

Abstract
Increases in prostaglandin E2 (PGE2) and cyclooxygenase-2 (COX-2) levels are features of colon cancer. Among the different E-type prostanoid receptor subtypes, EP4 receptors are considered to play a crucial role in carcinogenesis by, for example, inducing COX-2 when stimulated with PGE2. However, EP4 receptor levels and PGE2-induced cellular responses are inconsistent among the cellular conditions. Therefore, the connections responsible for the expression of EP4 receptors were investigated in the present study by focusing on cell density-induced hypoxia-inducible factor-1α (HIF-1α). The expression of EP4 receptors was examined using immunoblot analysis, quantitative polymerase chain reaction, and reporter gene assays in HCA-7 human colon cancer cells with different cellular densities. The involvement of HIF-1α and its signaling pathways were also examined by immunoblot analysis, reporter gene assays, and with siRNA. We here demonstrated that EP4 receptors as well as EP4 receptor-mediated COX-2 expression levels decreased with an increase in cellular density. In contrast, HIF-1α levels increased in a cellular density-dependent manner. The knockdown of HIF-1α by siRNA restored the expression of EP4 receptors and EP4 receptor-mediated COX-2 in cells at a high density. Thus, the cellular density-dependent increase observed in HIF-1α expression levels reduced the expression of COX-2 by decreasing EP4 receptor levels. This novel regulation mechanism for the expression of EP4 receptors by HIF-1α may provide an explanation for the inconsistent actions of PGE2. The expression levels of EP4 receptors may vary depending on cellular density, which may lead to the differential activation of their signaling pathways by PGE2. Thus, cellular density-dependent PGE2-mediated signaling may determine the fate/stage of cancer cells, i.e., the surrounding environments could define the fate/stage of malignancies associated with colon cancer.
Keywords: Cell density, COX-2, EP4 prostanoid receptor, GPCRs, HCA-7 cells, HIF-1α, human colon cancer, PGE 2
Introduction
One major pathophysiological feature of colon cancer is increased levels of prostaglandin E2 (PGE2) and cyclooxygenase-2 (COX-2), an enzyme that produces prostanoids including PGE2 (Eisinger et al. 2007). The actions of PGE2 are known to be mediated by its cognate receptors, namely EP receptors (Woodward et al. 2011). G-protein-coupled EP receptors consist of four subtypes: EP1 to EP4 receptors (Woodward et al. 2011). Based on the findings of knockout-mice studies and research using pharmacological reagents, all EP receptors are generally considered to play a role in carcinogenesis (Woodward et al. 2011). However, of the four EP receptor subtypes, EP4 receptors and their signaling pathways are suggested to be critically involved in carcinogenesis because of their wide distribution in the body and various pathophysiological roles (Konya et al. 2013; Yokoyama et al. 2013). In contrast to EP2 receptors, conventional Gαs protein-coupled EP4 receptors have been shown to activate a number of important signaling pathways including Gαi protein-mediated signaling (Konya et al. 2013; Yokoyama et al. 2013). These signaling pathways include phosphatidylinositol 3-kinase-, extracellular signal-regulated kinases (ERKs)-, β-catenin-, and/or β-arrestine-mediated pathways, which may be key regulators in development of cancer (Fujino and Regan 2003; Konya et al. 2013; Yokoyama et al. 2013). EP4 receptor signaling has also been shown to increase the expression of PGE2 synthase and cyclin D1 by inducing the expression of early growth response factor-1 (EGR-1) (Fujino et al. 2003; Fujino and Regan 2003) as well as COX-2 (Hull et al. 2004; Yoshida et al. 2013). Therefore, these signaling pathways may establish a positive feedback loop in which an increase in the synthesis of PGE2 may further drive the activation of EP4 receptors, as reported previously (Fujino and Regan 2003; Regan 2003).
A previous study indicated that the expression of EP4 receptors may be up-regulated in colorectal cancer and adenoma (Hawcroft et al. 2007). However, relatively higher mRNA expression levels for EP4 receptors have also been detected in normal colon tissue than those for the other EP receptor subtypes (Gustafsson et al. 2007). The intense stimulation of EP4 receptors in normal colonic epithelial cells provoked a loss in barrier integrity, which may play a role in the early onset and/or progression of colitis following inflammation (Lejeune et al. 2010). Colitis, especially ulcerative colitis, is a considerable risk factor for colon cancer. A previous study also reported that the expression of COX-2 was detected from a very early stage in carcinoma growth (Fujita et al. 1998). Since EP4 receptors are the key receptor subtype for the induction of colitis and COX-2 (Hull et al. 2004; Yoshida et al. 2013), EP4 receptors may play important roles in the early stages of development of colon cancer, in other words, in the pro-tumorigenic progression of intestinal epithelial cells, as has been suggested previously (Chell et al. 2006; Hawcroft et al. 2007).
Although increases in PGE2 and COX-2 expression levels have been documented extensively in the development of colon cancer, contradictory findings have been reported in terms of the effects of PGE2 in colon cancer cell lines. For example, the dose-dependent proliferative activity of PGE2 was previously observed in HT-29 human colorectal cancer cells (Qiao et al. 1995), whereas another study showed that PGE2 had no effect on the proliferation of HT-29 cells (Cassano et al. 2000). Moreover, a bell-shaped proliferation response was noted in SW1116 human colon cancer cells when PGE2 concentrations were increased (Qiao et al. 1995). These differences in the effects of PGE2 have been attributed to the cell-line-specific differential expression of EP receptor(s), use of different proliferation assays, and/or differential activation of EP receptors at different PGE2 concentrations (Lesuffleur et al. 1990; Cassano et al. 2000; Hull et al. 2004). Meanwhile, the positive effects of PGE2 on cell growth were observed in colorectal adenoma and carcinoma cell lines treated with lower concentrations of PGE2 (Chell et al. 2006). In contrast, the growth of colorectal adenoma cell lines, but not carcinoma cell lines, was inhibited at higher doses of PGE2 (Chell et al. 2006). These findings suggest that, at some stage of carcinogenesis, an increase in PGE2 levels may become inhibitory to tumor cells, i.e., PGE2 may be able to switch colon cancer cells from being tumor-promoting to growth-inhibitory, as described previously (Chell et al. 2006). However, the actual reason(s) and/or underlying mechanism(s) for these differences in cellular responses remain unknown.
Hypoxia-inducible factor-1α (HIF-1α) is an oxygen-sensitive transcriptional factor that has significant roles in the cellular physiology and pathophysiology of diseases such as the development of cancer (Semenza 2003; Mabjeesh and Amir 2007; Rohwer et al. 2013). Although the up-regulated expression of HIF-1α is known to be evoked by responses to a reduction in the availability of oxygen, the functional regulation of HIF-1α has also been reported under normoxic conditions (Kuschel et al. 2012). Thus, hypoxia may represent one of the factors among several HIF-1α-inducing stimuli that regulate the expression of HIF-1α (Paltoglou and Roberts 2005). In fact, in spite of the absence of hypoxic areas, increases in the expression of the HIF-1α protein have been reported in the most common cancer types (Kuschel et al. 2012). Furthermore, HIF-1α was previously shown to be induced in response to an increase in cellular density (Paltoglou and Roberts 2005), and this induction was even evoked when cells were grown under standard normoxic conditions (Wiesener et al. 1998).
In the present study, we demonstrated that the cellular density-dependent induction of HIF-1α protein expression was accompanied by and responsible for the decrease observed in the expression of the EP4 receptors in HCA-7 human colon cancer cells. PGE2-induced COX-2 expression was previously shown to be regulated by the activation of EP4 receptors (Yoshida et al. 2013); therefore, an increase in the expression of HIF-1α eventually reduced the expression of COX-2 by decreasing EP4 receptor levels. These results may provide one explanation for the discrepant actions of PGE2, such as its effects on proliferation, as described above, which may be due to the altered expression of EP4 receptors followed by alternations in the expression of COX-2 that may eventually change the levels of PGE2 produced as well as its actions. Although cellular proliferation may not be solely regulated by the turnover of PGE2/COX-2, some of the aforementioned conversions in cellular responses were triggered and regulated by cellular-density-dependent alternations in the expression of HIF-1α, which may cause cells to respond differently to PGE2 depending on the surrounding environment.
