

Highlights
-
•
X-linked female presenting with EDMD1 not explained by uneven X-inactivation.
-
•
First EDMD blood phenotype with highly lobulated lymphocytes in EDMD1 patient.
-
•
Found high incidence of spontaneous differentiation in cultured patient myoblasts.
-
•
Faster proliferation of emerin-null than emerin-positive EDMD1 patient myoblasts.
-
•
Loss of satellite cells from the above might explain EDMD pathology.
Keywords: Emery–Dreifuss muscular dystrophy, Emerin, EMD, Myoblast differentiation, X-inactivation
Abstract
Emery–Dreifuss muscular dystrophy (EDMD) is a neuromuscular disease characterized by early contractures, slowly progressive muscular weakness and life-threatening cardiac arrhythmia that can develop into cardiomyopathy. In X-linked EDMD (EDMD1), female carriers are usually unaffected. Here we present a clinical description and in vitro characterization of a mildly affected EDMD1 female carrying the heterozygous EMD mutation c.174_175delTT; p.Y59* that yields loss of protein. Muscle tissue sections and cultured patient myoblasts exhibited a mixed population of emerin-positive and -negative cells; thus uneven X-inactivation was excluded as causative. Patient blood cells were predominantly emerin-positive, but considerable nuclear lobulation was observed in non-granulocyte cells – a novel phenotype in EDMD. Both emerin-positive and emerin-negative myoblasts exhibited spontaneous differentiation in tissue culture, though emerin-negative myoblasts were more proliferative than emerin-positive cells. The preferential proliferation of emerin-negative myoblasts together with the high rate of spontaneous differentiation in both populations suggests that loss of functional satellite cells might be one underlying mechanism for disease pathology. This could also account for the slowly developing muscle phenotype.
1. Introduction
Emery–Dreifuss muscular dystrophy (EDMD) is a neuromuscular disease characterized by early contractures, slowly progressive muscular weakness and life-threatening cardiac arrhythmia that can develop into cardiomyopathy. Contractures affecting the elbows, Achilles tendons and post-cervical muscles usually occur as the first clinical manifestation [1]. EDMD is genetically variable and thus far ~50% of patients have been linked to EMD (STA), LMNA, FHL1, SYNE1, SYNE2, LUMA and SUN1 mutations [2]. Over 90% of linked patients have dominant mutations in LMNA [3] or recessive mutations in the X-chromosomal EMD gene [4]. The EMD gene encodes emerin, a 254 amino acid protein that is anchored in the nuclear envelope with a transmembrane span close to its C-terminus [5]. Most EMD mutations are predicted to cause loss of the protein, but missense mutations have also been reported. Out of 97 EMD mutations reported on http://www.umd.be only 6 mutations (affecting 5 codons) are missense mutations. Various reports have proposed that the disease results from defective emerin function affecting gene expression, cell proliferation and differentiation, or cellular susceptibility to mechanical stress damage [6].
Female carriers of EMD mutations are usually asymptomatic; however, cardiac involvement has been occasionally though rarely described [7]. In all thus far reported cases of symptomatic females the clinical manifestation has been associated with unequal X-inactivation. However, it is also possible that the phenotype in symptomatic carriers could be caused by modifying mutations similar to how modifying mutations have been previously shown to affect disease severity. For example, combinations of EMD and LMNA mutations [8] as well as EMD and DES (the gene encoding the muscle intermediate filament desmin) [9] can increase the severity of EDMD. Findings in tissue culture indicate that mutations in SUN1, SUN2 and SYNE1 also act as severity modifiers in muscular disease [10,11]. The remarkable intra- and inter-familial variability regarding onset and severity of EDMD [12–16] makes it likely that severity modifiers are frequently involved, possibly even involving mutations that on their own do not cause a noticeable phenotype.
Here we report a symptomatic female carrying an emerin mutation that has also been found in her affected father. We have excluded uneven X-inactivation as a causative factor, finding that the majority of muscle as well as blood cells express the emerin wild-type allele. This makes a modifying mutation likely and raises the question of the contribution of each mutation to the disease. Nonetheless, analysis of the growth and differentiation potential of emerin-positive and emerin-negative cells in the population suggests a model whereby the emerin mutation contributes to depletion of a functional satellite cell population. Finally, as the X-linked EMD gene would not normally have been sequenced for a female presenting with EDMD this study highlights the importance of extensive analysis of the pedigree when searching for disease-causing mutations.
