

Abstract
The gene regulatory network (GRN) that supports neural stem cell (NS cell) self-renewal has so far been poorly characterized. Knowledge of the central transcription factors (TFs), the noncoding gene regulatory regions that they bind to, and the genes whose expression they modulate will be crucial in unlocking the full therapeutic potential of these cells. Here, we use DNase-seq in combination with analysis of histone modifications to identify multiple classes of epigenetically and functionally distinct cis-regulatory elements (CREs). Through motif analysis and ChIP-seq, we identify several of the crucial TF regulators of NS cells. At the core of the network are TFs of the basic helix-loop-helix (bHLH), nuclear factor I (NFI), SOX, and FOX families, with CREs often densely bound by several of these different TFs. We use machine learning to highlight several crucial regulatory features of the network that underpin NS cell self-renewal and multipotency. We validate our predictions by functional analysis of the bHLH TF OLIG2. This TF makes an important contribution to NS cell self-renewal by concurrently activating pro-proliferation genes and preventing the untimely activation of genes promoting neuronal differentiation and stem cell quiescence.
Neural stem cells (NS cells) are the primary progenitors of both the developing and the adult central nervous system (CNS). They possess the cardinal stem cell properties of self-renewal and multipotency, being able to generate the neurons, astrocytes, and oligodendrocytes that populate the mature CNS (Temple 2001; Kriegstein and Alvarez-Buylla 2009; Fuentealba et al. 2012). NS cells, in the adult brain at least, can also enter a reversible growth-arrested state called quiescence (Fuentealba et al. 2012). These key cellular properties are served by a gene regulatory network (GRN) that must concurrently activate the genes required for self-renewal and prepare the cells for the timely and appropriate induction of genes required for differentiation or quiescence.
A crucial layer of control in all GRNs is provided by non-protein-coding cis-regulatory elements (CREs) that function to activate or repress target gene transcription or to prime genes for rapid induction following change of the cellular state. Transcription factors (TFs) with the ability to recognize and bind defined sequence motifs provide an important level of specificity in the control of CRE function. By binding CREs, frequently in combination with other TFs, regulatory and chromatin remodeling complexes are recruited to CREs that ultimately modulate the expression level of target genes (Davidson 2010; Spitz and Furlong 2012). A CRE’s epigenetic profile is thought to closely reflect its activity state and also its ability to recruit TF complexes (Heintzman et al. 2007, 2009; Lupien et al. 2008; Creyghton et al. 2010; Rada-Iglesias et al. 2010; Hawkins et al. 2011; Li et al. 2011b; Bonn et al. 2012).
Relatively little is known about the repertoire of CREs that function in NS cells and few of the key regulatory TFs that control their activity have been described (Visel et al. 2009, 2013). Knowledge of the important TFs and their functions will be crucial for a full understanding of NS cells’ differentiation potential and will also inform the development of NS cell–based cellular therapies for human CNS disorders.
Here we set out to identify the nature of major TFs that bind and regulate NS cell CREs and to make specific predictions about the precise roles of different TFs within the NS cell GRN. We identified CREs throughout the genome of a well-characterized culture model of mouse NS cells (Conti et al. 2005) by identifying regions of relatively open chromatin that reflect the binding of transcriptional regulatory complexes (Boyle et al. 2008). We then used ChIP-seq data for a wide range of histone modifications to classify CREs according to their epigenetic profiles. We performed motif analysis in the different classes of CREs to identify the most important TF regulators, whose binding was confirmed by ChIP-seq. We then used a machine learning approach to deduce, from our rich collection of well-annotated CREs, which CREs and which TFs have specific functions in regulating genes required for NS cell self-renewal, differentiation, and quiescence. We validated our model’s predictions via the functional analysis of the basic helix-loop-helix (bHLH) TF OLIG2.
Results
Epigenetic signatures allow the precise classification of accessible regions of the NS cell genome
DNase I hypersensitive sites (DHSs) represent regions of relatively open chromatin that are associated with the binding of transcriptional regulatory complexes. DHSs are a hallmark of most known functional CREs, including promoters, enhancers, insulators, and silencers (Heintzman et al. 2007, 2009; Boyle et al. 2008; Natarajan et al. 2012; Thurman et al. 2012). We used DNase-seq (Boyle et al. 2008) to identify 25,770 high-confidence DHSs across the genome of self-renewing cultured mouse ES cell–derived NS cells (Supplemental Fig. S1; Conti et al. 2005; Martynoga et al. 2013). We used ChIP-seq data for seven well-characterized histone modifications (Mikkelsen et al. 2007; Meissner et al. 2008; Martynoga et al. 2013) to compartmentalize DHSs into exclusive classes with distinct chromatin signatures. DNase-seq and histone modification ChIP-seq are a potent combination in the identification of putative CREs, since the well-described discriminatory power, but low spatial resolution, of histone modification ChIP-seq is complemented by the increased spatial resolution of DNase-seq (DHSs identified here range from 46 to 3344 bp, median = 470 bp) to allow the precise identification of binding sites of important regulatory complexes within the broader histone mark-defined blocks.
We considered transcription start site (TSS) proximal (within 2 kb of a TSS, n = 10,791) and TSS distal (n = 14,979) DHSs separately and used both the presence/absence and relative abundance of ChIP-seq signals for H3K27ac, H3K4me1,me2,me3, H3K27me3, H3K36me3, and H3K9me3 (Mikkelsen et al. 2007; Meissner et al. 2008; Martynoga et al. 2013) to computationally cluster the DHS regions. We defined five distinct TSS-proximal clusters and six TSS-distal clusters of DHSs, which varied widely according to the local presence and intensity of the seven histone modifications analyzed (Fig. 1; Supplemental Table S1).
Figure 1.

Clustering analysis reveals multiple classes of epigenetically distinct gene regulatory regions in NS cells. (A) Schematic depicting the classification and regulatory annotation of DHS regions employed in this study. (B,C) Heatmaps of TSS-proximal (B) and TSS-distal (C) DHS regions generated by k-means clustering analysis according to the local ChIP-seq signal for seven histone modifications (x-axis). Five distinct proximal clusters and six distal clusters were identified. Regulatory designations (y-axis) are given according to our downstream analyses, as described in the Results section. (D,E) Aggregate plots of histone modification signal centered upon the point of maximal DNase-seq enrichment for each proximal (D) and distal (E) putative regulatory element.
Proximal clusters 1 and 2 both exhibited the key features of active promoters, being positive for H3K4me3, H3K4me2, and H3K27ac (Heintzman et al. 2007), and were distinguishable from one another by a H3K4me1 signal at proximal cluster 1 regions (Fig. 1B,D). Unexpectedly, proximal cluster 3 regions displayed the profile of an active distal enhancer (H3K4me1-high, H3K4me3-low, H3K27ac-high) (Creyghton et al. 2010; Rada-Iglesias et al. 2010) despite being tightly associated with TSSs. As described in the analysis below, we designate these proximal elements as a class of “poised” promoters. Proximal cluster 4 showed prominent repression-associated H3K27me3 modification. Finally, proximal cluster 5 elements showed no significant enrichment for the epigenetic marks analyzed here (Fig. 1B,D). The majority of proximal clusters 1, 2, and 4, but the minority of cluster 3 and 5 promoters, contained CpG islands (Supplemental Fig. S2A).
