

Abstract
The major cause of death in patients with chronic kidney disease (CKD) is cardiovascular disease. Here, p-Cresyl sulfate (PCS), a uremic toxin, is considered to be a risk factor for cardiovascular disease in CKD. However, our understanding of the vascular toxicity induced by PCS and its mechanism is incomplete. The purpose of this study was to determine whether PCS enhances the production of reactive oxygen species (ROS) in vascular endothelial and smooth muscle cells, resulting in cytotoxicity. PCS exhibited pro-oxidant properties in human umbilical vein endothelial cells (HUVEC) and aortic smooth muscle cells (HASMC) by enhancing NADPH oxidase expression. PCS also up-regulates the mRNA levels and the protein secretion of monocyte chemotactic protein-1 (MCP-1) in HUVEC. In HASMC, PCS increased the mRNA levels of alkaline phosphatase (ALP), osteopontin (OPN), core-binding factor alpha 1, and ALP activity. The knockdown of Nox4, a subunit of NADPH oxidase, suppressed the cell toxicity induced by PCS. The vascular damage induced by PCS was largely suppressed in the presence of probenecid, an inhibitor of organic anion transporters (OAT). In PCS-overloaded 5/6-nephrectomized rats, plasma MCP-1 levels, OPN expression, and ALP activity of the aortic arch were increased, accompanied by the induction of Nox4 expression. Collectively, the vascular toxicity of PCS can be attributed to its intracellular accumulation via OAT, which results in an enhanced NADPH oxidase expression and increased ROS production. In conclusion, we found for the first time that PCS could play an important role in the development of cardiovascular disease by inducing vascular toxicity in the CKD condition.
Keywords: NADPH oxidase, oxidative stress, p-cresyl sulfate, uremic toxin, vascular damage
Introduction
The major cause of death in patients with chronic kidney disease (CKD), especially those who are undergoing dialysis, is cardiovascular disease (Anavekar and Pfeffer 2004). Endothelial dysfunction and vascular calcification play a pivotal role in the development of cardiovascular morbidity and the subsequently increased mortality (Ross 1999). In addition, endothelial dysfunction/vascular calcification and oxidative stress are prominent features in patients with CKD. Although the relation between this vascular damage and oxidative stress in CKD patients (Annuk et al. 2001) suggests that common causative factors or mechanisms that are interrelated could be involved, but the potential causes of this are not fully understood.
In CKD, metabolic changes and an impaired urinary excretion of metabolites leads to the accumulation of uremic toxins in the body (Duranton et al. 2012). Recent clinical studies have shown that the accumulation of a uremic toxin, p-cresyl sulfate (PCS) is related to the progression of CKD. Indeed, the buildup of PCS appears to be associated with the renal survival and cardiovascular survival rate for CKD patients (Bammens et al. 2006; Liabeuf et al. 2010; Wu et al. 2012). As such, PCS has the potential to function as a good predictor for the prognosis of CKD patients. For this reason, the biological activities of PCS have been the subject of considerable research interest, especially the toxicity of PCS in the kidney and the vascular system. We and other researchers recently proposed that several pathological mechanisms such as oxidative stress, inflammatory reaction, and renin-angiotensin-aldosterone system activation well contribute to the chronic kidney injury induced by PCS (Sun et al. 2012; Watanabe et al. 2013a). However, the effect of PCS on vascular endothelial and smooth muscle cells remains largely unknown. In fact, our previous investigations showed that PCS serves as a substrate for the organic anion transporter (OAT) and accumulates in the kidney via these transporters (Miyamoto et al. 2011; Watanabe et al. 2011, 2013b). Since it has also been reported that OAT is also expressed in vascular endothelial and smooth muscle cells (Yamamoto et al. 2006; Muteliefu et al. 2009; Ito et al. 2010), PCS would be expected to accumulate in vascular cells via OAT. If PCS causes vascular damage by similar mechanisms observed in kidney, PCS may accumulate in vascular endothelial and smooth muscle cells and induce oxidative stress by virtue of the production of radical oxygen species (ROS), thereby resulting in the development of cytotoxicity.
In the present study, we report on the characterization of the pro-oxidant properties of PCS and its vascular toxicity, such as the induction of the monocyte chemotactic protein-1 (MCP-1) and the osteoblast-specific protein, using human umbilical vein endothelial cells (HUVEC) and human aortic smooth muscle cells (HASMC). Moreover, to further confirm the action of PCS in vivo, PCS was administrated to 5/6-nephrectomized rats and its vascular toxicity was evaluated.
Materials and Methods
Chemicals and materials
PCS was synthesized following the method described by Feigenbaum and Neuberg. The product was >99% pure and its structure was confirmed by nuclear magnetic resonance spectroscopy (Feigenbaum and Neuberg 1941). Probenecid, diphenylene iodonium (DPI), wortmannin, calphostin C, dulbecco's phosphate-buffered saline (D-PBS), the mouse monoclonal anti-human β-actin antibody were purchased from Sigma-Aldrich (St Louis, MO). N-acetyl-l-cysteine (NAC) was purchased from Nacalai Tesque (Kyoto, Japan). 5-(and 6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), and OPTI-MEM I reduced serum medium were purchased from Gibco (Invitrogen, Grand Island, NY). Lipofectamine RNAiMAX, Human Nox4 siRNA, control siRNA, Alexa Fluor 546 goat anti-rabbit IgG (H+L) was purchased from Invitrogen. Human MCP-1, human alkaline phosphatase (ALP), human osteopontin (OPN), and human core-binding factor alpha 1 (Cbfα1) primers were purchased from Hokkaido System Science (Hokkaido, Japan). The rabbit polyclonal anti-human and rat Nox4 (clone #PAB12094) antibody was purchased from Abnova (Taipei, Taiwan). Mouse monoclonal anti-OPN (clone #AKm2A1) antibody was purchased from Santa Cruz Biotechnology (Dallas, USA). SuperSignal West Pico Chemiluminescent substrate was purchased from Thermo Scientific (Yokohama, Japan). All reagents obtained from commercial sources were of the highest grade available.