2. Materials and methods
2.1. Patient and controls
The patient attended the clinic followed by routine diagnostic mutational analysis of the EMD (MIM *300384) and LMNA (MIM *150330) genes. All materials (blood and muscle biopsies to generate myoblast lines) included in this study were taken with informed consent of the donors and with approval of the local ethics board.
2.2. Mutational analysis and tissue culture
Sanger sequencing was used to sequence the coding areas and exon/intron boundaries of the LMNA and EMD genes. Myoblasts were gained from a biopsy of biceps brachii muscle performed in the index patient at age 16. These myoblasts as well as myoblasts from an age matched control were grown in tissue culture using skeletal muscle cell growth medium (PromoCell, Heidelberg, Germany). Cells were kept from reaching confluency to avoid differentiation. For differentiation DMEM (containing 0.1% FBS, 5 mg/ml insulin and 5 mg/ml transferin) was used. Cells were grown at 37 °C in a 5% CO2 incubator.
2.3. Analysis of leucocyte populations by flow cytometry
A typically principally mononuclear leucocyte fraction was isolated from heparinized blood using Histopaque®-1077 (Sigma-Aldrich®) following the provided protocol. To determine composition of the fraction, cells were analysed by flow cytometry using fluorescently labelled antibodies to CD19 (Becton Dickinson, 555413) for B-cells, CD3 (Beckman Coulter, A07746) for T-cells, CD66b (BioLegend, 305102) for granulocytes, and CD14 (Becton Dickinson, 560349) for macrophages and myeloid cells. After staining, cells were analysed on an LSR II flow cytometer (BD Bioscience, UK) equipped with 488 nm and 350 nm lasers and appropriate filters. Cell debris and cell aggregates were excluded from analysis by application of electronic gates and numbers of the live singlet cells in each gate calculated using FlowJo software (TreeStar, Inc).
2.4. Immunohistochemistry
Myoblasts were fixed with methanol (−20 °C). As a marker for proliferation Ki-67 (Thermo Scientific, RM-9106-S0), antibody was used [17]. Relocalization from the centrosome to the nuclear envelope of PCM1 has been found to be an early and reliable marker of differentiation [18] and so was used to assess myoblast differentiation. For emerin immunofluorescence staining of myoblasts and biopsy sections MANEM1 antibody was used which recognizes emerin amino acids 89–96 (GYNDDYYE) [19]. For stainings with leucocyte type-specific antibodies the same fluorescently conjugated antibody set that was used for flow cytometry was employed; however, because the laser lines of the flow cytometer did not match the filter sets of the microscope, secondary antibodies were also used. All secondary antibodies were Alexafluor conjugated and generated in donkey with minimal species cross-reactivity. DNA was visualized with DAPI (4,6-diamidino-2 phenylindole, dihydrochloride). To determine if the small fragment encoded by the mutant allele is stably expressed, Western blot analysis optimized for small MW proteins was performed using Millipore Immobilon® PSQ Transfer Membranes and following the manufacturer's instructions. The blot was probed with monoclonal antibody MANEM14 against emerin amino acids 7–14 (LSDTELTT) [19].
2.5. Microscopy and image analysis
Most images were obtained using a Nikon TE-2000 microscope equipped with a 1.45 NA 100× objective or a 20× objective and CoolSnapHQ High Speed Monochrome CCD camera (Photometrics, Marlow, UK). Image analysis was performed using ImageJ software.
2.6. RNA depletion of emerin in control myoblasts
The previously published siRNA against emerin 5′GGUGGAUGAUGACGAUCUUtt-3′ [20] was transfected into myoblasts using jetPRIME® (Polyplus) following the manufacturer's instructions.