As expected, most TSS-distal clusters had different profiles compared to the proximal regions. DHSs in distal clusters 1–5 all carried a characteristic enhancer signature, being enriched for H3K4me1 and depleted for the promoter-associated H3K4me3 (Heintzman et al. 2007), while distal cluster 6 sites lacked significant enrichment for any of the histone marks analyzed (Fig. 1C,E). Of the five putative enhancer clusters, distal clusters 1 and 2 both showed strong enrichment of the active enhancer-associated mark H3K27ac (Creyghton et al. 2010; Rada-Iglesias et al. 2010), with distal cluster 1 additionally showing a higher signal for H3K4me2 and H3K4me3 than distal cluster 2 (Fig. 1C,E; Supplemental Fig. S2B,C). Distal cluster 5 regions were the only distal DHSs marked with H3K27me3. The remaining clusters (distal clusters 3 and 4) possessed a H3K4me1+, H3K27ac-low profile that has previously been designated as a poised or intermediate enhancer configuration in mouse embryonic stem cells (Fig. 1C,E; Creyghton et al. 2010; Zentner et al. 2011). Here we adopt the poised enhancer nomenclature for these clusters. All the putative enhancer clusters demonstrated a distinct “valley” shape, focused on the maximal point of DNase I hypersensitivity (Heintzman et al. 2007; Bonn et al. 2012). None of the proximal or distal DHS clusters showed a consistent presence of the repressive mark H3K9me3 or the transcript-elongation-associated mark H3K36me3.
Patterns of coassociation between different groups of distal and proximal CREs regulate genes expressed at different levels
We next explored the gene regulatory properties of the different categories of putative CREs. We associated the expression level, assayed by RNA-seq, of the closest annotated gene in Ensembl (Flicek et al. 2013) v61 to each CRE. Both for proximal and distal elements, the regions with an active epigenetic profile tend to be associated with genes expressed at higher levels, poised regions with genes expressed at medium levels, and repressed and unmarked regions with genes expressed at lower levels (Fig. 2A,B).
Figure 2.
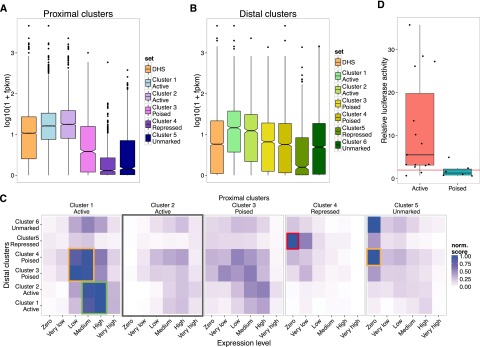
DHS clusters exist in a range of activity states. (A,B) Boxplots of the expression levels (RNA-seq) of the genes associated with proximal (A) and distal (B) DHS clusters. (C) Heatmap showing the strength of association between each class of distal DHS cluster (y-axis) and each proximal cluster (x-axis). Proximal cluster-associated genes have been further subdivided according to their expression level. Enrichment values are row-normalized and represent the P-values as calculated by GREAT (see Methods). Active distal elements are strongly enriched around expressed genes with active proximal elements (green box). Poised enhancers associate with less expressed or nonexpressed genes (yellow boxes), and repressed enhancers are most enriched around repressed promoters of silent genes (red box). Active proximal cluster 2 elements are weakly associated with all distal DHS classes. (D) Increased luciferase activity is driven by active (distal cluster 1 and 2) compared to poised (distal cluster 3 and 4) enhancers. Values presented are the fold change in normalized luciferase signal compared to the enhancerless parent vector containing only a minimal promoter.
We hypothesized that different classes of distal CREs would preferentially interact with TSS-proximal CREs that were in an equivalent activity state. We tested this by asking whether the different distal CREs were associated with genes with distinct proximal CRE configurations and expression levels (see Methods). Distal cluster 1 and 2 active enhancers were indeed strongly associated with proximal cluster 1 active promoters, particularly those expressed at medium and high levels (Fig. 2C, green box). Putative poised enhancers of distal cluster 3 were also strongly associated with genes with proximal cluster 1 active-promoters, but mainly those expressed at low to medium levels (Fig. 2C, orange boxes). In contrast, poised cluster 4 enhancers were most strongly associated with silent genes with unmarked promoters (Fig. 2C, orange box in proximal cluster 5). On the basis of this and our closest gene analysis and epigenomic profiling, we designate distal cluster 4 elements as “poised low” and distal cluster 3 elements as “poised high” enhancers.
There was a strong enrichment of repressed enhancers in the vicinity of genes with repressed promoters (Fig. 2C, red box), while unmarked distal elements were strongly associated with unmarked promoters of nonexpressed genes.
When we used CTCF peaks from ChIP-seq data (Phillips-Cremins et al. 2013) to define gene regulatory domains as intervals between two adjacent CTCF binding sites, we observed very similar trends. With this alternative domain definition, once again there was a clear tendency for promoters to be associated with distal elements in an equivalent activity state, without an intervening CTCF site to act as a putative boundary element (Supplemental Fig. S2D).
We were surprised to observe that the second class of active proximal elements (proximal cluster 2), which lacked any enrichment of the enhancer mark H3K4me1, was not strongly associated with any of the distal CREs defined here (Fig. 2C, gray box; Supplemental Fig. S2D), suggesting that the associated genes are mainly regulated at the proximal promoter level and/or by distal elements not detected by our strategy.
In an independent test of enhancer function, we cloned and tested the intrinsic enhancer activity of putative active and poised enhancers in a luciferase reporter gene assay. As predicted, putative active enhancer elements (distal cluster 1 or 2) possessed stronger activation potential in NS cells than those with a poised enhancer signature (distal clusters 3 and 4) (mean fold change of active enhancers 11.5 ± 11.8 vs 1.9 ± 1.6 for poised enhancers; Welch’s t-test, P < 0.01) (Fig. 2D).
Taken together, our fine-grained analysis of chromatin patterns, target gene expression analysis, and reporter gene assays support the existence of multiple distinct classes of distal and proximal CREs that vary greatly in their ability to influence gene expression in NS cells.
Different CRE classes associate with genes that are regulated when NS cells exit self-renewal
Following periods of active self-renewal, NS cells can either differentiate to generate glial or neuronal progeny or can enter a cell cycle-arrested state known as quiescence (Bonaguidi et al. 2011; Fuentealba et al. 2012). These state changes all involve rapid and extensive rewiring of the GRN underpinning NS cell self-renewal (Ohtsuka et al. 2011; Bracko et al. 2012; Martynoga et al. 2013). We hypothesized that genes whose expression is regulated during these different fate decisions would be associated with distinct functional classes of CREs. To address this question, we treated NS cells with three different growth factor regimes to induce cell cycle exit and stimulate differentiation or quiescence, and we used microarrays to detect significantly up- and down-regulated genes. We used BMP4 to induce astrocytic differentiation (Sun et al. 2011a), treatment with B27- and FGF2-containing medium to induce an early neuronal progenitor fate (Spiliotopoulos et al. 2009), and BMP4 + FGF2 to induce NS cell quiescence (Sun et al. 2011a; Martynoga et al. 2013). In all three experiments, 1340–2054 genes were significantly down-regulated and 1442–2054 genes were up-regulated, showing the extent of transcriptional rewiring. The responsive genes were independently validated and enriched for relevant functional categories and known marker genes (Supplemental Table S2; Supplemental Figs. S1, S3).
We then computed the statistical significance of the associations between each class of CRE and the sets of significantly regulated genes in each array. Repressed and unmarked promoters were enriched in the set of genes up-regulated in all three array experiments and showed no association with down-regulated genes, showing that many silent genes with these promoter classes are activated as NS cells exit self-renewal, as expected (Fig. 3A). Proximal cluster 3 promoter genes showed the same trend, indicating that this enhancer-like chromatin signature may mark a class of genes poised for induction during differentiation or quiescence. Active proximal cluster 2 elements appear to be strongly associated with NS cell self-renewal, since the linked genes showed a strong tendency to be down-regulated in differentiation and quiescence. Active proximal cluster 1 elements showed more complex associations and many linked genes were actually further up-regulated upon exit from self-renewal. Similar patterns were observed for the active distal enhancer–associated genes, which were both up- and down-regulated (Fig. 3B). Thus even genes with active proximal and distal CREs can be further activated following changes of cell state. As predicted, genes associated with poised enhancers were much more likely to be up- than down-regulated in differentiation or quiescence, as was also seen for repressed enhancer–associated genes.
Figure 3.
