1260–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7: 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden HS, Lim R, Brittnacher MJ, Sims EH, Ramage ER, Fong C, Wu Z, Crist E, Chang J, Zhou Y, et al. 2012. Evolution of Burkholderia pseudomallei in recurrent melioidosis. PLoS ONE 7: e36507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden MT, Titball RW, Peacock SJ, Cerdeño-Tárraga AM, Atkins T, Crossman LC, Pitt T, Churcher C, Mungall K, Bentley SD, et al. 2004. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci 101: 14240–14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskisson PA, Smith MC. 2007. Hypervariation and phase variation in the bacteriophage ‘resistome’. Curr Opin Microbiol 10: 396–400. [DOI] [PubMed] [Google Scholar]
- Howard K, Inglis TJ. 2003. Novel selective medium for isolation of Burkholderia pseudomallei. J Clin Microbiol 41: 3312–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. 1996. R: a language for data analysis and graphics. J Comput Graph Stat 5: 299–314. [Google Scholar]
- Janscak P, MacWilliams MP, Sandmeier U, Nagaraja V, Bickle TA. 1999. DNA translocation blockage, a general mechanism of cleavage site selection by type I restriction enzymes. EMBO J 18: 2638–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahramanoglou C, Prieto AI, Khedkar S, Haase B, Gupta A, Benes V, Fraser GM, Luscombe NM, Seshasayee AS. 2012. Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat Commun 3: 886. [DOI] [PubMed] [Google Scholar]
- Kasarjian JK, Iida M, Ryu J. 2003. New restriction enzymes discovered from Escherichia coli clinical strains using a plasmid transformation method. Nucleic Acids Res 31: e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ. 2009. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods 6: 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung VL, Ozer EA, Hauser AR. 2010. The accessory genome of Pseudomonas aeruginosa. Microbiol Mol Biol Rev 74: 621–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- Larsen E, Smith JJ, Norton R, Corkeron M. 2013. Survival, sublethal injury, and recovery of environmental Burkholderia pseudomallei in soil subjected to desiccation. Appl Environ Microbiol 79: 2424–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lu W, Han Y, Ping S, Zhang W, Chen M, Zhao Z, Yan Y, Jiang Y, Lin M. 2009. A novel RPMXR motif among class II 5-enolpyruvylshikimate-3-phosphate synthases is required for enzymatic activity and glyphosate resistance. J Biotechnol 144: 330–336. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Koonin EV. 2013. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res 41: 4360–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo M, Kaesti M, Harrington G, Cheng AC, Ward L, Karp D, Jolly P, Godoy D, Spratt BG, Currie BJ. 2011. Burkholderia pseudomallei in unchlorinated domestic bore water, Tropical Northern Australia. Emerg Infect Dis 17: 1283–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. 2012. The methylomes of six bacteria. Nucleic Acids Res 40: 11450–11462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi T, Ong C, Singh AP, Boddey J, Atkins T, Sarkar-Tyson M, Essex-Lopresti AE, Chua HH, Pearson T, Kreisberg JF, et al. 2010. A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathog 6: e1000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrokovski S, Hirshon J, Trifonov EN. 1990. Linguistic measure of taxonomic and functional relatedness of nucleotide sequences. J Biomol Struct Dyn 7: 1251–1268. [DOI] [PubMed] [Google Scholar]
- Pitt TL, Trakulsomboon S, Dance DA. 2007. Recurrent melioidosis: possible role of infection with multiple strains of Burkholderia pseudomallei. J Clin Microbiol 45: 680–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestman MA, Funke T, Singh IM, Crupper SS, Schönbrunn E. 2005. 5-Enolpyruvylshikimate-3-phosphate synthase from Staphylococcus aureus is insensitive to glyphosate. FEBS Lett 579: 728–732. [DOI] [PubMed] [Google Scholar]
- Puigbò P, Bravo IG, Garcia-Vallve S. 2008. CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct 3: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relman DA, Domenighini M, Tuomanen E, Rappuoli R, Falkow S. 1989. Filamentous hemagglutinin of Bordetella pertussis: nucleotide sequence and crucial role in adherence. Proc Natl Acad Sci 86: 2637–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DT, Dybvig K, et al. 2003. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31: 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, Macelis D. 2007. REBASE—enzymes and genes for DNA restriction and modification. Nucleic Acids Res 35: D269–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Turner S, Kasarskis A. 2010. A window into third-generation sequencing. Hum Mol Genet 19: R227–R240. [DOI] [PubMed] [Google Scholar]
- Sheppard SK, Didelot X, Jolley KA, Darling AE, Pascoe B, Meric G, Kelly DJ, Cody A, Colles FM, Strachan NJ, et al. 2013. Progressive genome-wide introgression in agricultural Campylobacter coli. Mol Ecol 22: 1051–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim SH, Yu Y, Lin CH, Karuturi RK, Wuthiekanun V, Tuanyok A, Chua HH, Ong C, Paramalingam SS, Tan G, et al. 2008. The core and accessory genomes of Burkholderia pseudomallei: implications for human melioidosis. PLoS Pathog 4: e1000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague LD, Neubauer H. 2004. Melioidosis in animals: a review on epizootiology, diagnosis and clinical presentation. J Vet Med B Infect Dis Vet Public Health 51: 305–320. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. 2005. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21: 456–463. [DOI] [PubMed] [Google Scholar]
- Stevens MP, Wood MW, Taylor LA, Monaghan P, Hawes P, Jones PW, Wallis TS, Galyov EE. 2002. An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol Microbiol 46: 649–659. [DOI] [PubMed] [Google Scholar]
- Tang DJ, He YQ, Feng JX, He BR, Jiang BL, Lu GT, Chen B, Tang JL. 2005. Xanthomonas campestris pv. campestris possesses a single gluconeogenic pathway that is required for virulence. J Bacteriol 187: 6231–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernikos GS, Parkhill J. 2006. Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics 22: 2196–2203. [DOI] [PubMed] [Google Scholar]
- Waldron DE, Lindsay JA. 2006. Sau1: a novel lineage-specific type I restriction-modification system that blocks horizontal gene transfer into Staphylococcus aureus and between S. aureus isolates of different lineages. J Bacteriol 188: 5578–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersinga WJ, Currie BJ, Peacock SJ. 2012. Melioidosis. N Engl J Med 367: 1035–1044. [DOI] [PubMed] [Google Scholar]
- Wootton JC, Federhen S. 1993. Statistics of local complexity in amino acid sequences and sequence databases. Comput Chem 17: 149–163. [Google Scholar]
- Wuthiekanun V, Smith MD, Dance DA, White NJ. 1995. Isolation of Pseudomonas pseudomallei from soil in north-eastern Thailand. Trans R Soc Trop Med Hyg 89: 41–43. [DOI] [PubMed] [Google Scholar]
- Wuthiekanun V, Limmathurotsakul D, Chantratita N, Feil EJ, Day NP, Peacock SJ. 2009. Burkholderia pseudomallei is genetically diverse in agricultural land in Northeast Thailand. PLoS Negl Trop Dis 3: e496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.