Cell cultures
HUVEC (American Type Culture Collection, Manassas, VA) were cultured in MCDB131 w/o glutamine (Gibco, Invitrogen, NY) containing 10% fetal bovine serum (FBS) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B at 37°C in an atmosphere of 5% CO2. HASMC (DS Pharma Biomedical, Tokyo, Japan) were maintained in The Dulbecco's Modified Eagle's medium (DMEM) containing SmGM-2 SingleQuots (Lonza, CA) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B at 37°C under 5% CO2 humidified atmosphere. Only cells (HASMC) between passages 4 and 10 were used for experiments.
Measurement of ROS production
To measure the production of ROS (largely peroxide), CM-H2DCFCA, an ROS-sensitive fluorescent dye, was used as an ROS probe. HUVEC or HASMC were incubated in 96-well plates (1 × 104 cells/well) in their medium at 37°C for 24 h, and then with 5 μmol/L CM-H2DCFCA for 30 min in D-PBS. After removing the D-PBS from the wells, the HUVEC or HASMC were incubated with different concentrations of PCS (10, 100, 500, 1000 μmol/L) in D-PBS for 1 h, in the presence or absence of 4% HSA. To determine the effect of inhibitors of OATs and NADPH oxidase, or an antioxidant on PCS-induced ROS production, HUVEC or HASMC was incubated in 96-well plates (1 × 104 cells/well) in their medium at 37°C for 24 h, and then with 5 μmol/L CM-H2DCFCA for 30 min in D-PBS. After removing the D-PBS from the wells, the HUVEC or HASMC was incubated with probenecid (1000 μmol/L), DPI (50 μmol/L), wortmannin (5 μmol/L), calphostin C (1 μmol/L), and NAC (1000 μmol/L) for 30 min, and then incubated with 1000 μmol/L PCS for 1 h. CM-H2DCFCA and each inhibitor were dissolved in N,N-dimethylformamide (Nacalai tesque). Final concentration of N,N-dimethylformamide was <0.1% in the medium. The solvent did not show antioxidative effects. Fluorescence intensity was measured at excitation 485 nm and emission 535 nm using a fluorescence microplate reader SPECTRAfluor Plus; Tecan, Männedorf, Switzerland.
Quantitative RT-PCR
Total RNA was extracted from the cultured cells using a High Pure RNA Isolation Kit (RNAiso Plus; Takara Bio, Shiga, Japan). The purity and quantity of the RNA preparations were determined by measuring the optical densities of the solutions at 260 and 280 nm. To estimate the mRNA levels of MCP-1, ALP, OPN, and Cbfα1, quantitative RT-PCR was performed using the LightCycler system (BIO-RAD, Hercules, CA). Total RNA was reverse transcribed with random hexamers using a first Strand cDNA Synthesis Kit for RT-PCR (RT Master Mix; Takara Bio). The reaction was processed as follows: samples were incubated at 25°C for 10 min and then at 42°C for 60 min, followed by 99°C for 5 min and cooling to 4°C for 5 min. The quantitative PCR was performed with the LightCycler FastStart DNA Master SYBR Green I (Takara Bio), LightCycler-primerSet, human MCP-1 primers (forward: 5′-CAAGCAGAAGTGGGTTCA-3′; reverse: 5′-GGGAAAGCTAGGGGAAAATAAG-3′), human ALP primers (forward: 5′-AGGTCTTGGGGTGCACCATGATT-3′; reverse: 5′-AGCCACGTTGGTGTTGAGCTTC-3′), human OPN primers (forward: 5′-CAGAATGCTGTGTCCTCTGAAGA-3′; reverse: 5′-GTCAATGGAGTCCTGGCTGTC-3′), human Cbfα1 primers (forward: 5′-ATGCTTCATTCGCCTCACAAAC-3′; reverse: 5′-CCAAAAGAAGTTTTCCTGACATGG-3′). The LightCycler system consists of a rapid PCR cycler and fluorescence detection component, which allows the real-time monitoring of fluorescence during PCR amplification. SYBR Green I, a double-stranded DNA-binding fluorescent dye, was used to monitor the cDNA amplification. The mRNA levels were measured as the ratio of each mRNA to 18S ribosomal RNA.
Western blotting analysis
HUVEC or HASMC was solubilized in 1% Triton X-100/PBS buffer containing a 50× protease inhibitor cocktail (Nacalai Tesque). The aortic arch was homogenized in a homogenization buffer composed of 70 mmol/L sucrose, 10 mmol/L 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid sodium salt (HEPES), 210 mmol/L mannitol, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L ethylene glycol tetraacetic acid (EGTA), pH 7.5, 200 μmol/L dithiothreitol, and 1% protease inhibitor cocktail. The homogenate was centrifuged at 10,000g for 5 min at 4°C, the supernatants were recovered. After measurement of the protein content, each sample was suspended in a loading buffer (2% SDS, 62.5 mmol/L Tris-HCl and 1% 2-mercaptoethanol). These samples (40 μg) were run on 12.5% sodium dodecyl sulfate polyacrylamide gels, followed by electrophoretic transfer to nitrocellulose membranes. The membranes were blocked with 5% skimmed milk in PBS for 1 h at room temperature and then incubated with rabbit polyclonal anti-human or rat Nox4 (1:500) antibody, mouse polyclonal OPN (1:500) antibody or mouse monoclonal anti-human β-actin antibody (1:5000) overnight at 4°C. The membranes were washed with 0.05% Tween-20 (T-PBS) and then horseradish peroxidase-conjugated anti-rabbit IgG antibody (1:5000), and an anti-mouse IgG antibody (1:10000) was used for the detection of the target proteins. SuperSignal Western blotting detection reagents (Thermo Scientific, Rockford, IL) were used for immunodetection.
Knockdown of Nox4 by small interfering RNA
Nox4 Stealth™ RNAi or a Stealth™ RNAi negative control duplex was transfected into HUVEC or HASMC, according to the manufacturer's recommendations. Stealth siRNA targeting human Nox4 (5′-GGAGAACCAGGAGAUUGUUGGAUAA-3′) was synthesized by Invitrogen. Cells grown in six-well plates were transfected with 10 nmol/L of siRNA/well by using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. After 72 h incubation as indicated, the cells were used for the experiments. Nox4 Stealth™ RNAi or a Stealth™ RNAi negative control duplex did not alter Nox1 or Nox2 mRNA levels (data not shown).