3. Clinical report
The 25-year-old female index patient of this family (IV-1, Figs. 1 and 2A) had normal developmental milestones and did not complain of muscle weakness during childhood. Symptoms at onset, at the age of 12 years, were Achilles tendon contractures along with myalgia of proximal muscles, mainly after exercise. Since age 15 she complained about mild proximal weakness during longer walks or when climbing stairs. At examination at age 16, there was a mild proximal weakness of 4–5/5, predominantly in the dorsal leg muscles, mild scapular winging, mild scoliosis, no facial weakness, no muscle atrophy, mild calf hypertrophy. Walking on tiptoes was possible; walking on heels was moderately impaired due to Achilles tendon contractures. At age 23, 4/5 weakness of small hand muscles occurred. Creatine kinase levels were mildly elevated with 180 U/l (normal <145). Electromyography was repeatedly performed and showed a mild myopathic pattern in proximal leg muscles. Motor and sensory nerve conduction velocity was found normal. Starting at age 21, the patient complained of palpitations and extrasystoles of the heart. At age 23, ECG showed sinusarrhythmia, vertical heart, unspecific ST elevations of maximal 0.1 mV in II, III, aVF, V5 and V6. The 24 h ECG showed sinus rhythm, heart rate 74 (range 50–166), no relevant asystolia, 90 VES, 2523 SVES and no high-grade dysrhythmia. Cardiac ultrasound, nocturnal pulse oxymetry and lung function were found normal. Whole body muscle MRI at age 23 showed mild asymmetric atrophy of the shoulder girdle muscles, but no fatty replacement of muscle tissue (Fig. 2B).
Fig. 1.

Pedigree of the patient. Arrow indicates the index patient.
Fig. 2.
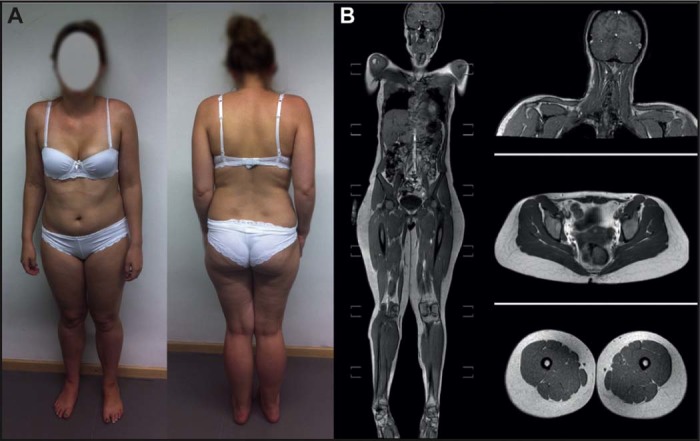
EDMD pathologies in the patient. A) The index patient shows a mild limb girdle phenotype with calf hypertrophy, Achilles tendon contractures and mild scapular winging. B) Whole body muscle MRI shows mild asymmetric atrophy of the shoulder girdle muscles, prominent calf muscles, but no fatty replacement of muscle tissue.
The father of the index patient (Fig. 1, III-2) was reported to suffer from muscular dystrophy, but had not been genetically characterized before. His symptoms started at age 4 with Achilles tendon contractures, during disease progression proximal weakness and atrophy occurred along with cardiac arrhythmia requiring pacemaker implantation at age 25. The father's affected brother (Fig. 1, III-4) died from cardiac arrest at age 35; he did not have a pacemaker. However, onset of symptoms in three affected uncles to the father of the index patient (Fig. 1, II-5, -6 and -7) was reported to have started not before age 20. Since family history had revealed only male affection through three generations, EDMD1 was suspected. The father of the index patient has nine further children (5 females, 4 males, age between eight and 20 years) with his second wife, who were unfortunately not available for examination. However, one of the five daughters (Fig. 1, IV-5) is reported to have Achilles tendon contractures. The other daughters have so far not shown any symptoms, but none of them was genetically or clinically tested and most are still younger than the index patient at the time of first clinical manifestation.