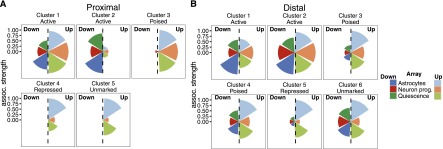
DHS cluster-associated genes have different expression patterns in differentiated and quiescent NS cells. (A,B) The relative strength of association between proximal (A) and distal (B) CREs and genes regulated when NS cells commit to astrocyte or neuronal differentiation, or enter quiescence. Wedge height is proportional to the statistical significance, and the associations with gene sets that are up- and down-regulated in each microarray experiment are presented in opposite wedges.
CRE-associated genes belong to different functional categories
Based on their association with different groups of expressed and nonexpressed genes, we predicted that different CREs would be associated with genes belonging to different functional categories. We used DAVID (Huang da et al. 2009) to discover enriched gene ontology (GO) biological processes associated with proximal CREs (Supplemental Fig. S4A). Both of the active proximal clusters were strongly enriched in the promoters of cell cycle genes and regulators of transcription, both fundamental cellular processes required for active NS cell self-renewal. Terms related to phosphate metabolism were more enriched in active proximal cluster 1 genes, and terms related to protein transport and localization were more enriched in proximal cluster 2 genes. Interestingly, repressed proximal cluster 4 promoters were very strongly enriched for genes associated with “neuron differentiation” and several related terms, suggesting that in self-renewing NS cells, this class of element marks and silences genes required for the generation of differentiated neuronal progeny.
We used GREAT to predict the functions of distal CRE-regulated genes (McLean et al. 2010). The six distal CRE clusters showed largely non-overlapping and frequently biologically relevant term enrichments (Supplemental Fig. S4B). For example, the terms “stem cell development,” “stem cell maintenance,” and “stem cell differentiation” were very highly enriched in active enhancer clusters 1 and 2 and in the “poised high” distal cluster 3, but not in less active or repressed enhancers. “Negative regulation of gene expression” and several related terms were among the most enriched terms in the repressed enhancer group, while the less active poised enhancers were most strongly enriched in cytoskeletal regulators (e.g., “actin cytoskeleton organization”), suggesting that some genes associated with poised elements may drive the extensive cellular remodeling associated with the differentiation and migration of NS cell progeny.
Altogether these analyses indicate that epigenetically defined CREs in different activity states associate with different functional classes of genes that are frequently highly responsive to NS cell state change. We predicted that these different properties would be driven by differential recruitment of TFs and set out to determine the most important TF regulators.
Motif enrichment analysis predicts key TFs binding and regulating the different classes of CRE
We used motif analysis to identify the enriched motifs of the key TFs binding the different functional classes of CRE. We used de novo search algorithms MEME (Machanick and Bailey 2011) and RSAT (Thomas-Chollier et al. 2012) to examine a narrow 400-bp window centered on the DHS summit, and also used CentriMo (Bailey and Machanick 2012) to identify previously described motifs with significant enrichment centered upon DHSs. We analyzed each cluster of DHSs separately and curated a core set of 16 distinct TF motifs that were each significantly enriched in at least one of the six distal and five proximal clusters of DHSs (Fig. 4A), and plotted the proportion of DHS sites in each cluster that showed significant motif matches (Fig. 4B,C).
Figure 4.

Differential TF motif enrichment within the different CRE classes. (A) The set of 16 TF motifs found to be enriched in at least one of the different DHS clusters (see Methods). Motif logos, the name employed in this study, and the motif database source and respective identifier are presented. (B,C) Heatmap representation of the abundance of the 16 TF motifs in each of the distal (B) and proximal (C) clusters of DHSs, binned in 10% intervals.
Among the enhancer clusters 1–5, a motif previously attributed to the nuclear factor I (NFI) TFs was most strongly enriched, as were two related E-box motifs, the predicted target motif for bHLH TFs. Among the active enhancer clusters 1 and 2, there was also clear enrichment of a novel motif consisting of two SOX-like motifs in opposite orientations (Fig. 4A, 2Sox motif). Motifs for TCFAP2A, SP1, and ZIC4 TFs were found more broadly across all enhancer classes but were particularly frequent in active enhancer cluster 1. Unmarked distal regions of cluster 6 and repressed enhancers were strongly enriched for the CTCF motif, suggesting that many of these sites may be bound by the CTCF regulatory factor.
Proximal CREs exhibited enrichment of a largely distinct set of TF motifs, with less representation of the NFI and SOX motifs. E-box1, a SMAD-like motif, and the TCFAP2A and ZIC4 motifs were very strongly enriched in both active and repressed proximal elements, and the SP1 motif was very prevalent in all proximal DHSs. E2F3 and ETS1 motifs were most specific for active proximal elements (Fig. 4B).
Motif enrichment is predictive of NFI, bHLH, and SOX TF binding to CREs
To directly test whether TF motif enrichment is indeed predictive of TF binding in NS cell CREs, we selected a set of TFs and performed genome-wide location analysis by ChIP-seq. We focused on enhancer CREs, since these were expected to contribute more to cell type–specific gene expression (Heintzman et al. 2009; Hawkins et al. 2011; Yu et al. 2013), and selected a set of TFs whose motifs were enriched in enhancers, which were robustly expressed in self-renewing NS cells (see FPKM expression values from RNA-seq, below) and for which we could obtain ChIP-grade antibodies. We selected an antibody that specifically recognizes all four NFI family members (NFIA [FPKM = 36], NFIB [FPKM = 28], NFIC [FPKM = 19], NFIX [FPKM = 28]) (Martynoga et al. 2013). We selected antibodies for four bHLH factors, each with distinct known and predicted functions in neural cells. Briefly, TCF3 (FPKM = 81) is a broadly expressed class I/E-protein bHLH factor (Ross et al. 2003); ASCL1 (FPKM = 16) is a potent inducer of neuronal differentiation that has recently been implicated in promoting NS cell proliferation (Castro et al. 2011; Andersen et al. 2014); OLIG2 (FPKM = 268) is implicated in driving NS cell proliferation and is also involved in context-dependent functions in oligodendrocyte and motor neuron specification (Meijer et al. 2012); and MAX (FPKM = 41) is a binding partner of the oncogenic MYC factors, which makes an important contribution to ES cell self-renewal (Rahl et al. 2010; Hishida et al. 2011). We also selected three SOX factors, each from different SOX factor subfamilies and all candidate regulators of NS cell fate (Sarkar and Hochedlinger 2013). SOX2 (FPKM = 116) is a known regulator of various classes of NS cells and non-NS cells (Liu et al. 2013), and SOX21 (FPKM = 27) is thought to predominantly act as a repressor that counteracts SOX1-3 function (Sandberg et al. 2005); SOX9 (FPKM = 54) has been linked to the establishment and maintenance of NS cell identity (Scott et al. 2010), although, like SOX21, its genomic binding targets have never been thoroughly described. We also obtained and reanalyzed published NS cell ChIP-seq data for the multifunctional transcriptional regulators CTCF (FPKM = 54) and SMC1A (FPKM = 64), a cohesin complex subunit (Phillips-Cremins et al. 2013), and also for FOXO3 (FPKM = 6), since it is a known regulator of NS cell homeostasis and therefore is likely to have important input to the NS cell GRN (Paik et al. 2009; Renault et al. 2009; Webb et al. 2013). FOXO3 ChIP-seq was conducted in mitogen-inhibited cells in order to induce nuclear localization of this TF.
We used the MACS2 peak caller (Zhang et al. 2008) to generate genome-wide maps of robust and specific binding sites for each TF (Supplemental Table S3; Supplemental Fig. S5). At least 80% of all distal CREs and 60% of all proximal CREs overlapped binding sites for at least one of our selected TFs (Fig. 5A), and frequently multiple different TF peaks mapped to the same CRE (Fig. 5B). Active enhancer regions were particularly densely bound, with an average co-occupancy of five different TFs and 468 sites showing binding of nine or more of the 11 TFs studied here. Thus, even the narrow genomic windows identified by DNase-seq frequently contain multiple binding sites for TFs belonging to distinct TF families. Of the distal CREs, the regions lacking histone modifications tended to be bound by fewer distinct factors (average < 2 TFs) (Fig. 5B). Proximal CREs were also less frequently bound by the set of TFs studied here, which was expected since the selection was based on factors whose motifs were more enriched in distal CREs. The exception to this was poised proximal cluster 3, which, as well as exhibiting an enhancer-like chromatin signature, was frequently bound by more than three of the TFs studied (Fig. 5B).