Measurement of MCP-1 protein
The MCP-1 protein released into the medium from HUVEC and rat plasma MCP-1 levels were measured with human and rat CCL2/MCP-1 ELISA (R&D Systems) (Minneapolis, USA), respectively, according to the manufacturer's instructions.
Measurement of ALP activity
HASMC was solubilized in 1% Triton X-100/PBS buffer containing a 50× protease inhibitor cocktail (Nacalai Tesque). The aortic arch of a rat was removed and homogenized in 0.5 mL of buffer (PBS, 1% protease inhibitor cocktail, 10 mmol/L EDTA, and 0.05% Tween-20). After centrifugation at 21,000g for 10 min at 4°C (two times), the supernatants were recovered. The amount of ALP activity in the supernatant was measured by enzyme immunoassay LabAssay ALP (Wako, Tokyo, Japan).
Animal study
Five-week-old male Sprague Dawley rats were purchased from Kyudo Co., Ltd. (Saga, Japan). At 7 weeks of age, the rats were subjected to a 5/6-nephrectomy or sham operation. After 9 weeks, the rats were randomly divided into two groups that is, CKD and PCS administrated CKD (PCS-CKD) rats. The PCS-CKD rats received an intraperitoneal injection with PCS (n = 9) at a dose of 50 mg kg−1 day−1 for 4 weeks. The CKD rats (n = 11) and sham rats (n = 4) received a daily injection of phosphate buffered saline for 4 weeks using the same volume as was administered to the PCS-CKD rats. At 16 and 20 weeks of age, various parameters were recorded for each rat, that is, body weight, serum levels of BUN and creatinine, urinary protein levels, and creatinine clearance. The serum PCS concentrations were measured by HPLC. All animal experiments were approved by the experimental animal ethics committee at the Kumamoto University.
Immunohistochemical analyses
At 20 weeks of age, the rats were anesthetized with diethyl ether, and euthanized by a single blood collection. To evaluate the aortic arch for abnormalities, an aortic arch was removed, weighed and fixed with 10% phosphate buffered formalin. The tissue was then dehydrated at room temperature through a graded ethanol series and embedded in paraffin. The prepared tissue was cut into 4.0 μm sections. After the slide had dried completely, a solution containing 50 mmol/L Tris/HCl + 0.1% Tween-20 (T-TB) was used to solubilize the aortic arch slice, followed by blocking with Block Ace (Dainippon Pharmaceutical Co., Ltd., Osaka, Japan) at room temperature for 15 min. Next, the primary antibody, a rabbit polyclonal anti-rat Nox4 (1:50) was conducted overnight at 4°C. The aortic arch slices were then washed with 50 mmol/L Tris/HCl (TB) and T-TB, followed by secondary antibody reaction at room temperature for 90 min. The secondary antibody for Nox4 was Alexa Fluor 546 goat anti-rabbit IgG (H+L). In each case, the secondary antibody was diluted 200-fold. The slide was then examined by fluorescence microscopy using a model BZ-8000 microscope (Keyence, Osaka, Japan).
Statistical analysis
All data are expressed as the mean ± SD. The means for groups were compared by analysis of variance followed by Tukey's multiple comparison. A probability value of P < 0.05 was considered to be significant.
Results
Effect of PCS on ROS production in HUVEC and HASMC
To determine whether PCS induces oxidative stress in HUVEC and HASMC, we examined the effect of PCS on ROS production in these cells. The findings indicated that PCS significantly increased ROS production, after a 1 h period of incubation and that the increase was concentration-dependent (10, 100, 500, 1000 μmol/L) (Fig.1A and B). Because PCS binds tightly to albumin in the circulating blood (>90%) (Watanabe et al. 2012), the effect of PCS on ROS production was also examined in the presence of 4% human serum albumin (HSA). The enhanced ROS production in HUVEC and HASMC by PCS was slightly reduced in the presence of 4% HSA. (80% (in the case of HUVEC) and 90% (in the case of HASMC) compared to the absence of HSA; see Figure1C and D).
Figure 1.
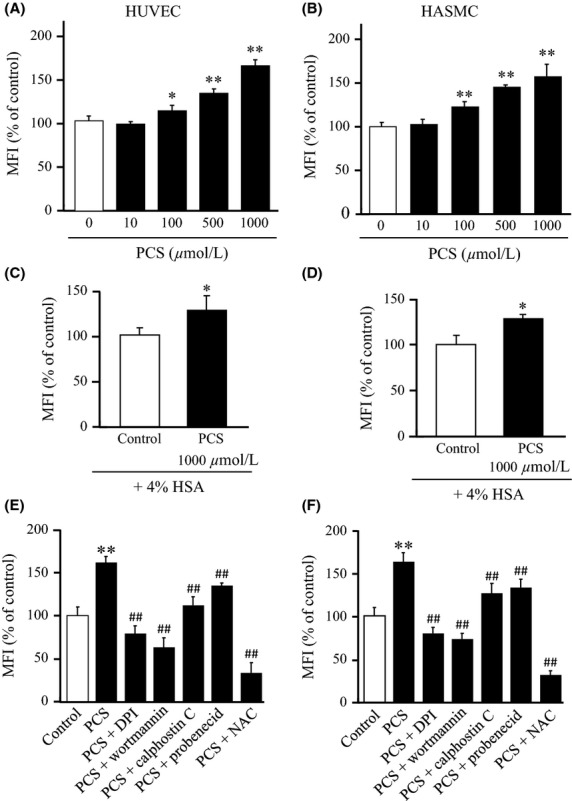
Effect of PCS on ROS production in HUVEC and HASMC: Concentration-dependency (for 1 h incubation) for PCS-induced ROS production in (A) HUVEC and (B) HASMC. Effect of 4% HSA on PCS (1000 μmol/L)-induced ROS production in (C) HUVEC and (D) HASMC. Effect of an NADPH oxidase inhibitor (50 μmol/L DPI), PI3K inhibitor (5 μmol/L wortmannin), PKC inhibitor (1 μmol/L calphostin C), OAT inhibitor (1000 μmol/L probenecid) and NAC (1000 μmol/L) on PCS (1000 μmol/L)-induced ROS production in (E) HUVEC and (F) HASMC. Results are expressed as percentages of fluorescence intensity compared to control (0 μmol/L PCS) (mean ± SD, n = 6, *P < 0.05 vs. control, **P < 0.01 vs. control, ##P < 0.01 vs. PCS).
