

Abstract
The p38 mitogen-activated protein kinase (MAPK) intracellular signaling pathway responds to a variety of extracellular stimuli, including cytokines, Toll-like receptor agonists, and components of cigarette smoke to influence the expression of proinflammatory mediators. Activation of p38 MAPK is increased within the lungs of chronic obstructive pulmonary disease (COPD) patients. In clinical trials, treatment of COPD patients with p38 MAPK inhibitors has been shown to reduce systemic inflammation plasma biomarkers C-reactive protein (CRP) and fibrinogen. As CRP and fibrinogen have been associated with poor clinical outcomes in COPD patients, such as mortality, exacerbation, and hospitalization, we analyzed gene expression data from COPD subjects treated with dilmapimod with the aim of understanding the effects of p38 MAPK inhibition on the inflammatory genome of immune cells within the systemic circulation. Whole blood and induced sputum samples were used to measure mRNA levels by gene array and PCR. Pathway and network analysis showed STAT1, MMP-9, CAV1, and IL-1β as genes regulated by dilmapimod that could also influence fibrinogen levels, while only IL-1β was identified as a gene regulated by dilmapimod that could influence CRP levels. This suggests that p38 MAPK inhibits specific inflammatory pathways, leading to to differential effects on CRP and fibrinogen levels in COPD patients.
Keywords: C-reactive protein, dilmapimod, gene expression, P38 mitogen activated protein kinase
Introduction
The p38 mitogen-activated protein kinase (MAPK) intracellular signaling pathway influences the expression of proinflammatory mediators in many different cell types. Extracellular stimuli, including cytokines, toll-like receptor (TLR) agonists, and components of cigarette smoke trigger the activation of a cascade of kinases that converge to activate p38 MAPK (Kumar et al. 2003; Cuadrado and Nebreda 2010). This kinase promotes inflammation in different ways, either by activating transcription factors that enhance inflammatory gene transcription (Whitmarsh 2010), increasing the posttranscriptional stabilization of mRNAs or by increasing protein translation (Kumar et al. 2003).
Chronic obstructive pulmonary disease (COPD) is characterized by an abnormal inflammatory response caused by the inhalation of noxious particles, most commonly from cigarette smoke. It has been demonstrated that p38 MAPK activation is increased within the lungs of COPD patients compared to controls (Renda et al. 2008; Gaffey et al. 2013), suggesting an important role for this kinase in the pathogenesis of inflammation in COPD. Cell culture studies using drugs that inhibit p38 MAPK have demonstrated a decrease in the secretion of proinflammatory mediators from COPD alveolar macrophages (Smith et al. 2008; Armstrong et al. 2011) lymphocytes, and bronchial epithelial cells (Gaffey et al. 2013). P38 MAPK inhibitors are currently in clinical development for the treatment of COPD, and a recent study has shown a significant improvement in forced expiratory volume in 1 sec (FEV1) after treatment for 6 weeks (Macnee et al. 2013).
Many COPD patients suffer with systemic inflammation, which is associated with clinical manifestations such as muscle wasting and cardiovascular disease. There is evidence that p38 MAPK plays a role in COPD systemic inflammation (Chung 2011). The p38 MAPK inhibitor PH-797804 reduced C-reactive protein (CRP) levels over 6 weeks (Macnee 2013), while a different inhibitor, losmapimod, reduced fibrinogen levels over 12 weeks but the effect on CRP levels was not sustained over 12 weeks (Lomas et al. 2012). Recently, it has been reported that losmapimod reduced CRP and fibrinogen levels, but at 24 weeks the effects did not reach statistical significance (Watz et al. 2014). Both CRP and fibrinogen are biomarkers of systemic inflammation which are associated with worse clinical outcomes in COPD patients, such as increased mortality and exacerbation rates (Faner et al. 2013). Orally administered p38 MAPK inhibitors may, therefore, achieve some therapeutic benefits through their systemic effects.
We have previously reported (Singh et al. 2010) a clinical trial in COPD patients of the effects of a single dose of the p38 MAPK inhibitor dilmapimod on systemic biomarkers of p38 activation; whole blood samples were stimulated “ex-vivo” to evaluate inhibition of tumor necrosis factor-alpha (TNF-α) secretion and phosphorylation of the p38 MAPK-dependent molecule heat shock protein 27 (HSP-27). We now report gene expression analysis of blood and sputum samples from this study. The primary aim of this analysis was to elucidate the effects of p38 MAPK inhibition on the inflammatory genome of immune cells within the systemic circulation.