Materials and Methods
Cell culture and imaging
The human colon cancer cell line HCA-7 was cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma, St Louis, MO) containing 10% fetal bovine serum (Thermo Scientific, Waltham, MA), 100 IU/mL penicillin (Meiji Seika, Tokyo, Japan), and 100 μg/mL streptomycin (Meiji Seika). All materials were obtained from Wako Pure Chemical (Osaka, Japan) unless otherwise stated. Cell images were obtained using a Nikon Diaphot microscope (Nikon, Tokyo, Japan) with a Nikon D70 digital camera (Nikon) and processed using Nikon Capture 4 (Nikon).
Western blotting
Cells were cultured at low (3 × 104 cells/each 6 well), middle (1 × 105 cells/each 6 well), and high (3 × 105 cells/each 6 well) densities in 6-well plates, and medium was replaced with fresh Opti-MEM prior to immunoblotting experiments (Invitrogen, Carlsbad, CA) containing 100 IU/mL penicillin and 100 μg/mL streptomycin. Cells were then treated with 1 μmol/L PGE2 (Cayman, Ann Arbor, MI) or 50 ng/mL epidermal growth factor (EGF) (Higeta Shoyu, Tokyo, Japan) for 6 h to induce COX-2 and phosphorylated ERKs. In experiments using HIF-1α siRNA, cells were transiently transfected using Lipofectamine RNAi MAX Reagent (Invitrogen) and 15 μmol/L HIF-1α siRNA (sc-35561; Santa Cruz Biotechnology, Santa Cruz, CA) for 8 h, and medium was then replaced with fresh Opti-MEM containing 100 IU/mL penicillin and 100 μg/mL streptomycin for 40 h. To induce COX-2, cells were treated with 1 μmol/L PGE2 (Cayman) for another 6 h. Cells were scraped into lysis buffer consisting of 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 8.0), 5 mmol/L EDTA (pH 8.0), 1% Igepal CA-630 (MP Biomedicals, Aurora, OH), 0.5% sodium deoxycholate, 10 mmol/L sodium fluoride, 10 mmol/L disodium pyrophosphate, 0.1% SDS, 0.1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L sodium orthovanadate, 10 μg/mL leupeptin (Sigma), and 10 μg/mL aprotinin and transferred to microcentrifuge tubes. Samples were rotated for 30 min at 4°C and centrifuged at 16,000g for 15 min. Aliquots of samples containing 40–100 μg of protein were electrophoresed on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes as described previously (Fujino et al. 2003). The membranes were incubated for 1 h at room temperature in 5% nonfat milk. Incubations were conducted for 16 h at 4°C in 5% bovine serum albumin (BSA) (Sigma) containing a 1:2000 dilution of an anti-COX-2-antibody (sc-1745; Santa Cruz Biotechnology); 1:1000 dilution of an anti-β-tubulin-antibody (T4026; Sigma); 1:1000 dilution of an anti-phospho-ERK1/2-antibody (#9106; Cell Signaling Technology, Danvers, MA); a mixture of 1:500 dilution of an anti-ERK1-antibody and 1:20,000 dilution of an anti-ERK2-antibody (sc-93 and sc-154, Santa Cruz Biotechnology); 1:2000 dilution of an anti-human EP4 receptor-antibody (#101775; Cayman); an anti-EGF receptor-antibody (sc-003, Santa Cruz Biotechnology); or 1:1000 dilution of an anti-HIF-1α-antibody (#3761; Cell Signaling Technology). After being incubated with primary antibodies, membranes were washed three times and incubated for 2 h at room temperature with a 1:4000 dilution of appropriate secondary antibodies conjugated with horseradish peroxidase under the same blocking conditions as those for the primary antibodies (Fujino et al. 2003). After washing three times, immunoreactivity was detected and visualized with chemiluminescence imaging system, LAS-1000 (Fuji Film, Tokyo, Japan). To ensure the equal loading of proteins, membranes were stripped and re-probed with the anti-β-tubulin-antibody or anti-ERK1/2-antibodies under the conditions described above.
Quantitative polymerase chain reaction
Cells were cultured at low, middle, and high densities in 6-well plates, and medium was replaced with fresh Opti-MEM containing 100 IU/mL penicillin and 100 μg/mL streptomycin prior to immunoblotting experiments. Total RNAs were isolated using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. After quantities were determined, cDNAs were prepared from 5 μg of total RNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA), and the expression of mRNA was determined using TaqMan Gene Expression Assay primers for the human EP4 receptor (Context Sequence, TCCATCCCGCTCGTGGTGCGAGTAT; Target Exon, 2; Chromosome, 5; NCBI gene reference, NM_000958.2) (Applied Biosystems, Foster City, CA). Polymerase chain reaction (PCR) reactions consisted of 40 cycles of 95°C for 15 sec and 60°C for 45 sec in an ABI 7500 thermal cycler (Applied Biosystems). Threshold values (Ct) were determined automatically by the system software and relative mRNA expression levels were analyzed using the comparative ΔΔCt method, as described previously (Ji et al. 2010). Data were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Luciferase assay
The human EP4 promoter luciferase reporter plasmid was constructed using the same methods as described previously (Kambe et al. 2008). Briefly, -1236 to -42 base pairs of the human EP4 promoter region were amplified by PCR from human genomic DNA (Promega, Madison, WI) using the primers 5′-GCAGATGGGAAGAGGTTTTT-3′ (sense) and 5′-TTCTCCTCCTCCAAGTTTCC-3′ (antisense), as reported previously (Kambe et al. 2008). Using the product of the first PCR (1197 bp) as a template, a second round of PCR was then performed to introduce restriction enzyme sites; Nhe I (upstream) and Hind III (downstream), using the following; 5′-GGGCTAGCCTGCAGATGGGAAGAGGTTTTTCCAGGAATTTAAA-3′ (sense) and 5′-GGAAGCTTTGGAGCTCGCGTGCTGCGGCCTTTCCACCCTCTGTACAAACTTTTCTCCTCCT-3′ (antisense) (Kambe et al. 2008). To construct the human EP4 promoter luciferase plasmid, pGL3/EP4R-Promoter-Luc, the product of the second round of PCR (1248 bp), and the pGL3-basic luciferase vector (Promega) were digested with Nhe I and Hind III restriction enzymes (TOYOBO, Osaka, Japan) and then purified and ligated, followed by sequencing and verification. Cells were cultured at low, middle, and high densities in 6-well plates, and medium was replaced with fresh Opti-MEM containing 100 IU/mL penicillin and 100 μg/mL streptomycin prior to the experiments. Cells were transiently transfected with firefly pGL3/EP4R-Promoter-Luc, the hypoxia response element (HRE) luciferase reporter plasmid (pGL3/HRE-Luc27) (Ji et al. 2010), or Renilla human EP2 or EP3 receptor promoter luciferase reporter plasmids (S713225, S722267; SwitchGear Genomics, Menlo Park, CA), and either with human HIF-1α siRNA, renilla luciferase control plasmids, pRL-CMV (Promega), or the firefly luciferase control plasmid; pGL3-CMV (CMV promoter of pRL-CMV was replaced with the pGL3 vector) using GeneJuice (Novagen/Merck, Darmstadt, Germany) reagent. After approximately 8 h, the transfection reagents were removed by a medium change and cells were incubated for a further 8 h. Cells were then lysed and assayed using the Dual luciferase reporter assay system (Promega) according to the manufacturer's instructions with the GL-200 luminometer (Microtech Nichion, Chiba, Japan). Data were normalized by calculating the ratios of the firefly luciferase scores to the corresponding renilla luciferase values or vice versa.
cAMP assay
Cells were cultured at low, middle, and high densities in 6-well plates and were then switched to Opti-MEM containing 100 IU/mL penicillin and 100 μg/mL streptomycin 16 h prior to the experiments. Cells were treated with 0.1 mg/mL isobutylmethylxanthine (Sigma) for 20 min followed by either vehicle (Me2SO) or 1 nmol/L to 10 μmol/L PGE2 for 60 min at 37°C. Experiments were terminated by removing medium and placing the cells on ice. The formation of cAMP was measured as described previously (Fujino et al. 2002). The amount of cAMP present was calculated from a standard curve prepared using unlabeled cAMP. The amounts of cAMP produced by HCA-7 cells in Opti-MEM were approximately 0.5 to 1 pmol/3 × 104 cells within 24 h of changing medium (vehicle-treated control of low density cells).