4. Results and discussion
Looking only at the last two generations of the family, inheritance of the disease appeared to be dominant, yet except for the index patient the extended inheritance matches that for an X-linked gene. Sequencing the index patient (Fig. 1, IV-1) for changes in the LMNA and EMD genes identified no LMNA mutation, but did identify the heterozygous EMD mutation c.174_175delTT (p.Y59*). The same mutation was found to be hemizygous in the affected father (Fig. 1, III-2). This mutation has not been reported before, but another mutation causing an early stop codon at the same position (c.177T > A; p.Y59*) has been found in an affected male suffering EDMD1 [21]. Other EMD mutations resulting in an early stop codon are known to cause EDMD1 in hemizygous patients and, in fact, emerin insufficiency accounts for the majority of EDMD1 (http://www.umd.be [22]). Therefore, the EMD p.Y59* mutation is most likely causing disease in the father and uncle through loss of emerin. However, according to this mechanism, the heterozygous patient should not be affected. In emerinopathies, a wide clinical intrafamilial and interfamilial variability is reported, independent of the type or localization of the mutation [23]. Varying heart block or cardiomyopathy can occur in otherwise healthy female EDMD1 carriers [1,22,24], but muscle weakness and contractures due to EMD mutations have hitherto not been described in females except in one female carrier, heterozygous for an EMD mutation, where uneven X-inactivation favouring the mutant allele was shown to be the cause of the clinical manifestation [22].
A biopsy of biceps brachii muscle was performed on the index patient, showing single rounded fibres but no fibrosis or fatty degeneration, consistent with general appearance on MRI (Fig. 2B). Immunohistochemical staining of the biopsy material for emerin and lamin A showed that 87% of the cells were positive for emerin using an antibody that would not recognize the mutant fragment if expressed (Fig. 3A,B). Additional staining of patient mononuclear blood cells showed an even higher percentage of emerin-positive cells (>99% positive for emerin, Fig. 3C). Also the majority of myoblasts gained from this biopsy were expressing emerin (Fig. 3D). This excludes loss of emerin through unequal X-inactivation favouring the mutant allele as had been found for the other symptomatic X-linked female.
Fig. 3.

Emerin staining excludes uneven X-inactivation. A–D) Patient cells were stained using an emerin antibody that would not recognize the predicted short mutant version if expressed. A) Patient biopsy staining of biceps brachii muscle, arrows indicate emerin-negative nuclei. A higher magnification view of the boxed section of the merged image is shown below it. B) Percentage of emerin positive cells (at least 200 nuclei counted for every bar). *cells ceased to proliferate at passage 11. C) Immunofluorescence staining of emerin in patient mononuclear blood cells indicates that nearly all cells are emerin positive. D) Cultured patient myoblasts at passage 2. Arrows indicate emerin-negative nuclei. E) To determine if the predicted short mutant version of emerin was expressed in the patient cells an antibody to an N-terminal peptide overlapping with the truncated sequence was used. A Western blot of patient and control myoblasts indicates the presence of only full length emerin (MW 34 kDa).
A possible explanation could be that the pathology is achieved in the presence of the wild-type allele if the predicted truncated protein, too short to be functional in the male family members that completely lack the wild-type protein, is expressed and functions as a dominant-negative in the index patient. The expression of the truncated protein (amino acids 1–59) was tested for by Western blot using an antibody to emerin amino acids 7–14 and conditions for detection of small peptides. This revealed no evidence of a possible truncated protein (Fig. 3E).
Yet another possibility that cannot be excluded is a second mutation modifying the disease. Modifying effects in EDMD1 have been shown for additional mutations in desmin or lamin A [8,9]. As the onset of the disease in the father occurred earlier than in his affected uncles, and apart from the index patient another of his daughters seems to be affected too, a modifying mutation seems likely. Additionally, the grandmother of the index patient (Fig. 1; II-2), who, based on the pedigree, is an obvious carrier of the disease allele, is reported as unaffected, thus excluding a dominant effect of the emerin mutation.