We next investigated the mechanism of the pro-oxidant properties of PCS. This was done by conducting the same set of experiments in the presence of various inhibitors, such as, an NADPH oxidase inhibitor (DPI), a PI3K inhibitor (wortmannin), a PKC inhibitor (calphostin C), an OAT inhibitor (probenecid), and antioxidant (NAC). As shown in Figure1E and F, the ROS induced by PCS in HUVEC or HASMC was significantly inhibited by all of these inhibitors, suggesting the contribution of NADPH oxidase system and cellular uptake of PCS via OAT. These data indicate that the intracellular accumulation of PCS and the activation of NADPH oxidase are involved in the process of ROS generation induced by PCS in HUVEC and HASMC.
Effect of PCS on the expression of an NADPH oxidase subunit in HUVEC or HASMC
We next examined whether NADPH oxidase plays an important role in PCS-induced ROS production in HUVEC and HASMC. To achieve this, we analyzed alterations in the expression levels of Nox4, an NADPH oxidase subunit after treatment with 1000 μmol/L PCS by means of Western blotting. We found that the presence of PCS resulted in a significant increase in the expression of Nox4 in HUVEC and HASMC cells in a time-dependent manner (Fig.2A and B).
Figure 2.
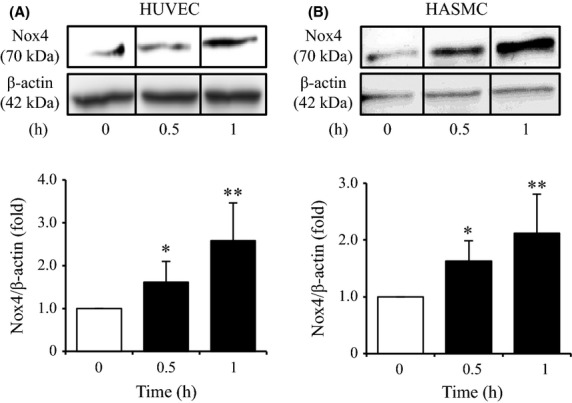
Effect of PCS on Nox4 protein expression in HUVEC and HASMC: Time-dependency for PCS (1000 μmol/L)-induced protein expression of Nox4 in HUVEC and HASMC was determined by western blotting. Representative immunoblots with anti-Nox4 antibody after stimulation with 1000 μmol/L PCS are shown. The densitometric analysis of Nox4 was normalized against β-actin. The results are shown as the fold in expression as compared to control (0 μmol/L PCS) (mean ± SD, n = 4, *P < 0.05, **P < 0.01 vs. control).
Effect of PCS on the expression of MCP-1 mRNA and MCP-1 protein secretion in HUVEC
We also examined this issue of whether the PCS-induced ROS production results in the development of vascular endothelial dysfunction. This was investigated by studying the effect of PCS on the expression of MCP-1, a protein that is involved in endothelial dysfunction. As shown in Figure3A, a 12 h incubation of HUVEC with 1000 μmol/L PCS resulted in an increase in the mRNA levels of MCP-1 by 2.5 times. Furthermore, the MCP-1 protein released into the culture medium was also measured using an enzyme immunoassay (24 h incubation of HUVEC with 1000 μmol/L PCS). PCS increased the secretion of the MCP-1 protein by 2.5 times (Fig.3B). In the presence of probenecid, these increases were suppressed to the control level.
Figure 3.
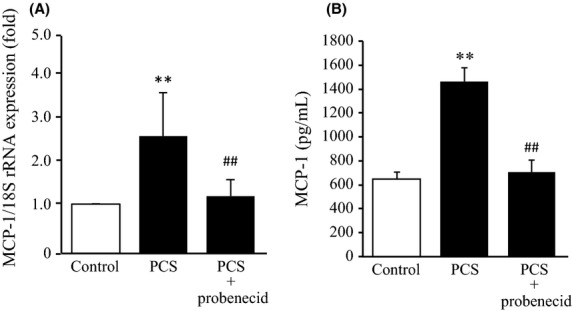
Effects of PCS on mRNA expression and protein secretion of MCP-1 in HUVEC: (A) PCS (1000 μmol/L)-induced mRNA expression of MCP-1 in HUVEC was normalized against 18S ribosomal RNA in the absence or presence of 1000 μmol/L probenecid. The results are shown as the fold change in expression as compared to the control (0 μmol/L PCS) (mean ± SD, n = 4, **P < 0.01 vs. control; ##P < 0.01 vs. PCS). (B) PCS (1000 μmol/L)-induced protein secretion of MCP-1 in medium from HUVEC was determined by ELISA in the absence or presence of 1000 μmol/L probenecid. HUVEC was incubated with PCS (1000 μmol/L) at 37°C for (A) 12 h and (B) 24 h (mean ± SD, n = 6, **P < 0.01 vs. control, ##P < 0.01 vs. PCS).
Effect of PCS on the expression of ALP, OPN and Cbfa1 mRNA and ALP activity in HASMC
To examine the effect of PCS on the osteogenic differentiation of vascular smooth muscle cells, the effect of PCS on the expression of ALP, OPN, and Cbfa1, which are osteoblast-specific proteins, in HASMC was investigated. As shown in Figure4A–C, a 24 h incubation of HASMC with 1000 μmol/L PCS resulted in a significant increase in the mRNA levels of ALP, OPN, and Cbfa1. Furthermore, PCS significantly increased ALT activity in HASMC (24 h incubation) (Fig.4D). These effects of PCS were suppressed to the control level in the presence of probenecid.
Figure 4.