Statistical analysis
Data are expressed as the mean ± SD or mean ± SEM. Statistical analyses were performed using Prism 5 for Mac OS X software (GraphPad Software, La Jolla, CA). The multiple comparison tests in the analysis of variance (ANOVA) were the Newman–Keuls multiple comparisons test. An ANOVA and t-test were used to evaluate three or more independent experiments. Significance was assumed at P < 0.05.
Results
PGE2-stimulated COX-2 expression and phosphorylation of ERKs was decreased by an increase in cellular density
HCA-7 human colon cancer cells were cultured at low, middle, and high densities, as described in the Materials and Methods (Fig.1A). To examine the effects of cellular density on PGE2-induced COX-2 expression, HCA-7 cells cultured of different densities were treated with 1 μmol/L PGE2 for 6 h. As shown in Figure1B, stimulating cells with PGE2 significantly induced the expression of COX-2 at the low cellular density. COX-2 was also significantly induced by the treatment with PGE2 at the middle cellular density, whereas the production of COX-2 was significantly lower at the high cellular density than at the low cellular density. The induction of COX-2 was previously reported via the activation of ERKs in HCA-7 cells (Yoshida et al. 2013); therefore, the PGE2-stimulated phosphorylation of ERKs was examined at different cellular densities. As with the induction of COX-2, the treatment with PGE2 significantly induced the phosphorylation of ERKs at the low cellular density (Fig.1C). However, PGE2 only slightly induced the phosphorylation of ERKs at the middle cellular density. In contrast, the phosphorylation of ERKs was significantly reduced by the PGE2 treatment at the high cellular density. To ensure the equal loading of proteins, blots were stripped and re-probed with antibodies to β-tubulin or ERKs in Figure1B or C, respectively. As shown in the lower panels of Figure1B and C, nearly identical amounts of proteins were present under both experimental conditions.
Figure 1.
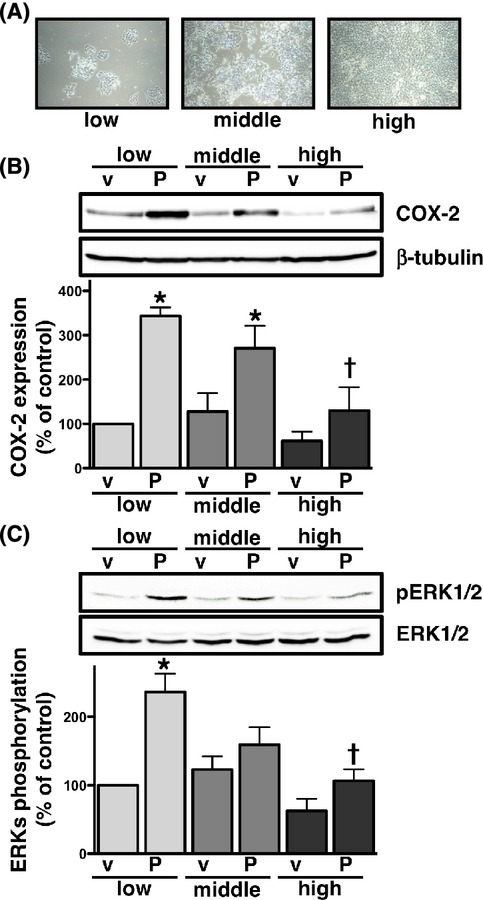
Effects of cellular density on PGE2-induced COX-2 expression (B) or phosphorylation of ERKs (C) in HCA-7 cells. Cells were cultured at low (3 × 104 cells/each 6 well), middle (1 × 105 cells/each 6 well), and high (3 × 105 cells/each 6 well) densities (A). Each cellular density was treated with either vehicle (v) or 1 μmol/L PGE2 (P) for 6 h (B) or 15 min (C) at 37°C and then subjected to immunoblot analysis with an antibody against COX-2 (B) or phospho-ERKs 1 and 2 (pERK1/2) (C), as described in the Materials and Methods. The blots were stripped and re-probed with an antibody against β-tubulin (B) or ERKs 1 and 2 (ERK1/2) (C). The bar graphs represent the ratio of COX-2 to β-tubulin (B) or pERK1/2 to total ERK1/2 (C) as assessed with pooled densitometric data (mean ± SD) from more than three independent experiments. Data are normalized to the ratio of COX-2 to β-tubulin (B) or pERK1/2 to total ERK1/2 (C) of vehicle-treated controls at the low cellular density as 100%. *P < 0.05, ANOVA, significantly different from the vehicle treatment. †P < 0.05, ANOVA, significantly different from PGE2-stimulated HCA-7 cells at the low cellular density.
COX-2 expression and phosphorylation of ERKs were not decreased by an increase in cellular density when cells were stimulated with EGF
Previous studies demonstrated that stimulating cells with EGF induced COX-2 expression as well as the phosphorylation of ERKs (Yoshida et al. 2013; Tomas et al. 2014). As shown in Figure2A, stimulating cells with EGF significantly induced the production of COX-2 at equivalent levels to the PGE2 stimulation at the low cellular density. However, in contrast to the PGE2 stimulation, the expression of EGF-stimulated COX-2 was also significantly induced at the middle and high densities to the same extent as that observed at the low cellular density. Similar results were found for the phosphorylation of ERKs when cells were treated with EGF, as shown in Figure2B. Although basal levels increased slightly in a cellular density-dependent manner, cellular density may not be involved in the induction of COX-2 expression or phosphorylation of ERKs in HCA-7 cells with the EGF treatment, unlike the PGE2 treatment.
Figure 2.