Figure 5.
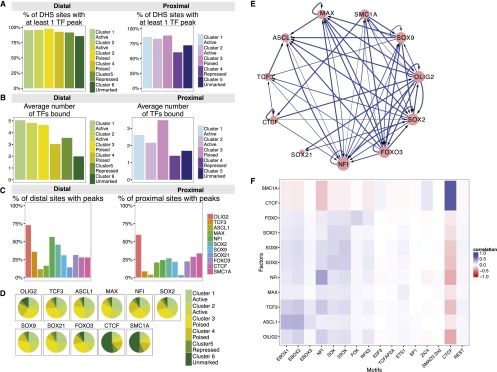
TFs whose motifs are enriched in distal enhancers show distinct patterns of CRE-binding. (A) Percentage of DHSs belonging to each cluster that contains a significant ChIP-seq binding peak for one or more of the 11 TFs studied here. (B) The mean number of different TFs bound to each class of DHS elements. (C) Percentage of distal and proximal DHSs that are bound by each TF. (D) Distribution of each factor's binding peaks within the different classes of distal DHSs. The majority of TF binding peaks are found in the more active (cluster 1 to 3) regions for all factors, apart from CTCF and SMC1A, which more frequently bind unmarked distal regions. (E) Network graph showing the predicted regulatory interactions between the key TFs analyzed in this study. Each arrow represents promoter-proximal binding from our ChIP-seq data. (F) Pairwise matrix showing the Pearson correlation coefficients between TF peaks (y-axis) and motifs (x-axis) within each distal DHSs.
The different TFs had remarkably different patterns of binding across the different classes of CRE, indicating that they have different regulatory functions (Fig. 5C,D). The bHLH factor OLIG2 bound promiscuously across an average of 75% of all distal elements and nearly 60% of all proximal elements. NFI factors were also very widely bound, particularly within the five distal enhancer CRE classes, suggestive of important enhancer-dependent functions of these TFs in NS cells. SOX2, SOX9, TCF3, and FOXO3 all bound fewer distal CREs overall and were more likely to bind within the more active classes of distal enhancer (distal clusters 1–3, Fig. 5D). ASCL1, SOX21, and MAX all bound an even smaller number of distal CREs in total and, when they were present, were most likely to be within active distal enhancers. Additionally, the bHLH factor MAX was the only TF that bound a greater proportion of proximal than distal elements and was found in ∼30% of both classes of active promoter elements (Fig. 5C). In contrast to the other factors, CTCF and SMC1A were observed to bind to only a small proportion of NS cell enhancers overall, as reported previously (Phillips-Cremins et al. 2013), but to nearly 60% of unmarked distal elements and nearly 40% of all unmarked proximal promoters (Fig. 5D). This binding pattern fits with the enrichment of CTCF motifs and suggests that many such unmarked regions might function as transcriptional insulators and/or topological domain boundaries whose chromatin modification patterns have been poorly characterized (Wang et al. 2012). Less expected was the observation that around a third of repressed distal enhancers and nearly 40% of repressed promoters contained CTCF and SMC1A binding, potentially implicating these factors in the repressive activity of certain CREs in NS cells (Fig. 5C).
To gain insights into the topology of the core network of TFs, we examined the patterns of cross-regulation among the 11 TFs analyzed by ChIP-seq. The resulting network graph was highly interconnected and 7/11 factors appear to auto-regulate, both features of a robust transcriptional network (Fig. 5E).
Widespread motif-independent binding of TFs in CREs
The frequency of TF binding within CREs (Fig. 5C) often exceeded the motif frequency within individual DHSs (Fig. 4B,C). Therefore, we examined the correspondence between each factor’s binding within distal CREs and the presence of each CRE-enriched motif (Fig. 5F). CTCF and SMC1A demonstrated a very strong correlation between TF and CTCF motif presence, suggesting robust motif-dependent binding of these factors. For the NFI, SOX, and FOX TFs, the strongest positive factor/motif correlations observed were also for each TF’s predicted motif (or the 2Sox motif in the case of all three SOX factors); however, the correlation coefficients tended to be fairly small, indicating that a substantial proportion of each TF’s binding is not mediated by that factor’s cognate motif. Apart from ASCL1, which correlated well with E-boxes 1 and 2, this trend was even more marked for the bHLH factors. MAX showed little preference for any of the motifs studied, while TCF3 and OLIG2 were actually slightly better correlated with the NFI motif than with the E-box motifs, indicating that in some cases NFI factors may recruit or stabilize binding of bHLH factors to CREs. Interestingly, all non-CTCF/cohesin factors were anti-correlated with the CTCF motif, suggesting strong avoidance of those sequences by this group of TFs.
Expressed TFs whose motifs are enriched in CREs do indeed exhibit widespread binding in epigenomically defined CREs. However, there is no precise one-to-one mapping between the presence of TF motifs and binding sites for the cognate factor; instead, most factors are recruited to a large portion of their binding sites less directly, presumably via their contribution to multi-TF, enhancer-associated complexes, as has been observed in several other cellular systems (Biggin 2011; Ernst and Kellis 2012; Kvon et al. 2012).
Mathematical modeling highlights the most important genomic features regulating genes required for NS cell self-renewal, quiescence, and differentiation
Our characterization of 11 different classes of CRE, each containing different combinations of TF motifs and each populated by binding sites for the 11 different TFs, yields a wealth of genomic features, each of which could potentially be predictive of genes with different functions in NS cells. We used a machine learning approach, with the goal to identify, in an unbiased and data-driven manner, the regulatory features most predictive of genes required for NS cell self-renewal, differentiation, and quiescence.
To this end, we focused on the sets of genes whose expression is regulated when we induced glial or neuronal differentiation, or quiescence (Fig. 3A,B; Supplemental Figs. S1, S3; Supplemental Table S2). We reasoned that genes down-regulated in any of these three conditions were more likely required for active self-renewal of NS cells and that up-regulated genes would have functions in differentiation and/or quiescence. We then used a logistic regression framework (see Methods) to build models that we could use to classify genes as NS cell self-renewal genes versus differentiation or quiescence genes according to the presence of CREs with distinct TF occupancy and motif presence in their regulatory domains (Fig. 6A).
Figure 6.

Modeling highlights genomic features predictive of genes regulated when NS cells exit self-renewal. (A) The occurrence of different proximal and distal DHS classes, with different motif matches and factor peaks, within the regulatory domains of genes is tabulated and used to generate logistic regression models to classify genes as up- or down-regulated when NS cells are differentiated or enter quiescence. (B) Heatmap of the model coefficients for each of the genomic features used (x-axis) in each cellular state (y-axis). Coefficients contributing to predictions of up-regulated genes are shown in shades of blue; for down-regulated genes, in shades of red. (C–E) Top 15 coefficients predictive of genes up- (blue) or down-regulated (red) in the individual models predicting genes responsive to neuronal (C) or astrocyte (D) differentiation, or to quiescence (E). Asterisks denote the features highlighted in the text. Note that transcription factor names are in uppercase for better differentiation from the motif names (first letter capitalized).
We determined the accuracy of our models using the area under a receiver-operator curve (AUC) in a cross-validation scheme (see Methods). For all three of the microarray experiments, our models showed an accuracy of at least 0.65 at classifying genes as down-regulated self-renewal genes or up-regulated differentiation/quiescence genes, representing a 30% improvement over random classification (Fig. 6C–E). The model coefficients for each of the 65 genomic features considered and their predictive value for up- and down-regulated gene sets are shown in Figure 6B.