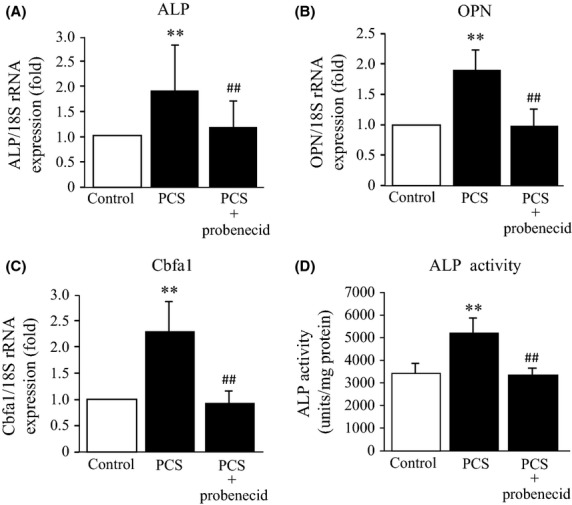
Effect of PCS on the mRNA expression of ALP, OPN, Cbfa1 and the ALP activity in HASMC: The mRNA levels corresponding to (A) ALP, (B) OPN and (C) Cbfa1 in HASMC were normalized against 18S ribosomal RNA in the absence or presence of 1000 μmol/L probenecid. The results are shown as the fold in expression as compared to control (0 μmol/L PCS) (mean ± SD, n = 4, **P < 0.01 vs. control; ##P < 0.01 vs. PCS). (D) ALP activity was determined in the absence or presence of 1000 μmol/L probenecid. (mean ± SD, n = 6, **P < 0.01 vs. control, ##P < 0.01 vs. PCS). HASMC was incubated with 1000 μmol/L PCS at 37°C for (A, B and C) 24 h and (D) 48 h.
Effect of Nox4 RNAi on the PCS-induced ROS production and MCP-1 protein secretion in HUVEC or ALP activity in HASMC
To better understand the role of NADPH oxidase in the development of PCS-induced vascular endothelial and smooth muscle cellular damage, a small interfering RNA strategy was used to knockdown Nox4 in HUVEC and HASMC. As shown in Figure5A and B, The expression of the Nox4 protein in HUVEC and HASMC wase almost completely inhibited by the introduction of Nox4 siRNA. The inhibition of Nox4 expression with the specific siRNA resulted in a significant reduction in PCS-induced ROS production in HUVEC and HASMC compared with control siRNA, respectively (Fig.5C and D). These data indicate that PCS-induced ROS production is largely controlled by the activation of NADPH oxidase. In addition, the knockdown of Nox4 expression with Nox4 siRNA blocked PCS-induced MCP-1 protein secretion or ALP activation to the control levels in HUVEC or HASMC, respectively (Fig.5E and F). In these experiments, baseline ROS generation remained nearly constant after the knockdown of Nox4, indicating that additional effects of Nox4 siRNA on the basal ROS levels can be excluded. Therefore, Nox4 may mediate PCS-induced ROS generation in HUVEC or HASMC but it has no effect on the basal ROS levels.
Figure 5.
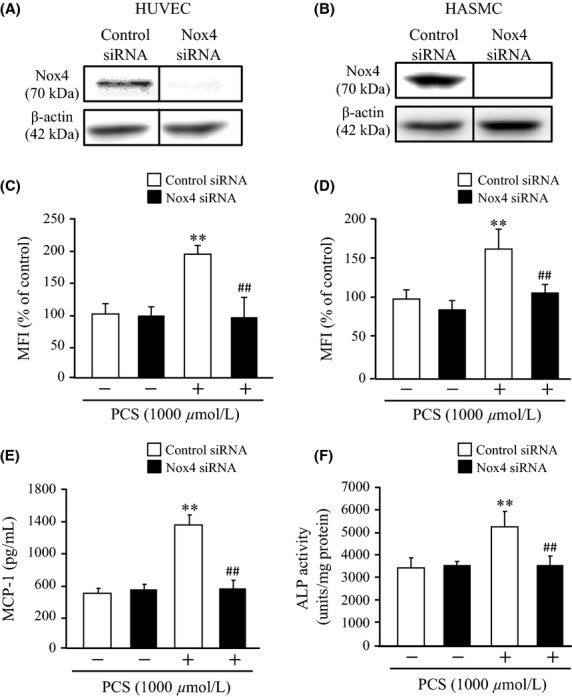
Effect of Nox4 RNAi on PCS-induced ROS production and MCP-1 protein expression in HUVEC or ALP activity in HASMC: Nox4 protein expression in (A) HUVEC and (B) HASMC. Effect of Nox4 RNAi on PCS-induced ROS production in (C) HUVEC and (D) HASMC. (E) MCP-1 expression in HUVEC and (F) ALP activity in HASMC (mean ± SD, n = 6, **P < 0.01 vs. control, ##P < 0.01 vs. PCS).
Effect of PCS administration on vascular damage in CKD model rats
To confirm the in vitro data, we further examined the issue of whether the administration of PCS to 5/6-nephrectomized rats (CKD rat) stimulated vascular damage by increasing oxidative stress. At 20 weeks of age, the serum PCS concentration in PCS-injected CKD rats (PCS-CKD rats) was approximately 7-fold higher than that in CKD rats (4.5 ± 5.3 μmol/L for CKD rats; 33.1 ± 18.8 μmol/L for PCS-CKD rats, P < 0.01). There were no significant differences in body weight (490.9 ± 33.8 g for CKD rats; 493.3 ± 58.9 g for PCS-CKD rats), serum creatinine (1.21 ± 0.16 mg/dL for CKD rats; 1.35 ± 0.15 mg/dL for PCS-CKD rats), blood urea nitrogen (43.5 ± 15.3 mg/dL for CKD rats; 45.9 ± 9.1 mg/dL for PCS-CKD rats), creatinine clearance (0.71 ± 0.16 mL/min for CKD rats; 0.63 ± 0.21 mL/min for PCS-CKD rats), urinary protein (218.5 ± 68.5 mg/day for CKD rats; 324.5 ± 137.5 mg/day for PCS-CKD rats), serum calcium (10.16 ± 0.29 mg/dL for CKD rats; 10.53 ± 0.23 mg/dL for PCS-CKD rats), serum phosphate (6.50 ± 0.73 mg/dL for CKD rats; 7.25 ± 0.73 mg/dL for PCS-CKD rats), calcium phosphate product (66.07 ± 8.00 mg2/dL2 for CKD rats; 76.38 ± 4.40 mg2/dL2 for PCS-CKD rats), and intact PTH (859.7 ± 102.9 pg/dL for CKD rats; 907.3 ± 203.3 mg/dL for PCS-CKD rats) between the CKD and PCS-CKD rats. Duranton et al. (2012) reviewed normal and pathologic concentrations of uremic toxins. In that study, they demonstrated that mean uremic concentrations of PCS in human was ∼90 μmol/L. In our study, at 20 weeks of age, the serum PCS concentration in PCS-injected CKD rats was an average of 33 μmol/L. This serum concentration in rats was comparable to those in human.