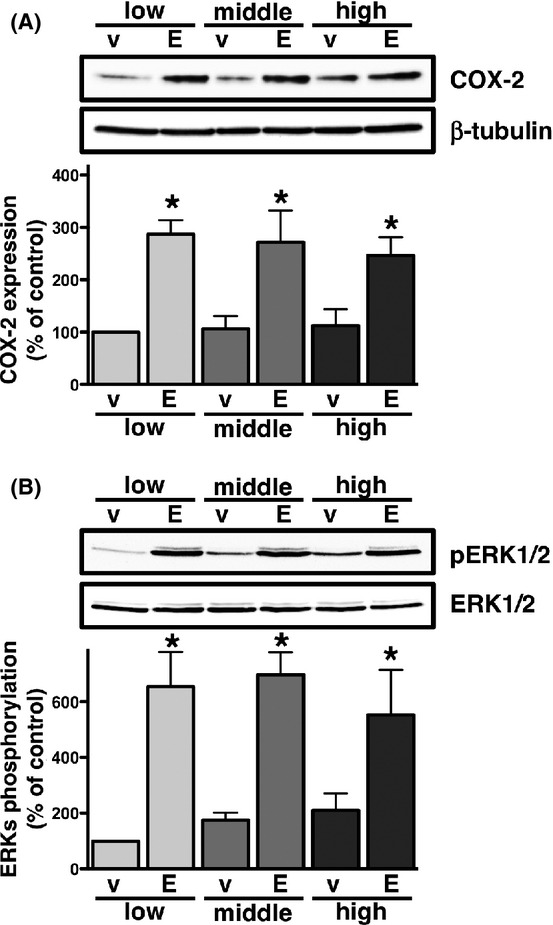
Effects of cellular density on EGF-induced COX-2 expression (A) or phosphorylation of ERKs (B) in HCA-7 cells. Cells were cultured at low (3 × 104 cells/each 6 well), middle (1 × 105 cells/each 6 well), and high (3 × 105 cells/each 6 well) densities. Each cellular density was treated with either vehicle (v) or 50 ng/mL of EGF (E) for 6 h (A) or 15 min (B) at 37°C and then subjected to immunoblot analysis with an antibody against COX-2 (A) or phospho-ERKs 1 and 2 (pERK1/2) (B) as described in the Materials and Methods. The blots were stripped and re-probed with an antibody against β-tubulin (A) or ERKs 1 and 2 (ERK1/2) (B). The bar graphs represent the ratio of COX-2 to β-tubulin (A) or pERK1/2 to total ERK1/2 (B) as assessed with pooled densitometric data (mean ± SD) from more than three independent experiments. Data are normalized to the ratio of COX-2 to β-tubulin (B) or pERK1/2 to total ERK1/2 (C) of vehicle-treated controls at the low cellular density as 100%. *P < 0.05, ANOVA, significantly different from the vehicle treatment.
The expression and function of EP4 receptors were cellular density-dependently down-regulated
The induction of COX-2 expression by PGE2 was previously shown to involve the activation of EP4 receptors, which was followed by the transactivation of EGF receptors (Yoshida et al. 2013). Since EGF receptor-mediated COX-2 expression was not affected by cellular density, the most plausible target that was functionally changed by cellular density was an upstream component from the EGF receptor, that is, the receptor for PGE2 in HCA-7 cells. Therefore, the expression of EP4 receptors was examined using HCA-7 cells cultured at different cellular densities. As shown in Figure3A, the expression of EP4 receptors in HCA-7 cells was significantly decreased in a cellular density-dependent manner. On the other hand, no significant differences were observed in the expression levels of EGF receptors at the different cellular densities (Fig.3B). To confirm that the reduction in the protein expression of the EP4 receptors was due to the down-regulation of EP4 receptors at the transcriptional level, we examined the expression levels of EP4 receptor mRNAs by quantitative PCR and the promoter activity of EP4 receptor genes by a luciferase assay using a reporter plasmid containing the EP4 receptor promoter region. As shown in Figure3C and D, mRNA expression levels as well as promoter activities of the EP4 receptors were significantly decreased in a cellular density-dependent manner. Since EP4 receptors conventionally couple to the Gαs protein, the effects of cellular density on the receptor function of EP4 was then examined using the cAMP assay. HCA-7 cells cultured at different cellular densities were treated with 1 nmol/L to 10 μmol/L of PGE2 for 1 h. As shown in Figure3E, the formation of cAMP was dose-dependently increased at the low cellular density; however, the effects of PGE2 on cAMP formation at both the middle and high cellular densities were negligible. Therefore, the PGE2-induced formation of cAMP was significantly reduced in a cellular density-dependent manner. Of the four EP receptor subtypes, EP4 receptors are reported to be expressed predominantly in HCA-7 cells (Yokoyama et al. 2013). However, HCA-7 cells have also been shown to express the mRNAs of EP2, EP3, and EP4 receptors (Fujino et al. 2011). Therefore, we attempted to confirm whether the reduction observed in the formation of cAMP at the middle and high cellular densities was due to the decreased expression of Gαs-coupled EP2 receptors and/or induction of Gαi-coupled EP3 receptors. EP2 and EP3 receptor promoter activities were examined at different cellular densities, using the luciferase reporter gene assay. As shown in Figure3F, neither a significant reduction in EP2 promoter activity nor significant induction of EP3 promoter activity were observed at the different cellular densities. These results suggested that the decrease observed in the formation of cAMP at the middle and high cellular densities may have been due to the reduced expression of EP4 receptors, as shown in Figure3A, C, and D.
Figure 3.
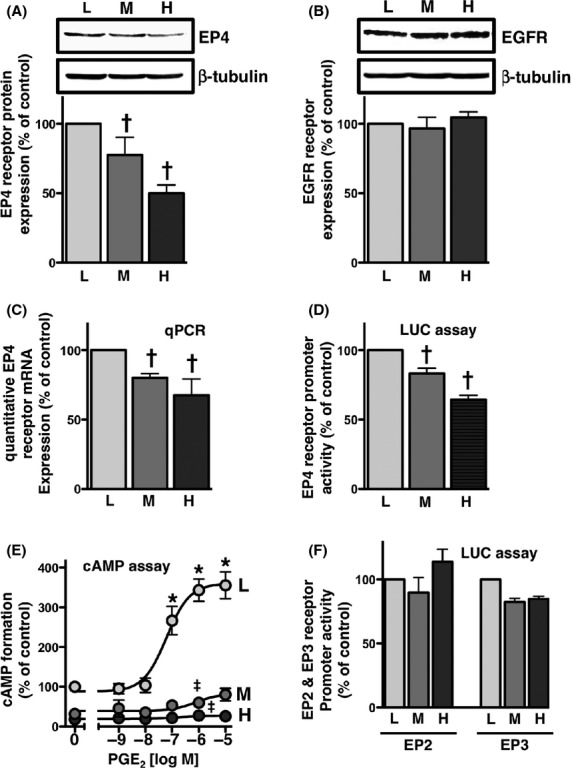
EP4 receptors were down-regulated in a cellular density-dependent manner in HCA-7 cells. Cells were cultured at low (L; 3 × 104 cells/each 6 well), middle (M; 1 × 105 cells/each 6 well), and high (H; 3 × 105 cells/each 6 well) densities. Each cellular density was then subjected to immunoblot analysis with an antibody against EP4 (A) or EGFR (B), quantitative PCR (C), the EP4 promoter-luciferase assay (D), PGE2-stimulated cAMP assay (E), or EP2 or EP3 promoter-luciferase assay (F), as described in the Materials and Methods. (A, B) The blots were stripped and re-probed with an antibody against β-tubulin. The bar graphs represent the ratio of COX-2 to β-tubulin as assessed with pooled densitometric data (mean ± SD) from more than three independent experiments. Data are normalized to the ratio of COX-2 to β-tubulin at the low cellular density as 100%. (C) The RNA of HCA-7 cells at each cellular density was isolated and used for quantitative real-time PCR with primers specific for either human EP4 or GAPDH, as described in the Materials and Methods. Data were analyzed by comparative methods relative to the expression of GAPDH and were normalized to the results obtained at the low cellular density as 100%. Data are the mean ± SD from three independent experiments. (D, F) Each cellular density was transiently transfected with human EP4 receptor promoter- (D), human EP2 promoter-, or EP3 promoter- (F) responsive luciferase reporter genes and control pRL-CMV or pGL3-CMV plasmids, and luciferase activity was determined as described in the Materials and Methods. Data were analyzed by comparative methods relative to the results of control luciferase activity levels, and were normalized to the results obtained at the low cellular density as 100%. Data are the mean ± SD from three independent experiments. (E) Each cellular density was treated with the indicated concentrations of PGE2 for 60 min. cAMP formation was determined as described in the Materials and Methods. Data are the means ± SEM of three independent experiments, with each being performed in duplicate. Data were normalized to the vehicle-treated control at the low cellular density as 100%. *P, †P < 0.05, ANOVA, significantly different from the vehicle-stimulated HCA-7 cells at the low cellular density. ‡P < 0.05, ANOVA, significantly different from PGE2-stimulated HCA-7 cells at the low cellular density (the analyzed results were shown for 1 μmol/L PGE2 only).