Two features were most predictive of genes associated with self-renewal and down-regulated in neuronal or glial differentiation or in quiescence. These were the presence of cluster 2 active promoters and the promoter-proximal binding of the TF MAX (Fig. 6B–E, green asterisks). Given the association of proximal cluster 2 with cell cycle regulators (Supplemental Fig. S4), this finding was expected and helps to validate our method. Interestingly, active proximal cluster 1–associated genes, which are also enriched for cell cycle regulators (Supplemental Fig. S4), were not predictive of self-renewal genes down-regulated in differentiation and quiescence, again supporting our segregation of these two different promoter types. The linking of MAX to self-renewal genes suggests that, as in ES cells (Rahl et al. 2010; Hishida et al. 2011), this TF promotes active stem cell self-renewal, presumably in collaboration with the MYC factors.
Further validation of our model came from the strong predictive value of repressed cluster 4 promoters for genes up-regulated in neuronal differentiation (Fig. 6C, purple asterisks), as this was also revealed by our previous GO analysis (Supplemental Fig. S4). Less expected was the association of double SOX motifs and proximal SOX2 binding with neuronal genes. It will be interesting to determine how SOX2 and other SOX factors regulate this class of neuron-associated genes.
Regarding the model for astrocytic differentiation, repressed proximal cluster 4–associated genes were also predicted to be up-regulated, but so too were the poised (proximal cluster 3) and unmarked (proximal cluster 5) promoters (Fig. 6D, brown asterisks). These last two groups of promoters were also predictive of genes up-regulated when NS cells enter quiescence (Fig. 6E). Therefore, genes with promoters in less active or repressed chromatin states but which possess significant DHSs in self-renewing NS cells, appear to be bookmarked for activation upon astrocytic differentiation or entry to quiescence.
All three models predicted involvement of FOXO TFs, via either the binding of FOXO3 itself or the presence of FOX motifs, in the activation of genes in neuronal and glial differentiation and in quiescence (Fig. 6C–E, orange asterisks). While FOXO factors’ role in promoting NS cell quiescence has been described (Paik et al. 2009; Renault et al. 2009), its precise involvement in glial and neuronal differentiation has been much less explored (Webb et al. 2013). We were also interested to observe that proximal binding of OLIG2 contributed to predictions of genes up-regulated in both quiescence and neuronal lineage commitment. Therefore, despite the very widespread binding of OLIG2 in NS cell CREs (Fig. 5C), our modeling strategy predicts specific regulatory functions for a subset of OLIG2 binding sites in controlling differentiation- and quiescence-associated genes.
Multiple roles for the TF OLIG2 in the maintenance of NS cell self-renewal
Due to its very widespread binding across almost all classes of distal and proximal CREs, and because our mathematical modeling approach indicated that some OLIG2 binding sites were predictive of genes associated with neuronal differentiation and quiescence, we explored the function of this TF in our cultured NS cells more directly. We derived NS cells from the ventral forebrain of embryonic day 16 mice homozygous for a conditional mutant allele of Olig2 (Cai et al. 2007). These cells had normal growth parameters and had a normal NS cell marker gene expression profile (data not shown). Upon adenoviral delivery of Cre recombinase, we were able to induce complete depletion of Olig2 transcripts, total protein, and DNA-bound protein within 48 h (Fig. 7A,G; Supplemental Fig. S6). Olig2-deleted cells remained undifferentiated according to morphological criteria and continued to proliferate, but at a reduced rate compared to control virus-transduced cells, as measured by EdU incorporation (Fig. 7B). We used microarrays to detect genes transcriptionally regulated by OLIG2 48 h after delivery of Cre (Supplemental Table S2). Deletion of Olig2 resulted in significant down-regulation of 616 genes (Fig. 7C), of which 558, or 90.6%, a highly significant proportion (P = 6 × 10−23, hypergeometric test), were associated with OLIG2 binding sites within one or more of the CREs defined in this study, suggesting that OLIG2 acts as a transcriptional activator for some of its functions in NS cells. OLIG2-activated genes were strongly enriched in categories relating to the “cell cycle,” “chromosome,” and “DNA replication” (Fig. 7D).
Figure 7.

OLIG2 is a multifunctional regulator of NS cell self-renewal. (A) Immunostaining shows the rapid and complete loss of OLIG2 protein from Olig2-conditional mutant NS cells following administration of Cre. (B) Reduced proliferation of Olig2-mutant NS cells 48 h after Cre delivery, as measured by 3 h exposure to EdU (P-value < 0.008 Wilcoxon test). (C) Venn diagram showing the large and significant (hypergeometric test) proportion of genes deregulated in Olig2-deleted cells that are associated with OLIG2-bound DHSs. (D,E) GO biological processes enriched among the genes down- (D) and up-regulated (E) by Olig2-deletion. DAVID P-values are shown. (F) Olig2-mutant cells normally arrest their proliferation when treated with quiescence-inducing medium, but are less likely to re-enter the cell cycle when returned to self-renewal medium, as measured by 3 h of EdU exposure (P-value < 0.008, Wilcoxon test). (G) Reduced expression of cell cycle regulators and active NS cell marker EGFR and inappropriate expression of quiescence-associated genes in mutant cells that have been stimulated to resume proliferation, as measured by quantitative RT-PCR. (H) Top 15 model coefficients predictive of genes up- (blue) or down-regulated (red) in the models predicting genes responsive to elimination of OLIG2 protein.
Seven hundred sixty genes were up-regulated in the absence of Olig2, a substantial and significant 93% of which (707 genes, P = 7 × 10−39) were associated with OLIG2-bound CREs (Fig. 7C), suggesting that OLIG2 also acts as a transcriptional repressor in NS cells. OLIG2-repressed genes were strongly enriched for gene ontology terms relating to neuronal differentiation (“neuron projection,” “neurogenesis,” “neuron projection development,” and “synapse organization”) (Fig. 7E). We were able to validate the up-regulation of multiple neuronal markers and neurogenic factors by qPCR (Supplemental Fig. S6). Therefore, part of Olig2’s function in self-renewing NS cells is to repress premature activation of the neurogenic gene expression program. This validates one of the predictions of our mathematical model: that proximal OLIG2 binding is predictive of genes up-regulated in neuronal progenitors.
Our model also showed that a subset of OLIG2 binding sites within proximal CREs was predictive of genes up-regulated during NS cell quiescence (Fig. 6E). We tested this directly by exploring whether Olig2 was required for NS cells to transition into quiescence or to re-enter the cell cycle from a quiescent state. Following exposure to quiescence-inducing medium (BMP4 + FGF2) (Martynoga et al. 2013), Olig2 mutant cells exited the cell cycle at the same rate as the controls and up-regulated the quiescent NS cell marker Gfap (Fig. 7F; data not shown). However, when stimulated to resume proliferation, the mutant cells incorporated EdU at a much lower rate than control cells (Fig. 7F), failed to up-regulate cell cycle regulators (e.g., Ccne2, Foxm1, and E2f2) and the activated stem cell marker Egfr, and failed to repress expression of quiescence-associated genes Anxa2, Cetn4, Gfap, and Id1 (Fig. 7G). Therefore, as well as activating proliferation genes, OLIG2 appears to repress quiescence genes. Consistently, OLIG2 binds and represses a substantial fraction of quiescence genes in self-renewing NS cells. Nine hundred thirty-two of 1854 (50.3%, P = 1.2 × 10−107, hypergeometric test) genes normally induced in quiescent NS cells had an OLIG2-bound CRE in self-renewing NS cells and 382 of the 760 (50.3%, P = 1.3 × 10−42, hypergeometric test) genes aberrantly up-regulated by OLIG2 depletion from self-renewing cells were genes normally only induced in quiescence and therefore were derepressed.