Figure6A shows a comparison of data for tissue samples taken from the aortal arch from sham, CKD, and PCS-CKD rats after immunostaining for Nox4. The expression of Nox4 was significantly elevated in PCS-CKD rats compared to the CKD group (Fig.6A and B). This is in agreement with the in vitro data obtained from HUVEC and HASMC experiments (Fig.2). We also determined plasma MCP-1 levels, vascular OPN expression and vascular ALP activity in the aortal arch tissue samples. As a result, PCS-CKD rats showed a significant increase in plasma MCP-1 levels, vascular OPN expression, and vascular ALP activity compared with the CKD rats (Fig.6C, D, and E). These data are entirely consistent with the results obtained from HUVEC and HASMC (Figs.3, 4). Our observations indicate that NADPH oxidase expression and oxidative stress are increased in the aortic arch of PCS-CKD rats compared with the other rats in this study. These data suggest that increased oxidative stress as a result of the action of PCS could be the cause of vascular damage in vivo.
Figure 6.
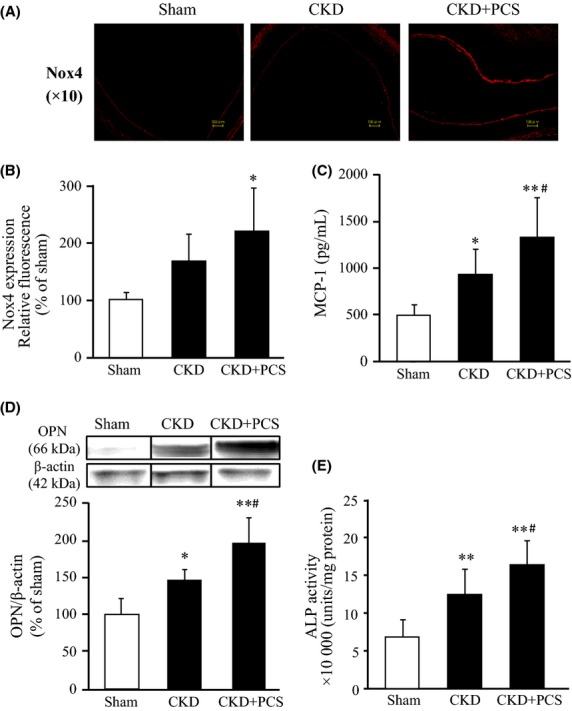
Effect of PCS administration on vascular Nox4 expression, serum MCP-1 level, vascular OPN expression and vascular ALP activity in CKD rats: (A) Representative vascular Nox4 expression in the arch of aorta, (B) Image analysis of the extent and intensity of Nox4 expression was performed, (C) Serum MCP-1 level, (D) vascular OPN expression and (E) ALP activity in the aortic arch samples were shown. Sham (n = 6), CKD (n = 10), and CKD+PCS (n = 10) rats were measured 4 weeks after PCS administration (50 mg kg−1 day−1 ip) (mean ± SD, *P < 0.05, **P < 0.01 vs. sham, #P < 0.05 vs. CKD.
Discussion
Oxidative stress is closely related to the progression of CKD and the onset of associated complications such as cardiovascular disease (Obrador et al. 2002; Cottone et al. 2008). Recent clinical studies have concluded that the accumulation of PCS is significantly correlated with the cardiovascular mortality of CKD patients (Bammens et al. 2006; Liabeuf et al. 2010; Wu et al. 2012). These findings strongly suggest that PCS may contribute to the pathogenesis of cardiovascular disease in patients with CKD. However, the mechanism responsible for the vascular toxicity induced by PCS is not well understood. To address this issue, we examined the pro-oxidant properties of PCS in causing vascular toxicity both in vitro and in vivo and the mechanism responsible for this using HUVEC and HASMC. By adopting this approach, we have arrived at the following conclusions; (1) PCS causes ROS production in HUVEC and HASMC, (2) PCS induced the expression level of Nox4, an NADPH oxidase subunit via a mechanism that involves the PKC and PI3K signaling pathways in HUVEC and HASMC, (3) PCS induces MCP-1 protein secretion in HUVEC, which contributes to endothelial dysfunction. (4) PCS induces the expression of osteoblast-specific proteins such as ALP, OPN, and Cbfa1 in HASMC, (5) PCS induces vascular damage, which is accompanied by an increase in Nox4 expression, as demonstrated using an in vivo CKD model. Thus, this study provides new evidence regarding the pro-oxidant properties of PCS and the relationships between this and vascular toxicity.
In the present study, the ROS production induced by PCS in HUVEC and HASMC was significantly suppressed when an inhibitor of NADPH oxidase, a source of intracelullar ROS generation, was present. Similarly, the inhibition of PKC and PI3K, important pathways for the activation of NADPH oxidase, also markedly decreased PCS-induced ROS production. It is well known that NADPH oxidase comprises p22phox and five members of the Nox family (Nox1 through 5) (Sirker et al. 2011). Among them, Nox4, in particular, is expressed in a wide variety of cells, including vascular endothelial cells and vascular smooth muscle cells (Shimoishi et al. 2007). In the present study, we found, for the first time, that PCS significantly increased the protein expression levels of Nox4 in HUVEC and HASMC. In addition, it was observed that Nox4 expression was increased in the aortic arch of PCS-overloaded CKD rats. In general, increased expression levels of NADPH oxidase subunit, such as Nox4, show increased NADPH oxidase activity (Watanabe et al. 2013a). Therefore, we propose that PCS activates NADPH oxidase by increasing the level of expression of NADPH oxidase itself via the PKC and PI3K signaling pathways. Such a mechanism would be predicted to lead to an increased level of ROS production. This was clearly demonstrated by the siRNA-mediated knockdown of Nox4 expression, which significantly suppressed PCS-induced ROS production in HUVEC and HASMC. Interestingly, we previously reported a similar phenomenon, in which PCS increased ROS production via the activation of NADPH oxidase in the kidney (Watanabe et al. 2013a). Further investigations using primary cells instead of HUVEC/HASMC or siRNA experiment would be needed for dissect the PKC or PI3K pathway leading to oxidative stress in response to PCS.