Materials and Methods
Study design
The design of this study has been described previously (Singh et al. 2010). Seventeen COPD patients aged between 40 and 75 years with postbronchodilator FEV1 of >50% and <80% predicted as well as serum CRP ≥ 1 mg/L participated; the demography of the patients is shown in Table1. Subjects were required to produce a viable sputum sample (≥50% viable cells) following induction with hypertonic saline at screening, and have serum CRP ≥ 1 mg/L at screening. Subjects with a history of active tuberculosis, lung cancer, or clinically overt bronchiectasis were excluded, as were patients with symptoms of any respiratory infection at the start of the study. Subjects with a history of rheumatoid arthritis, connective tissue disorders, and other conditions known to be associated with chronic inflammation were also excluded.
Table 1.
Patient demographics
| Age (years) | 63.2 (8.4) |
| Gender (M/F) | 12/5 |
| Pre-bronchodilator FEV1 (L) | 1.64 (0.35) |
| Pre-bronchodilator FEV1 (% predicted) | 56.3 (8.2) |
| Pre-bronchodilator FVC (L) | 3.31 (0.79) |
| FEV1 (L) | 1.86 (0.35) |
| FEV1 (% predicted) | 63.9 (7.2) |
| FVC (L) | 3.73 (0.82) |
| FEV1/FVC ratio | 0.51 (0.09) |
| BMI | 26.9 (4.2) |
| CRP (mg/L) | 2.4 (1–26.4) |
| Inhaled medications | |
| Tiotropium | 3 (18%) |
| Formoterol | 2 (12%) |
| Salbutamol | 6 (35%) |
| Ipratropium bromide | 2 (12%) |
Data shown are mean (SD), except for CRP which is median (range). Lung function data are post-bronchodilator unless otherwise stated. Other medications (used by at least 2 subjects); Acetylsalicylic acid (n = 4), bendrofluazide (n = 2), diltiazem (n = 2), nicotine (n = 2), oestradiol valerate (n = 2), omeprazole (n = 2), paracetamol (n = 2), simvastatin (n = 2).
We collected blood samples at pre-dose and 1, 2, and 6 h post-dose on each study day, and induced sputum at 2 h post-dose; this analysis compares the effects of a single oral dose of dilmapimod 25 mg compared to placebo in blood samples of 15 of the patients (no data were obtained from two subjects) and 14 sputum samples (no data from the same two patients as the blood samples and one additional patients where only baseline samples were obtained).
Blood and sputum sampling
Five milliliter of whole blood was collected into PAXgene blood RNA tubes (Qiagen, Valencia, CA). After gently inverting 8–10 times, these samples were incubated at room temperature for 2 h, followed by storage at −20°C. Sputum was induced using hypertonic saline and processed using dithiothreitol (DTT) to obtain cells as previously described (Pizzichini et al. 1996); these sputum cells were collected in to TRIZOL.
Whole-genome microarray analysis
Samples were randomized prior to RNA processing and the RNA was robotically extracted on a Biorobot 8000 (Qiagen). RNA quality was assessed by A260/A280 ratios from spectrophotometer readings and ribosomal RNA bands 28s/18s ratios using an Agilent Technologies Bioanalyzer (Agilent Technologies, Santa Clara, CA) high-resolution electrophoresis system.
Affymetrix microarray (Affymetrix, Santa Clara, CA) analysis of whole blood samples was performed using Nugen cDNA amplification and profiling (NuGEN Technologies, San Carlos, CA). For each sample, 50 ng of total RNA was converted to amplified antisense sscDNA using the NuGEN Ovation RNA Amplification System V2, including Ovation WB reagent, following manufacturer's instructions. cDNAs were purified using the Agencourt AMPure Magnetic Bead Purification System (Beckman Coulter, Indianapolis, IN). Amplified cDNA was fragmented and biotin-labeled using the FL-Ovation cDNA Biotin Module V2 (NuGEN) and hybridization cocktails were prepared in accordance with NuGEN protocols. Samples were then hybridized to the Affymetrix Human Genome HG-U133plus2 GeneChip. Arrays were washed and scanned on Genechip 3000 scanners (Affymetrix) according to Affymetrix protocols. Quality was assessed using report files generated in GCOS (GeneChip ® Operating System) software (Affymetrix) and checked for probe and hybridization quality.