The mixed population of emerin-positive and -negative myoblasts from the muscle biopsy provided a good model to investigate the effect of emerin loss in cells with identical genetic background. During passaging of the myoblasts the percentage of cells positive for emerin at first slightly increased from passage 2 (~74%) until passage 4 (~80%), but then steadily decreased until passage 10 (~38%). Inexplicably, the emerin-positive population increased again in passage 11 to 49%, but the cells ceased to proliferate at that point and may have entered senescence (Fig. 4A). Staining for the proliferation marker Ki-67 [17] showed a higher proliferation rate of emerin-negative cells compared to emerin-positive cells starting from passage four (Fig. 4A,B). Depletion of emerin by siRNA in control myoblasts similarly increased the cell proliferation rate compared to cells transfected with control siRNA (Fig. 4C). Emerin mutation or loss has been found to affect cell cycle regulation [25,26] and COS-7 cells transfected with an emerin EDMD mutation had increased cell-cycle length [27]. Correspondingly, emerin null fibroblasts have a rapid growth phenotype [28]. Also emerin-negative cells have previously been reported to become dominant in culturing of skin fibroblasts from a carrier of another EDMD emerin mutation [29]. Here we have shown this effect for the first time in muscle. Our data show that the emerin-negative cells had an advantage in proliferation beginning at passage four. The fact that this advantage was not observed until passage four might reflect acclimation after freezing or the number of divisions from a more complete satellite cell state. This could explain how over time the emerin-negative cells became predominant in the cultures.
Fig. 4.

Patient myoblasts preferentially undergo spontaneous differentiation in tissue culture. A) Percentage of patient emerin-positive and -negative cells expressing the proliferation marker Ki-67 (at least 200 nuclei counted for each bar). B) Example for Ki-67 staining in patient myoblasts. C) Percentage of Ki-67 positive cells in control myoblasts transfected with emerin siRNA or a scramble sequence control siRNA (at least 200 nuclei counted for each bar). D) Percentage of cells showing spontaneous differentiation in patient cells and an age matched control myoblast cell line (at least 200 nuclei counted for every passage). E) Example for PCM1 staining in patient myoblasts. F) Percentage of patient emerin-positive cells (at least 200 nuclei counted for each passage) and percentage of emerin-positive cells undergoing spontaneous differentiation (50 single nuclei outside myotubes counted for p2–7, 20 for 8 + 9 and 3 for p10). G) Percentage of patient emerin-positive cells (at least 200 nuclei counted for each passage) and percentage of emerin-positive cells undergoing differentiation (50 single nuclei outside myotubes counted for all passages).
Intriguingly, a high incidence of cells expressing morphological characteristics of differentiated myotubes was observed specifically in the patient myoblasts, even though cells were always maintained subconfluent in culture and with high serum medium. To investigate this phenomenon further, patient and control myoblast differentiation was assessed in addition to emerin status in exponentially growing cultures using PCM1 relocalization to the nuclear envelope as a marker of differentiating cells [18]. PCM1 has an advantage over markers that only appear upon differentiation because for these the level of intensity can confuse results for cells in early stages of differentiation whereas by being detectable at the centrosome in undifferentiated myoblasts this is not a problem with PCM1. This revealed a significantly higher differentiation rate of patient myoblasts compared to the control (Fig. 4D,E). The proportion of cells undergoing spontaneous differentiation in the population of emerin-positive cells matched that in emerin-negative cells through the first five passages; after that the majority of the cells undergoing spontaneous differentiation were emerin-positive (Fig. 4F). When differentiation was actively induced by moving cells to skeletal muscle differentiation medium for five days, a similar trend was observed: emerin-positive as well as emerin-negative cells were able to differentiate, but from passage six onwards there was a clear advantage of emerin-positive cells to differentiate both with and without induction (Fig. 4G). Moreover, all nuclei inside myotubes were positive for emerin.
Both the proliferation and spontaneous differentiation defects we observe in this female EDMD1 patient are consistent with various observations of cell cycle and myogenic effects in model systems. A mouse model lacking emerin showed no overt muscle phenotype although muscle regeneration seemed to be affected [30]. This lack of phenotype is thought to be the result of a higher expression level of the lamina-associated polypeptide1 (LAP1) in mouse cells compared to human myoblasts [31]. Despite this, muscle tissue from the emerin null mice showed abnormal cell-cycle parameters and delayed myogenic differentiation with many genes involved in MyoD and pRb pathways aberrantly expressed [30]. A role of emerin in normal myogenesis has also been shown where it inhibits binding of the transcription factor Lmo7 to the MyoD promoter in C2C12 mouse myoblasts [32]. Additionally, cell cycle defects have been reported for particular EDMD1 mutations when expressed in cultured cells [27]. The faster proliferation of the emerin-negative patient myoblasts and deficiencies in differentiation at later passages is consistent with the mouse data; however, the observation of spontaneous differentiation at early passages in both emerin-positive and emerin-negative myoblasts suggests an additional factor, such as an unknown modifying mutation, must contribute to this second characteristic we observe in the patient cells.