In HUVEC, PCS was found to up-regulate mRNA levels and protein secretion into the culture medium of MCP-1. In addition, PCS also increased the mRNA levels of osteoblast-specific proteins such as ALP, OPN, Cbfa1, and ALP activity in HASMC. Moreover, an increased plasma MCP-1 level and an increased ALP activity in the aortic arch were observed in PCS-overloaded CKD rats. The siRNA-mediated knockdown of Nox4 in HUVEC and HASMC significantly inhibited this increase in the MCP-1 secretion and ALP activation, suggesting that NADPH oxidase plays a crucial role in PCS-induced endothelial and smooth muscle cell toxicity. In a previous study, Tumur Z et al. reported that indoxyl sulfate upregulates the expression of the intercellular adhesion molecule-1 (ICAM-1) and MCP-1 by the ROS-induced activation of NF-κB in vascular endothelial cells (Tumur et al. 2010). Regarding the osteogenic differentiation of vascular smooth muscle cells, Byon et al. (2008) demonstrated that the AKT-mediated induction of Runx2 plays a critical role in oxidative stress-induced calcification in smooth muscle cells. Therefore, further investigations aimed at dissecting the NF-κB or Runx2 pathway in response to PCS-induced oxidative stress would be highly desirable.
We recently demonstrated that PCS is a good substrate for OAT and that the intracellular concentration of PCS was significantly decreased in the presence of probenecid (Miyamoto et al. 2011; Watanabe et al. 2013b). The findings reported herein clearly show that the increase in ROS production, mRNA levels, and the protein secretion of MCP-1 in HUVEC as well as an increase in the mRNA levels of ALP, OPN, Cbfa1, and ALP activity in HASMC induced by PCS were significantly suppressed in the presence of probenecid. These findings strongly suggest that PCS-induced endothelial damage and osteogenic differentiation of smooth muscle cells can be directly attributed to the intracellular accumulation of PCS. Moreover, our findings also showed that PCS-induced ROS production was largely reproduced, even in the presence of 4% HSA. We (and other groups) previously reported that in HUVEC, HASMC and HK-2 cells (a proximal tubular cell line), and indoxyl sulfate or PCS also increased ROS production, even in the presence of 4% HSA (Dou et al. 2007; Shimoishi et al. 2007; Muteliefu et al. 2009; Watanabe et al. 2013a). It has also been reported that the biological activity (IC50 value) of uremic toxins are not influenced by the presence or absence of HSA (Tsujimoto et al. 2008). These findings suggest that the presence of HSA in serum is unlikely to affect the ability of PCS to induce ROS production in endothelial and smooth muscle cells. In addition, plasma concentrations of PCS in PCS-overloaded CKD rats were around 33 μmol/L. Yet the concentrations used in the in vitro studies are supraphysiological and range from 10 to 1000 μmol/L, with 1000 μmol/L being the standard dose. Recently, Duranton et al. (2012) reviewed normal and pathologic concentrations of uremic toxins. In that study, they demonstrated that normal, mean uremic concentrations of PCS, and highest uremic concentrations of PCS in human were ∼8.0, ∼90, and 170 μmol/L, respectively. Figure1A and B showed that PCS significantly increased ROS production in HUVEC and HASMC in a dose-dependent manner. In fact, 100 μmol/L PCS, which is physiological uremic concentration, significantly increased ROS production. In clinical situation, uremic patients were exposed to pathological PCS levels for many years. But, in this cell culture study, the cells were exposed to PCS only for several hours. Therefore, higher PCS concentration (1000 μmol/L) used in this study does not seem to be inappropriate. In fact, many researchers have used 1∼5 mmol/L of uremic toxins in their cell culture studies (Shimoishi et al. 2007; Kawakami et al. 2010; Chiang et al. 2013; Watanabe et al. 2013a). In CKD patients, endothelial and smooth muscle cells are in constant contact with uremic toxins and could be a privileged target of these toxins. In fact, we also revealed that the pro-oxidant property of PCS in vivo caused by an increased Nox4 expression in the aortic arch of PCS-overloaded CKD rats. Collectively, these findings strongly suggest that PCS causes an increase in ROS production in vascular endothelial and smooth muscle cells of CKD patients. Our findings could support the conclusion that the accumulation of high levels of PCS is correlated with cardiovascular mortality rate in CKD patients.
Conclusion
PCS induces ROS production in endothelial and smooth muscle cells by increasing NADPH oxidase expression via the PKC or PI3K pathways. Enhanced ROS production, in turn, triggers the induction of MCP-1 and osteoblast-specific proteins that are involved in endothelial and smooth muscle cell dysfunction. Together, in addition to its role in renal fibrosis (Watanabe et al. 2013a), PCS also appears to have pro-oxidative effects on vascular endothelial and smooth muscle cells, which might be involved in cardiovascular disease in CKD patients. Thus, the removal of PCS and blocking its signaling pathway represents a potentially useful strategy for the treatment of the CKD-CVD connection.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) (KAKENHI 25460190) and a Grant for Pathophysiological Research Conference in CKD from The Kidney Foundation, Japan.
Glossary
Abbreviations
- ALP
alkaline phosphatase
- Cbfa1
core-binding factor alpha 1
- CKD
chronic kidney disease
- DPI
diphenylene iodonium
- HASMC
human aortic smooth muscle cells
- HUVEC
human umbilical vein endothelial cells
- MCP-1
monocyte chemotactic protein-1
- NAC
N-acetyl-l-cysteine
- OAT
organic anion transporter
- OPN
osteopontin
- PCS
p-cresyl sulfate
- ROS
reactive oxygen species
Author Contributions
H. W., Y. M., M. F., and T. M. participated in the research design and wrote or contributed to writing of the manuscript. H. W., Y. M., Y. E., and Y. M. conducted the experiments. H. W. and Y. M. analyzed the data. H. W., Y. M., Y. I., D. K., S. K., M. N., M. T., K. M., Y. M., T. K., M. F., and M. O. contributed new reagents or analysis tools.