The expression of HIF-1α was cellular density-dependently up-regulated
HIF-1α, a DNA binding complex generally shown to be regulated by oxygen, is also reported to be induced by a high cellular density (Wiesener et al. 1998; Paltoglou and Roberts 2005). As shown in Figure4A, the increase observed in the expression of the HIF-1α protein was confirmed to be dependent on cellular density. To determine if the increase in HIF-1α protein levels was involved in activating the target gene as a transcriptional factor, HIF-1α-mediated transcriptional activity was examined using the HRE-contained luciferase reporter plasmid (Ji et al. 2010). Figure4B shows that the luciferase transcriptional activity of HRE was dependent on increases in cellular density; therefore, the increase observed in HIF-1α levels was associated with an enhancement in the activity of its transcriptional potential in HCA-7 cells cultured at a high cellular density.
Figure 4.
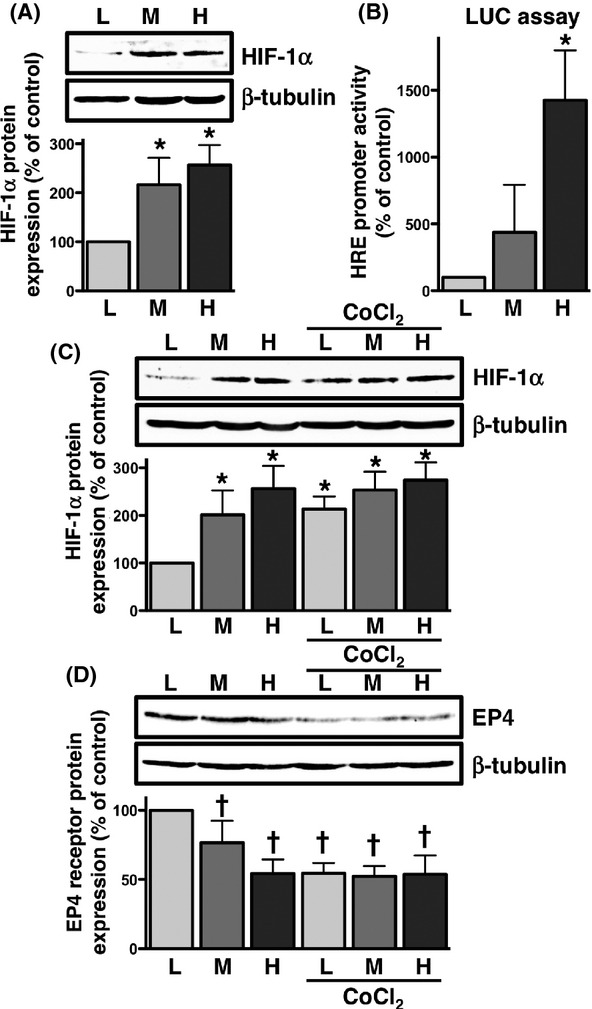
HIF-1α was cellular density- and CoCl2-dependently up-regulated, and EP4 receptors were down-regulated by CoCl2 in HCA-7 cells. Cells were cultured at low (L; 3 × 104 cells/each 6 well), middle (M; 1 × 105 cells/each 6 well), and high (H; 3 × 105 cells/each 6 well) densities. Each cellular density was then subjected to immunoblot analysis with antibody against HIF-1α (A, C) or the EP4 receptor (D), or the HIF-responsive HRE luciferase assay (B), as described in the Materials and Methods. A, The blots were stripped and re-probed with an antibody against β-tubulin. The bar graphs represent the ratio of HIF-1α to β-tubulin as assessed with pooled densitometric data (mean ± SD) from more than three independent experiments. Data were normalized to the ratio of HIF-1α to β-tubulin at the low cellular density as 100%. (B) Each cellular density was transiently transfected with the HRE luciferase reporter genes and control pRL-CMV plasmid, and luciferase activity was determined as described in the Materials and Methods. Data were analyzed by comparative methods relative to the results of control renilla luciferase activity levels, and were normalized to the results obtained at the low cellular density as 100%. Data are the mean ± SD from three independent experiments. (C, D) Each cellular density was treated with 100 μmol/L CoCl2 for 16 h, then subjected to immunoblot analysis with an antibody against HIF-1α (C) or the EP4 receptor (D), as described in the Materials and Methods. The blots were stripped and re-probed with an antibody against β-tubulin. The bar graphs represent the ratio of HIF-1α (C) or EP4 receptors (D) to β-tubulin as assessed with pooled densitometric data (mean ± SD) from more than three independent experiments. Data are normalized to the ratio of HIF-1α (C) or EP4 receptors (D) to β-tubulin at the low cellular density as 100%. *P < 0.05, ANOVA, significantly different from the vehicle treatment. †P < 0.05, ANOVA, significantly different from the low cellular density control.
Enhanced expression of HIF-1α by CoCl2 was accompanied by a decrease in EP4 receptor expression
Cobalt chloride (CoCl2) is known to be a chemical inducer of HIF-1 (Piret et al. 2002); therefore, the effects of CoCl2 on HIF-1α expression in HCA-7 cells cultured at different cellular densities were examined. As shown in Figure4C, the expression of HIF-1α was significantly increased by the treatment with CoCl2, especially at the low cellular density. Since certain levels of the HIF-1α protein were intrinsically expressed at the middle and high cellular densities, no further increment was observed in its expression by the treatment with CoCl2. The relationship between the increase in HIF-1α expression and decrease in EP4 receptor expression was then examined by western blot analysis using different densities of cells treated with CoCl2. Figure4D showed that the treatment with CoCl2 eliminated the cellular density-dependent decrease in EP4 receptor expression. Thus, as inversely correlated with the increment in HIF-1α as shown in Figure4C, the expression of EP4 receptors was significantly decreased by the treatment with CoCl2. Therefore, a negative correlation may exist between the expression levels of HIF-1α and EP4 receptors.
Knockdown of HIF-1α by siRNA reduced HRE luciferase activity and increased EP4 receptor expression and promoter activity
To confirm if the expression of HIF-1α was involved in the expression of EP4 receptors, we used siRNA for HIF-1α to examine the protein expression levels of EP4 receptors and their promoter transcriptional activities. The potency of siRNA for HIF-1α was initially evaluated using western blot analysis and HIF-1α-mediated transcriptional activity with the HRE luciferase reporter plasmid. As shown in Figure5A and B, the transfection of siRNA to HCA-7 cells significantly reduced HIF-1α protein expression at the middle and high cellular densities and HRE promoter activity at the high cellular density. These results confirmed the potency of siRNA; therefore, its effects on the expression and promoter transcriptional activity of EP4 receptors were subsequently examined. The results of western blotting showed that blocking the expression and activity of HIF-1α with siRNA inhibited the reduction in EP4 receptor protein expression and EP4 receptor promoter-mediated luciferase activity, and this increase was significant at the high cellular density, as shown in Figure5C and D, respectively. Thus, these results suggested that HIF-1α may inversely regulate the expression of EP4 receptors.
Figure 5.