To our knowledge no blood phenotypes or pathology has been previously reported for EDMD patients; however, in analysing the patient blood to also test for uneven X-inactivation, it was observed that a large number of cells had highly lobulated nuclei (Fig. 5A,B). This would typically indicate amplification of neutrophils, but the patient did not show signs of any infection at the time the blood was taken. Moreover, isolation of white blood cells was performed roughly 1 week after the blood was taken and neutrophils would not be expected to survive that long, even when maintained in the cold. Thus, the cells were analysed by flow cytometry to determine and quantify the leucocyte cell types present in the population. This analysis showed an unexpectedly large percentage of cells staining positive with the granulocyte marker CD66b (Fig. 5C). In all 27% of the cells analysed in the patient sample were positive for CD66b compared to only 1% of cells in a control donor. The patient exhibited a corresponding loss of cells in other populations, principally the CD3 positive cells which dropped from 53% in the control donor to 8% in the patient. Granulocytes, particularly neutrophils, are the only white blood cell population defined by extensive nuclear lobulation. To determine if the lobulated cells were all CD66b (granulocyte) positive, immunofluorescence staining was performed using the same leucocyte type-specific antibody set. Surprisingly, this revealed that lobulated patient nuclei stained positive for several different markers, including CD3 that should recognize principally T-cells and CD14 that should recognize principally macrophages, so that many highly lobulated cells were not granulocytes (Fig. 6A,B). As most of the lobulated cells were also positive for emerin using the antibody that would only recognize the wild-type allele, this effect is likely the result of the unknown modifying mutation we predict from the other data. Modifying mutations identified previously in EDMD patients have included both other nuclear envelope proteins and non-nuclear envelope proteins. We postulate that this modifier must be another nuclear envelope protein to have such a striking effect on nuclear morphology in white blood cells.
Fig. 5.

Blood cell distributions and nuclear lobulation in the patient. A) Dapi staining of nuclei from cells isolated from patient blood, 20× magnification. B) Dapi staining of nuclei from cells isolated from patient blood, 100× magnification. C) FACS staining of cells isolated from patient and control blood for following markers: CD3 (T-cells), CD14 (macrophages), CD66b (granulocytes) and CD19 (B-cells).
Fig. 6.

Highly lobulated nuclei in patient blood include non-granulocyte cells. A) Immunofluorescence staining of control blood cells for the following markers: CD3 (T-cells), CD14 (macrophages), CD66b (granulocytes). B) Immunofluorescence staining of patient blood cells for the same markers. The right panels are magnifications of the boxed areas in the merged images.
5. Conclusions
This case study underscores the importance of extended family history in choosing which genes to sequence first in EDMD and potentially other genetically heterogeneous diseases caused by both autosomal and X-linked genes. By focusing on dominant inherited disease alleles the emerin mutation could have easily been missed in this patient.
Most likely a second mutation is necessary to cause the full phenotype observed in the female patient, but, if the findings in culture reflect the situation in the patient tissue, the emerin mutation plays the leading role. We postulate that the growth advantage of the emerin-negative cells enables them to eventually dominate the muscle cell population while emerin-positive satellite cells are further depleted because of their tendency to undergo spontaneous differentiation, likely due to the second mutation. Thus, in the course of muscle use and ageing the two mutations work together to deplete emerin-positive cells from the population of muscle satellite cells, resulting in the later onset dystrophic phenotype. However, tissue differences must also affect the way the mutations work together because, in the blood, the near absence of emerin-negative cells suggests a significant survival disadvantage from the emerin mutation. This is the first finding of blood cell effects in a case of EDMD. While difficulties in accessing samples from other family members hinder identification of the second mutation, the considerable nuclear lobulation in non-granulocyte cells together with the aberrant cell distributions suggests that the second mutation is also a nuclear envelope protein. At the same time, as the father did not exhibit unusually severe symptoms and the mother was completely unaffected this second mutation is unlikely to be in one of the existing genes linked to EDMD.