We used our logistic regression paradigm to identify genomic features that can discriminate between genes activated or repressed by OLIG2 in self-renewing NS cells (Fig. 7H). Proximal SOX9, MAX, and SP1 regulation were associated with activating functions of OLIG2, while repressed CREs and SOX21, SOX2, and FOXO3 binding were most strongly predictive of OLIG2-repressed genes. Altogether, these experimental data indicate that despite very widespread binding, OLIG2 has very specific functions in self-renewing NS cells and its deletion results in very specific cellular defects. Therefore, OLIG2 binding sites in different regulatory contexts can have very different functions.
Discussion
Identifying the repertoire of CREs controlling NS cell self-renewal
As an important first step toward identifying the TF regulators of NS cell self-renewal, we first charted the landscape of TF-accessible CREs in the NS cell genome. We married the discriminatory power of combinatorial analysis of histone modifications with the spatial resolution of DNase-seq to identify multiple epigenetically distinct classes of CRE.
CREs, both proximal and distal to gene TSSs, exist in a spectrum of different activity states. In particular, we observe distal enhancers in five distinct epigenetically encoded activity states, which associate with different functional classes of genes that are expressed at different levels. This is generally in agreement with other recent studies in other cellular systems (Creyghton et al. 2010; Rada-Iglesias et al. 2010; Zentner et al. 2011). We extend this insight by showing strong coassociations between distal and proximal elements that are in matched activity states. Several of the proximal CREs that we define have the characteristics of super-enhancers (Whyte et al. 2013). These regions consisted of clusters of DHS sites and overlapped almost exclusively with our active proximal clusters types. We could not find clear functional distinctions between active regions with and without super-enhancer features (data not shown).
We also characterize a set of TSS-proximal CREs with a histone signature more akin to an active distal enhancer than an active promoter, and propose that this is a novel class of poised promoters. A similar poised promoter signature has recently been reported at a subset of promoters in mouse ES cells that becomes active during cardiomyocyte differentiation (Wamstad et al. 2012), and promoters with the same H3K4me1+/H3K4me3− signature have recently been linked to transcriptional repression in a range of cell types (Cheng et al. 2014). In contrast to ES cells where many lineage-specific genes are marked by a bivalent H3K4me3+/H3K27me3+ (Bernstein et al. 2006), we do not observe a coherent group of promoters with this pattern in NS cells, suggesting that these cells tend not to use this mechanism to poise genes for later expression.
The core set of TFs regulating NS cell enhancers
Key to our CRE identification approach was the tight definition of DHS regions, which directed our motif analysis to the sites of maximal regulatory complex binding within CREs. Echoing our previous analysis of a specific class of active enhancers in self-renewing and quiescent NS cells (Martynoga et al. 2013), we see clear enrichment of NFI, bHLH, and SOX motifs within the NS cell enhancers defined in this study. We now show that these same motifs are also enriched in poised and repressed enhancers, and we add to this set of NS cell enhancer-specific motifs a TCFAP2A motif and also a novel motif consisting of a pair of oppositely oriented SOX motifs. This latter motif has recently been reported to be enriched in conserved noncoding elements of the human genome, suggesting that cobinding of SOX dimers may be an evolutionary ancient method to achieve binding specificity for these factors, which as monomers recognize a short motif that appears very frequently in the genome (Guturu et al. 2013). We were unable to identify motifs that completely distinguished enhancers with different activities, suggesting that an overlapping set of TFs regulates them all and that the sequence rules determining different activities are subtle and combinatorial and cannot be deciphered with the approach used here.
Machine learning reveals key regulatory features of self-renewal, differentiation, and quiescence genes
To begin to reveal the regulatory logic of CREs with defined motif content and TF binding, we used a modeling approach to learn which of these genomic features are predictive of genes that are up- or down-regulated when NS cells exit self-renewal. We found that a logistic regression classifier performed as well as more complex approaches, such as support vector machines (SVMs), and yielded more easily interpretable and biologically meaningful output. Our approach achieved good classification accuracy and yielded several important and unexpected predictive regulatory features worthy of further investigation.
Understanding and modeling how different CREs influence gene expression remains one of the biggest challenges in biology. Several recent studies have used modeling approaches to predict gene expression levels from genomic features within a single cell type (Karlic et al. 2010; Cheng et al. 2011; Dong et al. 2012). Some of these models achieve very high prediction accuracy, although they tend to focus on promoter proximal CREs, or at least give preference to these elements, and do not give full weight to more distal elements, as we have tried to do here. Other studies have set out to identify the genes expressed in different cell types or tissues based on CREs (Ouyang et al. 2009; Cheng et al. 2012; Natarajan et al. 2012; Wilczynski et al. 2012). Our approach is conceptually similar to these latter methods; with the key difference that we explain more subtle changes, namely, the differentiation or quiescence process in one cell type and its descendants, instead of gross differences between very diverse cell states.
OLIG2 is a multifunctional regulator of NS cell self-renewal
To test key predictions of our modeling strategy, we acutely deleted Olig2 from NS cells and showed that from the huge number of OLIG2-bound CREs, this TF has rather specific primary functions in proliferating NS cells. OLIG2 contributes to the maintenance of NS cell self-renewal by binding sets of genes associated with neuronal differentiation and quiescence and preventing their untimely induction. OLIG2 also appears to directly activate another set of genes, including several core cell cycle regulators, and by this means promotes NS cell proliferation. The contribution of Olig2 to the proliferation of both normal and malignant neural progenitors has been described previously, although this work has focused on OLIG2’s antagonism of Cdkn1a (also OLIG2-bound here) and the p53 pathway (Ligon et al. 2007; Mehta et al. 2011; Sun et al. 2011b), rather than the novel functions in directly inducing cell cycle activators and repressing quiescence genes that we describe here. It will be interesting to determine how previously described post-translational mechanisms such as phosphorylation of key residues of OLIG2 (Li et al. 2011a; Sun et al. 2011b) and interactions with TRP53 protein (Mehta et al. 2011) combine with chromatin-level epigenetic and co-factor-mediated mechanisms described here to explain how OLIG2 can be directed toward such context-specific functions to both activate and repress target gene expression.
In summary, our rich new compendium of TF binding data, motif analysis, and CRE annotations in NS cells represents a big step toward a full understanding of the structure and dynamics of the self-renewing NS cell GRN. This work was conducted in ES cell–derived NS cell cultures. Equivalent data sets are lacking from NS cells from other in vivo and in vitro sources, but we are confident that the detailed regulatory annotations reported and interpreted here are more broadly applicable. As well as providing a resource for the research community, we are optimistic that our strategy to define functional CREs and to home in on the critical TF regulators in well-defined and disease-relevant cell types such as NS cells will also have great utility in the development of new therapeutic tools. For example, high-quality CRE annotation and CRE-regulator identification are crucial in focusing attention on human genetic variants that are located in functional regulatory elements and therefore are more likely to be causally relevant to pathological phenotypes (Schaub et al. 2012; Weedon et al. 2013).
Methods
NS cell culture
ES cell–derived NS5 NS cells were cultured in Euromed-N medium (Oxford Biosystems Cadama) with N2, FGF-2, and EGF (both 10 ng/mL) supplements according to standard methods (Conti et al. 2005) with the following minor modification: Cells were plated onto uncoated tissue culture plastic with the addition of laminin (Sigma) at 2 μg/mL to the medium. Conditional mutant Olig2 NS cells were derived from the ventral telencephalon of embryonic day 16 Olig2flox/flox mice (a kind gift of Richard Lu) (Cai et al. 2007) as described previously (Conti et al. 2005) and cultured in identical conditions as NS5 cells. Olig2 was deleted by delivery of Cre recombinase-expressing adenoviruses at an MOI of 20–40 (Vector Biolabs). OLIG2 protein was detected by immunostaining with a rabbit anti-OLIG2 antibody (Millipore, 1:500).
DNase-seq
Nuclei for DNase I treatment were isolated from 20 million cells. After cell lysis and chromatin purification, chromatin was incubated with 4 units DNase I for 10 min at 37°C.