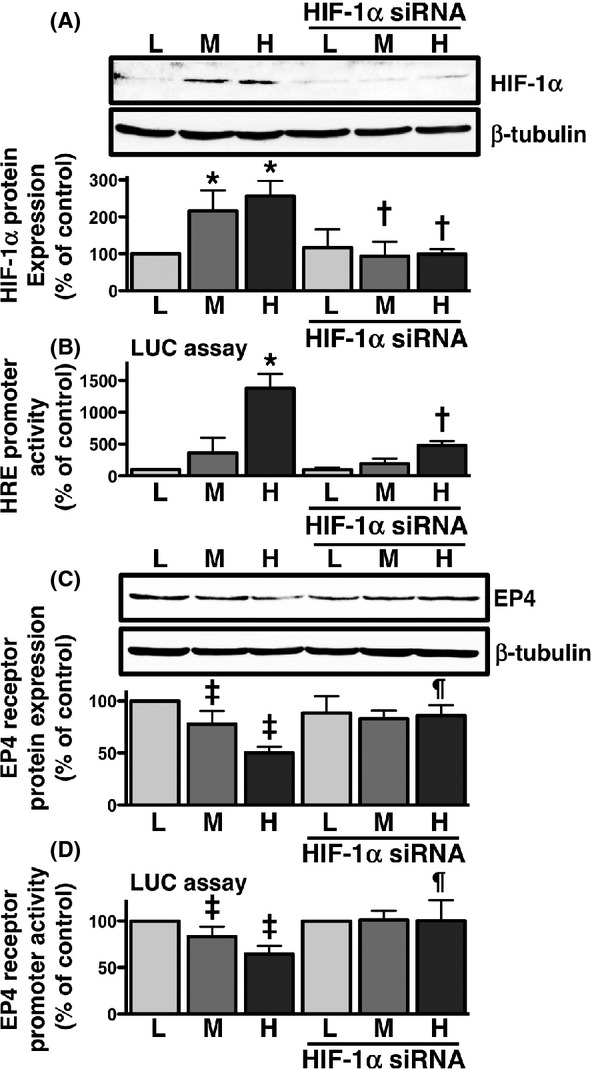
Effects of HIF-1α siRNA on HIF-1α protein expression, HIF-responsive luciferase activity, EP4 receptor expression, and EP4 receptor promoter-responsive luciferase activity in HCA-7 cells cultured at different cellular densities. Cells were cultured at low (L; 3 × 104 cells/each 6 well), middle (M; 1 × 105 cells/each 6 well), and high (H; 3 × 105 cells/each 6 well) densities. Each cellular density was transfected with HIF-1α siRNA for 48 h, and cells were then subjected to immunoblot analysis with antibody against HIF-1α (A) or EP4 receptors (C), or the luciferase assay for HIF-responsive HRE (B) and EP4 receptor promoter-responsive (D) activities, as described in the Materials and Methods. (A, C) The blots were stripped and re-probed with an antibody against β-tubulin. The bar graphs represent the ratio of HIF-1α (A) or EP4 receptors (C) to β-tubulin as assessed with pooled densitometric data (mean ± SD) from more than three independent experiments. Data were normalized to the ratio of HIF-1α (A) or EP4 receptors (C) to β-tubulin at the low cellular density as 100%. (B, D) Each cellular density was transiently transfected with HIF-responsive HRE luciferase reporter genes (B) or EP4 receptor promoter luciferase reporter genes (D) and the control pRL-CMV plasmid 20 h after siRNA transfection, and luciferase activity was determined, as described in the Materials and Methods. Data were analyzed by comparative methods relative to the results of control renilla luciferase activity levels, and were normalized to the result obtained at the low cellular density as 100%. Data are the mean ± SD from three independent experiments. *P, ‡P < 0.05, ANOVA, significantly different from control transfection at the low cellular density. †P, ¶P < 0.05, ANOVA, significantly different from each density control that was not transfected with siRNA.
Knockdown of HIF-1α by siRNA restored PGE2-induced reductions in COX-2 expression in high cellular density-cultured cells
The effects of blocking HIF-1α with siRNA on PGE2-induced COX-2 expression was confirmed, particularly in HCA-7 cells cultured at a high cellular density. As shown in Figure6, the stimulation with PGE2 did not induce the expression of COX-2 at a high density of HCA-7 cells (lane e vs. lane a), similar to that shown in Figure1B. However, when cells were transfected with HIF-1α siRNA, the expression level of COX-2 was increased (lane h vs. lane e) to a similar level to the PGE2-induced expression of COX-2 at the low cellular density (lane h vs. lane c). This effect of siRNA was also observed in cells cultured at the middle cellular density (lane g vs. lane d). In addition, the expression of COX-2 was unaffected in cells cultured at the high cellular density transfected with HIF-1α siRNA alone without the PGE2 stimulation (lane b vs. lane a). Thus, restoring the expression of COX-2 may reflect the restoration of EP4 receptor expression by offsetting the increment in cellular density-dependent HIF-1α expression by siRNA, especially in HCA-7 cells cultured at the high cellular density.
Figure 6.
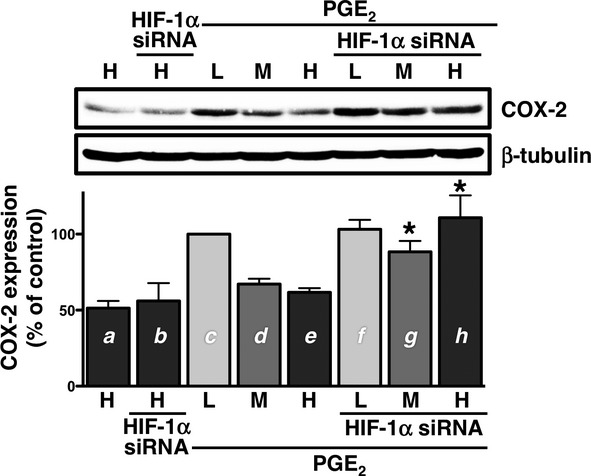
Effects of HIF-1α siRNA on PGE2-induced COX-2 expression in high density cultured HCA-7 cells. Cells were cultured at low (L; 3 × 104 cells/each 6 well), middle (M; 1 × 105 cells/each 6 well), and high (H; 3 × 105 cells/each 6 well) densities. Cells were transfected with HIF-1α siRNA for 48 h, treated with either vehicle or 1 μmol/L PGE2 for 6 h at 37°C, and then subjected to immunoblot analysis with an antibody against COX-2, as described in the Materials and Methods. The blots were stripped and re-probed with an antibody against β-tubulin. The bar graphs represent the ratio of COX-2 to β-tubulin as assessed with pooled densitometric data (mean ± SD) from more than three independent experiments. Data were normalized to the ratio of COX-2 to β-tubulin of PGE2-treated controls at the low cellular density (c) as 100%. *P < 0.05, t-test, significantly different from d (without HIF-1α siRNA at the middle cellular density) to g (with siRNA at the middle cellular density) and/or e (without HIF-1α siRNA at the high cellular density) to h (with siRNA at the high cellular density), respectively.
Discussion and Conclusions
The present study revealed a clear inverse correlation between cellular density/HIF-1α expression and EP4 receptor/PGE2-induced COX-2 expression, as depicted in Figure7. As shown in Figure1 and previously reported (Yoshida et al. 2013), PGE2 has the ability to strongly induce expression of COX-2 via EP4 receptors at a low cellular density. However, the effect of PGE2 on COX-2 induction was significantly reduced under the high cellular density condition. In addition, the results obtained at the middle cellular density were inconclusive, such that the outcomes were vague and unclear; they were similar to those at the high cellular density and other times to the low cellular density. Similar patterns have been also reported previously (Paltoglou and Roberts 2005).
Figure 7.