Authors' contributions
PM and ECS wrote the manuscript. PS and MCW evaluated the patient. PM, MW and ECS designed the project direction and experiments. PM, VS, NK and PLT did differentiation and microscopy experiments while GJMC and DRC performed flow cytometry experiments. All authors read and approved the final manuscript.
Acknowledgements
Work in the Schirmer lab is supported by Wellcome Trust Senior Research Fellowship 095209, providing also salaries for ECS, PM, VS, and NK, and Centre Grant 092076. Phú Lê Thành is supported by an MRC Studentship. Human myoblast cultures were obtained from the Muscle Tissue Culture Collection at the Friedrich-Baur-Institut (Department of Neurology, Ludwig-Maximilians-University, Munich, Germany). The Muscle Tissue Culture Collection is part of the German network on muscular dystrophies (MD-NET) and the German network for mitochondrial disorders (mito-NET, project D2, 01GM1113A) funded by the German Ministry of Education and Research (BMBF, Bonn, Germany). The Muscle Tissue Culture Collection is a partner of EuroBioBank (www.eurobiobank.org) and TREAT-NMD (www.treat-nmd.eu). The Muscular Dystrophy Association (USA) supports monoclonal antibody development in the laboratory of Glenn E. Morris, whom we want to thank for providing emerin antibodies.
Contributor Information
Eric C. Schirmer, Email: e.schirmer@ed.ac.uk.
Maggie C. Walter, Email: maggie.walter@lrz.uni-muenchen.de.
References
- 1.Emery A.E.H. Emery-Dreifuss muscular dystrophy – a 40 year retrospective. Neuromuscul Disord. 2000;10:228–232. doi: 10.1016/s0960-8966(00)00105-x. [DOI] [PubMed] [Google Scholar]
- 2.Wehnert M.S., Meinke P. John Wiley & Sons Ltd; Chichester: 2012. LiNC complex and human genetic muscular disease.http://www.els.net eLS. [Google Scholar]
- 3.Bonne G., Di Barletta M.R., Varnous S. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 4.Bione S., Maestrini E., Rivella S. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 5.Manilal S., Nguyen T.M., Sewry C.A., Morris G.E. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet. 1996;5:801–808. doi: 10.1093/hmg/5.6.801. [DOI] [PubMed] [Google Scholar]
- 6.Muchir A., Worman H.J. Emery-Dreifuss muscular dystrophy. Curr Neurol Neurosci Rep. 2007;7:78–83. doi: 10.1007/s11910-007-0025-3. [DOI] [PubMed] [Google Scholar]
- 7.Emery A.E. Emery-Dreifuss syndrome. J Med Genet. 1989;26:637–641. doi: 10.1136/jmg.26.10.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meinke P., Nguyen T.D., Wehnert M.S. The LINC complex and human disease. Biochem Soc Trans. 2011;39:1693–1697. doi: 10.1042/BST20110658. [DOI] [PubMed] [Google Scholar]
- 9.Muntoni F., Bonne G., Goldfarb L.G. Disease severity in dominant Emery Dreifuss is increased by mutations in both emerin and desmin proteins. Brain. 2006;129:1260–1268. doi: 10.1093/brain/awl062. [DOI] [PubMed] [Google Scholar]
- 10.Taranum S., Vaylann E., Meinke P. LINC complex alterations in DMD and EDMD/CMT fibroblasts. Eur J Cell Biol. 2012;91:614–628. doi: 10.1016/j.ejcb.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li P., Meinke P., Huong le T.T., Wehnert M., Noegel A.A. Contribution of SUN1 mutations to the pathomechanism in muscular dystrophies. Hum Mutat. 2014;35:452–461. doi: 10.1002/humu.22504. [DOI] [PubMed] [Google Scholar]
- 12.Bonne G., Mercuri E., Muchir A. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48:170–180. [PubMed] [Google Scholar]
- 13.Granger B., Gueneau L., Drouin-Garraud V. Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum Genet. 2011;129:149–159. doi: 10.1007/s00439-010-0909-1. [DOI] [PubMed] [Google Scholar]
- 14.Hoeltzenbein M., Karow T., Zeller J.A. Severe clinical expression in X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 1999;9:166–170. doi: 10.1016/s0960-8966(98)00120-5. [DOI] [PubMed] [Google Scholar]