Pulse field gel electrophoresis was performed to verify that the nuclei were fragmented to a desired fragment size of < 500 bp. DNase-seq libraries were generated as previously described (Boyle et al. 2008; Song and Crawford 2010) with a slight modification made to the linkers to increase ligation efficiency (Song et al. 2011). Libraries were sequenced on the Genome Analyzer IIx (Illumina).
Chromatin immunoprecipitation
NS cells were fixed sequentially with di(N-succimidyl) glutarate and 1% formaldehyde in phosphate-buffered saline and then lysed, sonicated, and immunoprecipitated as described previously (Castro et al. 2011) using material from ∼5 × 106 cells per sample. All antibodies used had all been previously used for ChIP and validated for their specificity. Immunoprecipitations were with mouse anti-ASCL1 (BD Pharmingen) (Castro et al. 2006), rabbit anti TCF3 (Santa Cruz, sc-349) (Lin et al. 2010), rabbit anti-OLIG2 (Millipore, AB9610) (Mazzoni et al. 2011), rabbit anti-MAX (Santa Cruz, sc-197) (Rahl et al. 2010), goat anti-NFI (Santa Cruz, sc-30918) (Pjanic et al. 2011; Martynoga et al. 2013), goat anti-SOX2 (Santa Cruz sc-17320) (Chen et al. 2008), rabbit anti-SOX9 (Millipore, AB5535) (Mead et al. 2013), and goat anti-SOX21 (R&D systems, AF3538) (Matsuda et al. 2012). Primers used for ChIP-PCR are listed in Supplemental Table S5. ChIP-seq data generation and analysis are described in the Supplemental Methods.
Computational analysis
We used R (R Core Team 2014) and Bioconductor for all computational analysis, unless otherwise stated. Full details of all analyses conducted are provided in the Supplemental Methods.
Motif analysis
To identify motifs overrepresented in the different genomic regions, we used three tools: MEME (Bailey and Elkan 1994) and RSAT peak-motifs (Thomas-Chollier et al. 2012) with 400 bases as input centered on the peak summit; and CentriMo (Bailey and Machanick 2012) with 2000 bases as input centered on the peak summit. Further details are provided in the Supplemental Methods.
Generation and analysis of microarray and RNA-seq data
For microarray analysis, total RNA from three biological replicates per condition was TRIzol extracted (Life Technologies, column purified [Qiagen] and hybridized to Illumina MouseRef-8 v2.0 expression BeadChips according to the manufacturer’s specifications). Normalization and statistical analysis were carried out with GeneSpring software (Agilent). Probes were reannotated (Barbosa-Morais et al. 2010), collapsed by gene, and considered regulated if there was ≥ 1.5-fold differential expression with Benjamini-Hochberg-corrected P-value < 0.05 (t-test). For RNA-seq, the sequencing library was prepared according to the TruSeq RNA sample preparation v2 protocol (Illumina) and sequenced on an Illumina HiSeq 2000. We obtained a total of 114 million 80-bp single end reads. We filtered out the first 9 bases of all reads using FASTX-toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/). We then mapped the filtered reads to the mm9 mouse genome using TopHat (Trapnell et al. 2012) version 2.0.9 (with Bowtie version 2.1.0). Next we used Cufflinks (Trapnell et al. 2012) version 2.1.1 to estimate expression level of the genes defined in Ensembl version 61, specifying in addition the following parameters: –frag-bias-correct–upper-quartile-norm –multi-read-correct.
Logistic regression modeling
For the task of predicting whether a gene will be up- or down-regulated in our three differentiation microarrays, we considered different levels of information: histone modifications, as encoded by the different types of clusters of DHS; motif matches within the DHS peaks; and factor peaks overlapping the DHS peaks. Each of these three types of features was divided into proximal and distal groups according to the DHS peaks. As depicted in Figure 6A, for each gene regulated in each array we tabulated this information, in a binary way, as the presence or absence of each feature in the regulatory domain of the gene (Supplemental Table S4). Each gene was labeled as up or down depending on the direction of regulation on the array, and this was the target variable for the classification task. For the classifier, we used a logistic regression paradigm from the data mining suite WEKA version 3.7.7 (Hall et al. 2009), specifically the implementation called SimpleLogistic. The performance of the model was measured in a cross-validation scheme with 10 folds and using the AUC statistic.
Data access
DNase-seq, ChIP-seq, and RNA-seq data generated in this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under accession numbers ERP004671, ERP004644, and ERP004633, respectively. The data are also available via ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession numbers E-MTAB-2270, E-MTAB-2228, and E-MTAB-2230. Processed high-throughput sequencing data can be visualized in our UCSC Genome Browser track hub: http://genome.ucsc.edu/cgi-bin/hgTracks?db=mm9&hubUrl=http://www.nimr.mrc.ac.uk/trackhub/cisstemhub/hub.txt.
Supplementary Material
Acknowledgments
We thank Abdul Sesay and the NIMR high-throughput sequencing facility for expert assistance with ChIP-seq and microarrays. We thank Cristina Minieri and Carole Hyacinthe for help with cloning and other members of the Guillemot laboratory for suggestions and comments on this work. We thank Q. Richard Lu for kindly providing Olig2 conditional mutant mice. This work was supported by a Small Collaborative Project Grant from the 7th Framework Programme of the European Commission (FP7-223210 to P.F., L.E., J.W., and F.G.), The Wellcome Trust (grant numbers WT095908 and WT098051 to P.F.), a FEBS Long-Term Fellowship to D.v.d.B., and a Grant-in-Aid from the Medical Research Council (U117570528 to F.G.).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.173435.114.
References
- Andersen J, Urbán N, Achimastou A, Ito A, Simic M, Ullom K, Martynoga B, Lebel M, Göritz C, Frisén J, et al. 2014. A transcriptional mechanism integrating inputs from extracellular signals to activate hippocampal stem cells. Neuron 83: 1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- Bailey TL, Machanick P. 2012. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40: e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Dunning MJ, Samarajiwa SA, Darot JF, Ritchie ME, Lynch AG, Tavare S. 2010. A re-annotation pipeline for Illumina BeadArrays: improving the interpretation of gene expression data. Nucleic Acids Res 38: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. [DOI] [PubMed] [Google Scholar]
- Biggin MD. 2011. Animal transcription networks as highly connected, quantitative continua. Dev Cell 21: 611–626. [DOI] [PubMed] [Google Scholar]
- Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ, Ming G-l, Song H. 2011. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczyński B, Riddell A, Furlong EEM. 2012. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat Genet 44: 148–156. [DOI] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. 2008. High-resolution mapping and characterization of open chromatin across the genome. Cell 132: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracko O, Singer T, Aigner S, Knobloch M, Winner B, Ray J, Clemenson GD, Suh H, Couillard-Despres S, Aigner L et al. 2012. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J Neurosci 32: 3376–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Chen Y, Cai W-H, Hurlock EC, Wu H, Kernie SG, Parada LF, Lu QR. 2007. A crucial role for Olig2 in white matter astrocyte development. Development 134: 1887–1899. [DOI] [PubMed] [Google Scholar]
- Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, Critchley JA, Nguyen L, Gossler A, Gottgens B, et al. 2006. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell 11: 831–844. [DOI] [PubMed] [Google Scholar]
- Castro DS, Martynoga B, Parras C, Ramesh V, Pacary E, Johnston C, Drechsel D, Lebel-Potter M, Garcia LG, Hunt C, et al. 2011. A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev 25: 930–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117. [DOI] [PubMed] [Google Scholar]
- Cheng C, Yan KK, Yip KY, Rozowsky J, Alexander R, Shou C, Gerstein M. 2011. A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets. Genome Biol 12: R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Alexander R, Min R, Leng J, Yip KY, Rozowsky J, Yan KK, Dong X, Djebali S, Ruan Y, et al. 2012. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res 22: 1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Blum R, Bowman C, Hu D, Shilatifard A, Shen S, Dynlacht BD. 2014. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell 53: 979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying Q-L, Cattaneo E, et al. 2005. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 3: e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH. 2010. Emerging properties of animal gene regulatory networks. Nature 468: 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Greven MC, Kundaje A, Djebali S, Brown JB, Cheng C, Gingeras TR, Gerstein M, Guigo R, Birney E, et al. 2012. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol 13: R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2012. Interplay between chromatin state, regulator binding, and regulatory motifs in six human cell types. Genome Res 22: 1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. 2013. Ensembl 2013. Nucleic Acids Res 41: D48–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba LC, Obernier K, Alvarez-Buylla A. 2012. Adult neural stem cells bridge their niche. Cell Stem Cell 10: 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guturu H, Doxey AC, Wenger AM, Bejerano G. 2013. Structure-aided prediction of mammalian transcription factor complexes in conserved non-coding elements. Philos Trans R Soc Lond B Biol Sci 368: 20130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH. 2009. The WEKA data mining software: an update. SIGKDD Explor Newsl 11: 10–18. [Google Scholar]