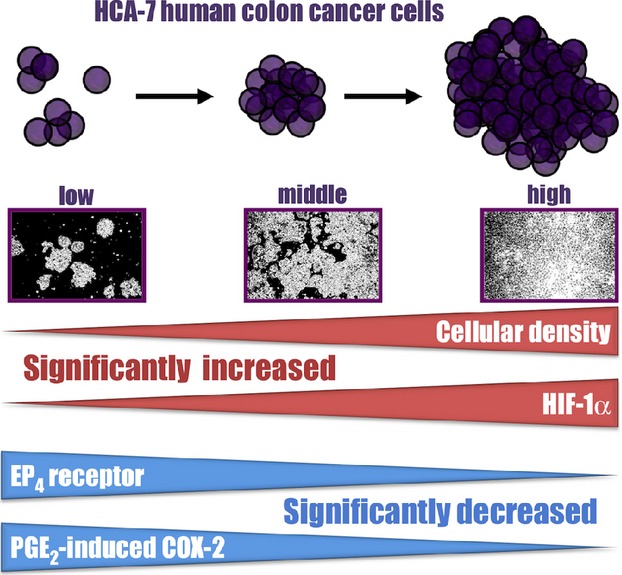
Schema for the cellular density-dependent regulation of HIF-1α, EP4 receptor, and PGE2-induced COX-2 expression in HCA-7 cells. The expression of HIF-1α increased in a cellular density-dependent manner, whereas that of the EP4 receptors and PGE2-induced COX-2 decreased.
Interestingly, even at higher, and possibly close to the maximum expression levels of HIF-1α at the high cellular density, quantitative PCR, luciferase assay, and western blotting revealed that the expression levels of EP4 receptors only decreased by 50% (Fig.3A, C, and D). HIF-1α has been implicated in the induction of SP-1 and EGR-1 transcriptional factors (Wong et al. 2010), which are known to be critical factors in regulation of the EP4 receptor promoters (Kambe et al. 2008). Thus, one possible explanation is that either the direct or indirect regulation of reductions in the expression of EP4 receptors by HIF-1α may be accompanied by the induction of SP-1 and EGR-1 expression, which counteractively induces receptor expression. Although the expression of EP4 receptors was not completely reduced at the high cellular density, their functions were eliminated because the reduction in COX-2 expression (Fig.1B), and also that in cAMP formation were almost completely ameliorated (Fig.3E) by the PGE2 stimulation. Therefore, although EP4 receptors may not be expressed on membranes as functional receptors at the high cellular density, they could be diffusely expressed in the cytoplasm, as reported previously in inflammatory cells (Lejeune et al. 2010). We do not yet have strong evidence for the localization of EP4 receptors in HCA-7 cells cultured with different cellular densities; therefore, the mechanism responsible for how the cellular density-dependent increases in HIF-1α regulated EP4 receptor expression currently remains unknown. However, as shown in Figure5, since the knockdown of HIF-1α mRNA by siRNA recovered the expression of EP4 receptors, even at the high cellular density, the involvement of HIF-1α in regulating the expression of EP4 receptors is clear.
An increase in the expression of COX-2 is generally considered to be a hallmark for the development of colon cancer; therefore, an increase in the expression of HIF-1α may induce the expression of COX-2 aside from the reduction in EP4 receptor expression. Indeed, HIF-1α was previously shown to up-regulate COX-2 expression by direct transcriptional regulation in HT-29 and HCT116 colon cancer cell lines under hypoxic conditions (Kaidi et al. 2006). In contrast, since COX-2 expression is down-regulated by transforming growth factor-β1 (TGF-β1), an increase in HIF-1α could reduce COX-2 expression by acting on the HRE of the TGF-β1 promoter region, followed by the production/secretion of that cytokine, as reported previously (Takai et al. 2013; Hung et al. 2013). However, in our experimental conditions, as shown in Figure6 lane b, the knockdown of HIF-1α by siRNA did not alter the expression of COX-2. Moreover, the EGF-induced expression of COX-2 was not altered by an increase in the expression of HIF-1α, as shown in Figure2A, the EP4 receptor-independent, but HIF-1α-mediated direct regulation of COX-2 expression may be not much involved in. Therefore, the PGE2-induced expression of COX-2 in HCA-7 cells may simply be regulated by EP4 receptor levels and their signaling pathways.
As described above, although the up-regulated expression of COX-2 is associated with the development of colon cancer, the underlying mechanism/meaning involving the cellular density-dependent loss of PGE2-induced COX-2 expression in HCA-7 cells remains unclear. COX-2 expression was previously detected from a very early stage in carcinoma growth (Fujita et al. 1998). Therefore, COX-2 expression may be induced in the early stages of colon cancer development, by PGE2-stimulated EP4 receptors, which may further induce COX-2 expression and cellular growth through a positive feedback loop. These results may be corroborated by previous findings in which an increment in HIF-1α occurred at the early stages of carcinogenesis and strongly correlated with malignant progression such as angiogenesis and metastasis (Mabjeesh and Amir 2007). Thus, an increase in HIF-1α may induce the expression of vascular endothelial growth factor (VEGF), thereby increasing neovascularzation for cancer growth, as reported previously (Semenza 2003; Mabjeesh and Amir 2007; Rohwer et al. 2013). We previously demonstrated that VEGF was constitutively secreted in HCA-7 cells, a condition corresponding to the middle to high cellular densities (Fujino et al. 2011). Meanwhile, the induction of HIF-1α by the increase in cellular growth should reduce the expression of EP4 receptors, as demonstrated in the present study. As we previously reported and stated earlier, HCA-7 cells also endogenously express EP3 prostanoid receptors (Fujino et al. 2011), and the expression level of EP3 receptors is likely remain unchanged by cellular density, as shown in Figure3F. Therefore, under these cellular conditions, exogenously applied PGE2 stimulated EP3 receptors, as shown previously, but not EP4 receptors (Fujino et al. 2011), and this may have been because EP4 receptors had already lost their functions. Moreover, the stimulation of EP3 receptors evoked cellular migration by enhancing the expression of VEGF receptor-1, which is known to play an important role in establishing cellular migration/metastasis, as we previously reported (Fujino et al. 2011). Thus, at the high cellular density, HCA-7 cells enhanced the expression levels of VEGF by increasing HIF-1α as well as VEGFR-1 expression, not by fewer EP4 receptors, but by the activation of stationary EP3 receptors that stimulate cellular migration such as invasion/metastasis. The exact meanings/reasons for a reduction in the expression of EP4 receptors by HIF-1α at the high cellular density currently remain unclear. However, the decrease in COX-2 levels may promote colon cancer cells from the cancer growth stage at the low cellular density to the metastatic stage at the high cellular density by switching the responsive primary EP receptor subtypes from EP4 receptors to EP3 receptors. EP3 receptors are known to have one of the highest affinities for PGE2 (Narumiya et al. 1999; Breyer et al. 2001); therefore, these receptors could be activated at even the low levels of PGE2 produced by the reduced expression of COX-2.
Although the increase observed in the expression of EP4 receptors during carcinogenesis has been widely accepted (Chell et al. 2006; Hawcroft et al. 2007), other studies demonstrated that EP4 receptor levels were significantly higher in normal colon tissue than in tumor tissue (Gustafsson et al. 2007). One possible explanation for these differences is that the expression of EP4 receptors could be changed under certain conditions such as cell density. The results of this study were exclusively obtained from HCA-7 cells; therefore, additional experiments need to be conducted using other colon cancer cell lines and/or other cancer cells. However, the results of the present study suggest that this novel regulation mechanism of EP4 receptor expression by HIF-1α may play important roles in determining the fate/stage of colon cancer via its surrounding environment.
Acknowledgments
This research was supported in part by a Grant-in-Aid for Scientific Research (22590079 and 25460091) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. In addition, we have no conflict of interest to declare.
Glossary
Abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- CoCl2
cobalt chloride
- COX
cyclooxygenase
- DMEM
Dulbecco's modified Eagle's medium
- EGF
epidermal growth factor
- EGR-1
early growth response factor-1
- EP
E-type prostanoid receptor
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HIF
hypoxia inducible factor
- HRE
hypoxia response element
- PCR
polymerase chain reaction
- PGE2
prostaglandin E2
- TGF
transforming growth factor
- VEGF
vascular endothelial growth factor
Disclosures
None declared.