Real-time PCR
Real-time PCR was performed on blood and sputum RNA; CCL5, HSPB1 (HSP27), IL-1β, IL-6, IL-8, MMP-9, and TNF-α were measured in blood and CCL4, CXCL1, CXCL10, HSPB1, IL-1β, IL-6, IL-8, PPARγ, and TNF-α were measured in sputum. The selection of these genes for analysis was made before the results of the microarray analysis were available. RL19, GAPDH, B-actin, and cyclophilin were analyzed as housekeeper genes for normalization. The sequences of the primers used in this study are shown in the Supporting Information. Real-time PCR results were generated using either the 5′ nuclease assay (TaqMan) or the SYBR Green assay. RNA was converted to cDNA by reverse transcription utilizing the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). For TaqMan, the equivalent of 10 ng mRNA per well was arrayed into 384-well plates using a Biomek FX robot (Beckman Coulter) and quantitative RT-PCR was carried out using a 7900HT Sequence Detector System (Applied Biosystems) in a 5 μL reaction volume. Primers were used at 900 and 100 nm (probes). TaqMan Universal PCR Master Mix 2X (Applied Biosystems) was used and universal PCR conditions recommended by the manufacturer were followed. For SYBR Green, 8 μL of PCR Master Mix was added to each well using Biomek robotics (Beckman Coulter). Primers were used at 400 nmol/L. The PCR reaction was carried out on an ABI7900 Sequence Detector using the PCR parameters: 50°C for 2 min, 95°C for 10 min, 33 cycles of 95°C for 15 sec, 60°C for 1 min.
Statistical analysis
Gene array data quality was assessed for homogeneity of quality control metrics by principal component analysis (PCA) using SIMCA-P+ software (Umetrics, Windsor, UK). Global analysis of gene expression was performed by normalizing probe intensity data by robust multichip average (RMA; Bolstad et al. 2003) using ArrayStudio software (OmicSoft Corporation, Cary, NC). A repeated measures random effects analysis of variance (ANOVA) was used to assess differences between treatments using SAS Software (SAS UK, Marlow, UK). The response used was log base 10 of the normalized intensity values. Other factors included in the model were visit, time point, and treatment. Subject was fitted as a random effect and time as a repeated measures effect. Significantly regulated probesets were defined as ≥1.3-fold change and P < 0.01.
For the real-time PCR data, outliers were removed if they deviated more than 4 robust standard deviations away from the overall median. The robust standard deviation was defined using the median absolute deviation (MAD) statistic. The effect of the housekeepers was investigated in ArrayStudio using PCA. The scores from the first principal component were used as a covariate in the subsequent SAS analysis. For the blood real-time PCR data, the same repeated measures random effects ANOVA as for the microarray analysis was performed. The response used was log base 2 of the abundance values. For the sputum real-time PCR data, repeated measures random effects ANOVA was used to assess differences between the treatment groups at the 2 h timepoint.
Pathway and network analysis
Pathways enriched in significantly regulated probesets (≥1.3-fold change and P < 0.01) were identified using the Fisher's exact test on pathways taken from GeneGo Metabase (Thomson Reuters, http://www.genego.com). Gene expression analysis results were uploaded into the Ingenuity Pathway Analysis (IPA) web-based tool (Qiagen Ingenuity, http://www.ingenuity.com). Significantly regulated probesets were included in Upstream Regulators analysis within the IPA tool to identify upstream transcriptional regulators or cytokines affected by dilmapimod treatment; this approach matches gene expression changes in the data set to factors which are known to be upstream of them. An activation z-score cut-off of 2 was applied. Network analysis was also performed within the IPA tool.
Results
Microarray analysis of the effects of dilmapimod in whole blood
Whole genome expression profiling showed that dilmapimod 25 mg caused a significant change compared to placebo, defined as >1.3-fold and P < 0.01, in the gene expression levels of 62 probesets at 1 h post-dose, 93 probesets at 2 h and 219 probesets at 6 h; all these probesets are shown in Supporting Information, while Table2 shows the ten most highly regulated genes at each time point. There was one gene (FAM174A) significantly changed at all three time points and six genes significantly changed at both the 2 and 6 h time points (ADM, CCL8, CXCL1, IL-1β, TLR1, and ZNF519).
Table 2.