- Hawkins RD, Hon GC, Yang C, Antosiewicz-Bourget JE, Lee LK, Ngo Q-M, Klugman S, Ching KA, Edsall LE, Ye Z, et al. 2011. Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res 21: 1393–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Calcar SV, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishida T, Nozaki Y, Nakachi Y, Mizuno Y, Okazaki Y, Ema M, Takahashi S, Nishimoto M, Okuda A. 2011. Indefinite self-renewal of ESCs through Myc/Max transcriptional complex-independent mechanisms. Cell Stem Cell 9: 37–49. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. 2010. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci 107: 2926–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. 2009. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32: 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvon EZ, Stampfel G, Yáñez-Cuna JO, Dickson BJ, Stark A. 2012. HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev 26: 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, de Faria JP, Andrew P, Nitarska J, Richardson WD. 2011a. Phosphorylation regulates OLIG2 cofactor choice and the motor neuron-oligodendrocyte fate switch. Neuron 69: 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Thomas S, Sabo PJ, Eisen MB, Stamatoyannopoulos JA, Biggin MD. 2011b. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol 12: R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, Bachoo RM, Kane M, Louis DN, Depinho RA, et al. 2007. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 53: 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. 2010. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11: 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lin B, Zhao M, Yang X, Chen M, Gao A, Liu F, Que J, Lan X. 2013. The multiple roles for Sox2 in stem cell maintenance and tumorigenesis. Cell Signal 25: 1264–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. 2008. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132: 958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, Bailey TL. 2011. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27: 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B, Mateo JL, Zhou B, Andersen J, Achimastou A, Urban N, van den Berg D, Georgopoulou D, Hadjur S, Wittbrodt J, et al. 2013. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev 27: 1769–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Kuwako K, Okano HJ, Tsutsumi S, Aburatani H, Saga Y, Matsuzaki Y, Akaike A, Sugimoto H, Okano H. 2012. Sox21 promotes hippocampal adult neurogenesis via the transcriptional repression of the Hes5 gene. J Neurosci 32: 12543–12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Iacovino M, Morrison CA, Mountoufaris G, Closser M, Whyte WA, Young RA, Kyba M, Gifford DK, et al. 2011. Embryonic stem cell-based mapping of developmental transcriptional programs. Nat Methods 8: 1056–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead TJ, Wang Q, Bhattaram P, Dy P, Afelik S, Jensen J, Lefebvre V. 2013. A far-upstream (−70 kb) enhancer mediates Sox9 auto-regulation in somatic tissues during development and adult regeneration. Nucleic Acids Res 41: 4459–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, Alberta JA, Kane MF, Theisen M, Ligon KL, et al. 2011. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell 19: 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer DH, Kane MF, Mehta S, Liu H, Harrington E, Taylor CM, Stiles CD, Rowitch DH. 2012. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat Rev Neurosci 13: 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U. 2012. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res 22: 1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka T, Shimojo H, Matsunaga M, Watanabe N, Kometani K, Minato N, Kageyama R. 2011. Gene expression profiling of neural stem cells and identification of regulators of neural differentiation during cortical development. Stem Cells 29: 1817–1828. [DOI] [PubMed] [Google Scholar]
- Ouyang Z, Zhou Q, Wong WH. 2009. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proc Natl Acad Sci 106: 21521–21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J-H, Ding Z, Narurkar R, Ramkissoon S, Muller F, Kamoun WS, Chae S-S, Zheng H, Ying H, Mahoney J, et al. 2009. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 5: 540–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, et al. 2013. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pjanic M, Pjanic P, Schmid C, Ambrosini G, Gaussin A, Plasari G, Mazza C, Bucher P, Mermod N. 2011. Nuclear factor I revealed as family of promoter binding transcription activators. BMC Genomics 12: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org.
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2010. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, Mccuine S, Burge CB, Sharp PA, Young RA. 2010. c-Myc regulates transcriptional pause release. Cell 141: 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault VM, Rafalski VA, Morgan AA, Salih DAM, Brett JO, Webb AE, Villeda SA, Thekkat PU, Guillerey C, Denko NC, et al. 2009. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5: 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SE, Greenberg ME, Stiles CD. 2003. Basic helix-loop-helix factors in cortical development. Neuron 39: 13–25. [DOI] [PubMed] [Google Scholar]
- Sandberg M, Källström M, Muhr J. 2005. Sox21 promotes the progression of vertebrate neurogenesis. Nat Neurosci 8: 995–1001. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Hochedlinger K. 2013. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 12: 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. 2012. Linking disease associations with regulatory information in the human genome. Genome Res 22: 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, Gomez Gaviro M-V, Booth S, Gao B, Cheah KSE, Lovell-Badge R, et al. 2010. SOX9 induces and maintains neural stem cells. Nat Neurosci 13: 1181–1189. [DOI] [PubMed] [Google Scholar]
- Song L, Crawford GE. 2010. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc 2010: pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, et al. 2011. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res 21: 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiliotopoulos D, Goffredo D, Conti L, Di Febo F, Biella G, Toselli M, Cattaneo E. 2009. An optimized experimental strategy for efficient conversion of embryonic stem (ES)-derived mouse neural stem (NS) cells into a nearly homogeneous mature neuronal population. Neurobiol Dis 34: 320–331. [DOI] [PubMed] [Google Scholar]
- Spitz F, Furlong EEM. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626. [DOI] [PubMed] [Google Scholar]
- Sun Y, Hu J, Zhou L, Pollard SM, Smith A. 2011a. Interplay between FGF2 and BMP controls the self-renewal, dormancy and differentiation of rat neural stem cells. J Cell Sci 124: 1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Meijer DH, Alberta JA, Mehta S, Kane MF, Tien A-C, Fu H, Petryniak MA, Potter GB, Liu Z. 2011b. Phosphorylation state of Olig2 regulates proliferation of neural progenitors. Neuron 69: 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple S. 2001. The development of neural stem cells. Nature 414: 112–117. [DOI] [PubMed] [Google Scholar]
- Thomas-Chollier M, Herrmann C, Defrance M, Sand O, Thieffry D, van Helden J. 2012. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res 40: e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, McKinsey GL, Pattabiraman K, Silberberg SN, Blow MJ, et al. 2013. A high-resolution enhancer atlas of the developing telencephalon. Cell 152: 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. 2012. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. 2012. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res 22: 1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AE, Pollina EA, Vierbuchen T, Urbán N, Ucar D, Leeman DS, Martynoga B, Sewak M, Rando TA, Guillemot F, et al. 2013. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep 4: 477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, Rodriguez-Segui SA, Shaw-Smith C, Cho CH, Allen HL, et al. 2013. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet 46: 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczynski B, Liu YH, Yeo ZX, Furlong EE. 2012. Predicting spatial and temporal gene expression using an integrative model of transcription factor occupancy and chromatin state. PLoS Comput Biol 8: e1002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Xiao S, Xin X, Song CX, Huang W, McDee D, Tanaka T, Wang T, He C, Zhong S. 2013. Spatiotemporal clustering of the epigenome reveals rules of dynamic gene regulation. Genome Res 23: 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.