References
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MA. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- Cassano G, Gasparre G, Susca F, Lippe C, Guanti G. Effect of prostaglandin E2 on the proliferation, Ca2+ mobilization and cAMP in HT-29 human colon adenocarcinoma cells. Cancer Lett. 2000;152:217–222. doi: 10.1016/s0304-3835(00)00339-6. [DOI] [PubMed] [Google Scholar]
- Chell SD, Witherden IR, Dobson RR, Moorghen M, Herman AA, Qualtrough D. Increased EP4 receptor expression in colorectal cancer progression promotes cell growth and anchorage independence. Cancer Res. 2006;66:3106–3113. doi: 10.1158/0008-5472.CAN-05-3702. , et al. ( [DOI] [PubMed] [Google Scholar]
- Eisinger AL, Prescott SM, Jones DA, Stafforini DM. The role of cyclooxygenase-2 and prostaglandin in colon cancer. Prostaglandins Other Lipid Mediat. 2007;82:147–154. doi: 10.1016/j.prostaglandins.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Fujino H, Regan JW. Prostanoid receptors and phosphatidylinositol 3-kinase: a pathway to cancer? Trends Pharmacol Sci. 2003;24:335–340. doi: 10.1016/S0165-6147(03)00162-7. [DOI] [PubMed] [Google Scholar]
- Fujino H, West KA, Regan JW. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J Biol Chem. 2002;277:2614–2619. doi: 10.1074/jbc.M109440200. [DOI] [PubMed] [Google Scholar]
- Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem. 2003;278:12151–12156. doi: 10.1074/jbc.M212665200. [DOI] [PubMed] [Google Scholar]
- Fujino H, Toyomura K, Chen XB, Regan JW, Murayama T. Prostaglandin E2 regulates cellular migration via induction of vascular endothelial growth factor receptor-1 in HCA-7 human colon cancer cells. Biochem Pharmacol. 2011;81:379–387. doi: 10.1016/j.bcp.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Fujita T, Matsui M, Takaku K, Uetake H, Ichikawa W, Taketo MM. Size- and invasion-dependent increase in cyclooxygenase 2 levels in human colorectal carcinomas. Cancer Res. 1998;58:4823–4826. , et al. ( [PubMed] [Google Scholar]
- Gustafsson A, Hansson E, Kressner U, Nordgren S, Andersson M, Wang W. EP1-4 subtype, COX and PPARγ receptor expression in colorectal cancer in prediction of disease-specific mortality. Int J Cancer. 2007;121:232–240. doi: 10.1002/ijc.22582. , et al. ( [DOI] [PubMed] [Google Scholar]
- Hawcroft G, Ko CWS, Hull MA. Prostaglandin E2-EP4 receptor signaling promotes tumorigenic behaviour of HT-29 human colorectal cancer cells. Oncogene. 2007;26:3006–3019. doi: 10.1038/sj.onc.1210113. [DOI] [PubMed] [Google Scholar]
- Hull MA, Ko SCW, Hawcroft G. Prostaglandin EP receptors: targets for treatment and prevention of colorectal cancer? Mol Cancer Ther. 2004;3:1031–1039. [PubMed] [Google Scholar]
- Hung SP, Yang MH, Tseng KF, Lee OK. Hypoxia-induced secretion of TGF-β1 in mesenchymal stem cell promotes breast cancer cell progression. Cell Transplant. 2013;22:1869–1882. doi: 10.3727/096368912X657954. [DOI] [PubMed] [Google Scholar]
- Ji R, Chou CL, Xu W, Chen XB, Woodward DF, Regan JW. EP1 prostanoid receptor coupling to Gi/o up-regulates the expression of hypoxia-inducible factor-1α through activation of phosphoinositide-3 kinase signaling pathway. Mol Pharmacol. 2010;77:1025–1036. doi: 10.1124/mol.110.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006;66:6683–6691. doi: 10.1158/0008-5472.CAN-06-0425. [DOI] [PubMed] [Google Scholar]
- Kambe A, Iguchi G, Moon Y, Kamitani H, Watanabe T, Eling TE. Regulation of EP4 expression via Sp-1 transcription factor: inhibition of expression by anti-cancer agents. Biochim. Biophys. Acta. 2008;1783:1211–1219. doi: 10.1016/j.bbamcr.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther. 2013;138:485–502. doi: 10.1016/j.pharmthera.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschel A, Simon P, Tug S. Functional regulation of HIF-1α under normoxia-is there more than post-translational regulation? J Cell Physiol. 2012;277:514–524. doi: 10.1002/jcp.22798. [DOI] [PubMed] [Google Scholar]
- Lejeune M, Leung P, Beck PL, Chadee K. Role of EP4 receptor and prostaglandin transporter in prostaglandin E2-induced alteration in colonic epithelial barrier integrity. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1097–G1105. doi: 10.1152/ajpgi.00280.2010. [DOI] [PubMed] [Google Scholar]
- Lesuffleur T, Barbat A, Dussaulx E, Zweibaum A. Growth adaptation to methotrexate of HT-29 human colon carcinoma cells is associated with their ability to differentiate into columnar absorptive and mucus-secreting cells. Cancer Res. 1990;50:6334–6343. [PubMed] [Google Scholar]
- Mabjeesh NJ, Amir S. Hypoxia-inducible factor (HIF) in human tumorigenesis. Histol Histopathol. 2007;22:559–572. doi: 10.14670/HH-22.559. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Paltoglou SM, Roberts BJ. Role of the von Hippel-Lindau tumour suppressor protein in the regulation of HIF-1α and its oxygen-regulated transactivation domains at high cell density. Oncogene. 2005;24:3830–3835. doi: 10.1038/sj.onc.1208531. [DOI] [PubMed] [Google Scholar]
- Piret JP, Mottet D, Raes M, Michiels C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann N Y Acad Sci. 2002;973:443–447. doi: 10.1111/j.1749-6632.2002.tb04680.x. [DOI] [PubMed] [Google Scholar]
- Qiao L, Kozoni V, Tsioulias GJ, Koutsos MI, Hanif R, Shiff SJ. Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocyte in vivo. Biochim. Bipphys. Acta. 1995;1258:215–223. doi: 10.1016/0005-2760(95)00100-q. , et al. ( [DOI] [PubMed] [Google Scholar]
- Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–153. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Rohwer N, Zasada C, Kempa S, Cramer T. The growing complexity of HIF-1α's role in tumorigenesis: DNA repair and beyond. Oncogene. 2013;32:3569–3576. doi: 10.1038/onc.2012.510. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Takai E, Tsukimoto M, Kojima S. TGF-β1 downregulates COX-2 expression leading to decrease of PGE2 production in human lung cancer A549 cells, which is involved in fibrotic response to TGF-β1. PLoS ONE. 2013;8:e76346. doi: 10.1371/journal.pone.0076346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24:26–34. doi: 10.1016/j.tcb.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1α. Blood. 1998;92:2260–2268. , et al. ( [PubMed] [Google Scholar]
- Wong DL, Tai TC, Wong-Faull DC, Claycomb R, Siddall BJ, Bell RA. Stress and adrenergic function: HIF1α, a potential regulatory switch. Cell Mol Neurobiol. 2010;30:1451–1457. doi: 10.1007/s10571-010-9567-z. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward DF, Jones RL, Narumiya S. International union of basic and clinical pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013;65:1010–1052. doi: 10.1124/pr.112.007195. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Fujino H, Otake S, Seira N, Regan JW, Murayama Y. Induction of cyclooxygenase-2 expression by prostaglandin E2 stimulation of the prostanoid EP4 receptor via coupling to Gαi and transactivation of the epidermal growth factor receptor in HCA-7 human colon cancer cells. Eur J Pharmacol. 2013;718:408–417. doi: 10.1016/j.ejphar.2013.08.002. [DOI] [PubMed] [Google Scholar]