- 15.Mercuri E., Poppe M., Quinlivan R. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: from congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch Neurol. 2004;61:690–694. doi: 10.1001/archneur.61.5.690. [DOI] [PubMed] [Google Scholar]
- 16.Rankin J., Auer-Grumbach M., Bagg W. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644C. Am J Med Genet A. 2008;146A:1530–1542. doi: 10.1002/ajmg.a.32331. [DOI] [PubMed] [Google Scholar]
- 17.Scholzen T., Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Srsen V., Fant X., Heald R., Rabouille C., Merdes A. Centrosome proteins form an insoluble perinuclear matrix during muscle cell differentiation. BMC Cell Biol. 2009;10:28. doi: 10.1186/1471-2121-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manilal S., Sewry C.A., Pereboev A. Distribution of emerin and lamins in the heart and implications for Emery-Dreifuss muscular dystrophy. Hum Mol Genet. 1999;8:353–359. doi: 10.1093/hmg/8.2.353. [DOI] [PubMed] [Google Scholar]
- 20.Salpingidou G., Smertenko A., Hausmanowa-Petrucewicz I., Hussey P.J., Hutchison C.J. A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J Cell Biol. 2007;178:897–904. doi: 10.1083/jcb.200702026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yates J.R., Bagshaw J., Aksmanovic V.M. Genotype-phenotype analysis in X-linked Emery-Dreifuss muscular dystrophy and identification of a missense mutation associated with a milder phenotype. Neuromuscul Disord. 1999;9:159–165. [PubMed] [Google Scholar]
- 22.Manilal S., Recan D., Sewry C.A. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum Mol Genet. 1998;7:855–864. doi: 10.1093/hmg/7.5.855. [DOI] [PubMed] [Google Scholar]
- 23.Wehnert M.S., Bonne G. The nuclear muscular dystrophies. Semin Pediatr Neurol. 2002;9:100–107. doi: 10.1053/spen.2002.33806. [DOI] [PubMed] [Google Scholar]
- 24.Sakata K., Shimizu M., Ino H. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation. 2005;111:3352–3358. doi: 10.1161/CIRCULATIONAHA.104.527184. [DOI] [PubMed] [Google Scholar]
- 25.Dabauvalle M.C., Muller E., Ewald A., Kress W., Krohne G., Muller C.R. Distribution of emerin during the cell cycle. Eur J Cell Biol. 1999;78:749–756. doi: 10.1016/S0171-9335(99)80043-0. [DOI] [PubMed] [Google Scholar]
- 26.Ellis J.A., Craxton M., Yates J.R.W., Kendrick-Jones J. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J Cell Sci. 1998;111:781–792. doi: 10.1242/jcs.111.6.781. [DOI] [PubMed] [Google Scholar]
- 27.Fairley E.A.L., Riddell A., Ellis J.A., Kendrick-Jones J. The cell cycle dependent mislocalisation of emerin may contribute to the Emery-Dreifuss muscular dystrophy phenotype. J Cell Sci. 2002;115:341–354. doi: 10.1242/jcs.115.2.341. [DOI] [PubMed] [Google Scholar]
- 28.Markiewicz E., Tilgner K., Barker N. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25:3275–3285. doi: 10.1038/sj.emboj.7601230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris G.E. Protein interactions, right or wrong, in Emery-Dreifuss muscular dystrophy. Symp Soc Exp Biol. 2004;(56):57–68. [PubMed] [Google Scholar]
- 30.Melcon G., Kozlov S., Cutler D.A. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet. 2006;15:637–651. doi: 10.1093/hmg/ddi479. [DOI] [PubMed] [Google Scholar]
- 31.Shin J.Y., Mendez-Lopez I., Wang Y.X. Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev Cell. 2013;26:591–603. doi: 10.1016/j.devcel.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dedeic Z., Cetera M., Cohen T.V., Holaska J.M. Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J Cell Sci. 2011;124:1691–1702. doi: 10.1242/jcs.080259. [DOI] [PubMed] [Google Scholar]