Disclosures
None declared.
References
- Anavekar NS, Pfeffer MA. Cardiovascular risk in chronic kidney disease. Kidney Int Suppl. 2004;92:S11–S15. doi: 10.1111/j.1523-1755.2004.09203.x. [DOI] [PubMed] [Google Scholar]
- Annuk M, Zilmer M, Lind L, Linde T, Fellström B. Oxidative stress and endothelial function in chronic renal failure. J Am Soc Nephrol. 2001;12:2747–2752. doi: 10.1681/ASN.V12122747. [DOI] [PubMed] [Google Scholar]
- Bammens B, Evenepoel P, Keuleers H, Verbeke K, Vanrenterghem Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006;69:1081–1087. doi: 10.1038/sj.ki.5000115. [DOI] [PubMed] [Google Scholar]
- Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CK, Nangaku M, Tanaka T, Iwawaki T, Inagi R. Endoplasmic reticulum stress signal impairs erythropoietin production: a role for ATF4. Am J Physiol Cell Physiol. 2013;304:C342–C353. doi: 10.1152/ajpcell.00153.2012. [DOI] [PubMed] [Google Scholar]
- Cottone S, Lorito MC, Riccobene R, Nardi E, Mulè G, Buscemi S. Oxidative stress, inflammation and cardiovascular disease in chronic renal failure. J Nephrol. 2008;21:175–179. , et al. ( [PubMed] [Google Scholar]
- Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, Dignat-George F. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost. 2007;5:1302–1308. doi: 10.1111/j.1538-7836.2007.02540.x. , et al. ( [DOI] [PubMed] [Google Scholar]
- Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R. Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol. 2012;23:1258–1270. doi: 10.1681/ASN.2011121175. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenbaum J, Neuberg CA. Simplified method for the preparation of aromatic sulfuric acid esters. J Am Chem Soc. 1941;63:3529–3530. [Google Scholar]
- Ito S, Osaka M, Higuchi Y, Nishijima F, Ishii H, Yoshida M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J Biol Chem. 2010;285:38869–38875. doi: 10.1074/jbc.M110.166686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Inagi R, Wada T, Tanaka T, Fujita T, Nangaku M. Indoxyl sulfate inhibits proliferation of human proximal tubular cells via endoplasmic reticulum stress. Am J Physiol Renal Physiol. 2010;299:F568–F576. doi: 10.1152/ajprenal.00659.2009. [DOI] [PubMed] [Google Scholar]
- Liabeuf S, Barreto DV, Barreto FC, Meert N, Glorieux G, Schepers E, et al. (EUTox) EUTWG. Free p-cresyl sulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol Dial Transplant. 2010;25:1183–1191. doi: 10.1093/ndt/gfp592. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Watanabe H, Noguchi T, Kotani S, Nakajima M, Kadowaki D. Organic anion transporters play an important role in the uptake of p-cresyl sulfate, a uremic toxin, in the kidney. Nephrol Dial Transplant. 2011;26:2498–2502. doi: 10.1093/ndt/gfq785. , et al. ( [DOI] [PubMed] [Google Scholar]
- Muteliefu G, Enomoto A, Jiang P, Takahashi M, Niwa T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. 2009;24:2051–2058. doi: 10.1093/ndt/gfn757. [DOI] [PubMed] [Google Scholar]
- Obrador GT, Pereira BJ, Kausz AT. Chronic kidney disease in the United States: an underrecognized problem. Semin Nephrol. 2002;22:441–448. doi: 10.1053/snep.2002.2002.35962. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Shimoishi K, Anraku M, Kitamura K, Tasaki Y, Taguchi K, Hashimoto M. An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm Res. 2007;24:1283–1289. doi: 10.1007/s11095-007-9248-x. , et al. ( [DOI] [PubMed] [Google Scholar]
- Sirker A, Zhang M, Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol. 2011;106:735–747. doi: 10.1007/s00395-011-0190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CY, Chang SC, Wu MS. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE. 2012;7:e34026. doi: 10.1371/journal.pone.0034026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto M, Kinoshita Y, Hirata S, Otagiri M, Ohtani H, Sawada Y. Effects of uremic serum and uremic toxins on hepatic uptake of digoxin. Ther Drug Monit. 2008;30:576–582. doi: 10.1097/FTD.0b013e3181838077. [DOI] [PubMed] [Google Scholar]
- Tumur Z, Shimizu H, Enomoto A, Miyazaki H, Niwa T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am J Nephrol. 2010;31:435–441. doi: 10.1159/000299798. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Miyamoto Y, Otagiri M, Maruyama T. Update on the pharmacokinetics and redox properties of protein-bound uremic toxins. J Pharm Sci. 2011;100:3682–3695. doi: 10.1002/jps.22592. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Noguchi T, Miyamoto Y, Kadowaki D, Kotani S, Nakajima M. Interaction between two sulfate-conjugated uremic toxins, p-cresyl sulfate and indoxyl sulfate, during binding with human serum albumin. Drug Metab Dispos. 2012;40:1423–1428. doi: 10.1124/dmd.112.045617. , et al. ( [DOI] [PubMed] [Google Scholar]
- Watanabe H, Miyamoto Y, Honda D, Tanaka H, Wu Q, Endo M. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013a;83:582–592. doi: 10.1038/ki.2012.448. , et al. ( [DOI] [PubMed] [Google Scholar]
- Watanabe H, Sakaguchi Y, Sugimoto R, Kaneko KI, Iwata H, Kotani S. Human organic anion transporters function as a high-capacity transporter for p-cresyl sulfate, a uremic toxin. Clin Exp Nephrol. 2013b doi: 10.1007/s10157-013-0902-9. , et al. ( DOI: 10.1007/s10157-013-0902-9. [DOI] [PubMed] [Google Scholar]
- Wu IW, Hsu KH, Hsu HJ, Lee CC, Sun CY, Tsai CJ. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients–a prospective cohort study. Nephrol Dial Transplant. 2012;27:1169–1175. doi: 10.1093/ndt/gfr453. , et al. ( [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Tsuruoka S, Ioka T, Ando H, Ito C, Akimoto T. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006;69:1780–1785. doi: 10.1038/sj.ki.5000340. , et al. ( [DOI] [PubMed] [Google Scholar]