Most highly regulated genes in response to dilmapimod
| Time point | Gene name | Description | P-value | Fold change |
|---|---|---|---|---|
| 1 h | LPP | LIM domain containing preferred translocation partner in lipoma | 0.0064 | 1.62 |
| LYVE1 | Lymphatic vessel endothelial hyaluronan receptor 1 | 0.0014 | 1.61 | |
| PLCB2 | Phospholipase C, beta 2 | 0.0002 | 1.60 | |
| UBR2 | Ubiquitin protein ligase E3 component n-recognin 2 | 0.0045 | 1.58 | |
| CACNA1D | Calcium channel, voltage-dependent, L type, alpha 1D subunit | 0.0057 | 1.57 | |
| IL1RAP | Interleukin 1 receptor accessory protein | 0.0057 | −1.56 | |
| TMX1 | Thioredoxin-related transmembrane protein 1 | 0.0045 | 1.51 | |
| TPM1 | Tropomyosin 1 (alpha) | 0.0001 | −1.48 | |
| BICD1 | Bicaudal D homolog 1 | 0.0026 | 1.47 | |
| SEPT2 | Septin 2 | 0.0003 | 1.46 | |
| 2 h | TCF7L2 | Transcription factor 7-like 2 | 0.0011 | 2.07 |
| LPP | LIM domain containing preferred translocation partner in lipoma | 0.0003 | 1.97 | |
| TRMT1L | TRM1 tRNA methyltransferase 1-like | 0.0069 | −1.88 | |
| EIF4G2 | Eukaryotic translation initiation factor 4 gamma, 2 | 0.0062 | 1.82 | |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 | 0.0023 | −1.80 | |
| SLCO4C1 | Solute carrier organic anion transporter family, member 4C1 | 0.0032 | −1.73 | |
| C4B | Complement component 4B | 0.0058 | 1.65 | |
| CAPRIN1 | Cell cycle-associated protein 1 | 0.0047 | −1.64 | |
| ZNF519 | Zinc finger protein 519 | 0.0072 | −1.63 | |
| WSB1 | WD repeat and SOCS box-containing 1 | 0.0061 | −1.60 | |
| 6 h | MMP9 | Matrix metallopeptidase 9 | 0.0002 | −2.22 |
| LOC731424 | Hypothetical LOC731424 | 0.0058 | −1.98 | |
| PLK4 | Polo-like kinase 4 | 0.0020 | 1.87 | |
| HELLS | Helicase, lymphoid-specific | 0.0019 | 1.85 | |
| TRIM73/TRIM74 | Tripartite motif-containing 73/tripartite motif-containing 74 | 0.0015 | 1.77 | |
| BCL6 | B-cell CLL/lymphoma 6 | 0.0017 | −1.70 | |
| QPCT | Glutaminyl-peptide cyclotransferase | 0.0018 | −1.69 | |
| PADI4 | Peptidyl arginine deiminase, type IV | 0.0006 | −1.69 | |
| GATAD1 | GATA zinc finger domain containing 1 | 0.0078 | 1.68 | |
| CLUAP1 | Clusterin-associated protein 1 | 0.0083 | 1.67 |
Pathway analysis
Pathways within the GeneGo Metabase enriched in genes changing in response to dilmapimod treatment were identified; Table3 shows regulation of the following cytokine-associated inflammatory pathways; IL-17, inflammasome, and interferon signaling.
Table 3.
Pathways enriched in genes responding to dilmapimod treatment
| Time point | Pathway | P-value | No. of genes in pathway | Genes in pathway regulated by dilmapimod treatment1 |
|---|---|---|---|---|
| 1 h | Meiosis | 0.00040 | 153 | PMS1,PPP2R3A, HSPA14, ANAPC5 |
| Adiponectin signaling | 0.00531 | 47 | PLCB2,APPL1 | |
| Chromatin modification | 0.00756 | 178 | SMARCE1, SAP18, PBRM1 | |
| Glutamate regulation of Dopamine D1A receptor signaling | 0.00891 | 62 | PLCB2,PPP2R3A | |
| Regulation of G1/S transition | 0.00891 | 62 | PPP2R3A,ANAPC5 | |
| 2 h | IL-17 signaling pathways | 0.00015 | 68 | CEBPD, CXCL1, ICAM1, IL1B |
| Inflammasome in inflammatory response | 0.00025 | 30 | IL1B, CARD8, CARD9 | |
| Th17-derived cytokines | 0.00063 | 101 | CEBPD, CXCL1, ICAM1, IL1B | |
| Innate inflammatory response | 0.00743 | 203 | C4B, IL1B, TLR1, TLR6 | |
| Interferon signaling | 0.00905 | 112 | ICAM1, IL1B, CCL8 | |
| 6 h2 | Th17-derived cytokines | 0.00002 | 98 | CXCL1, IL1B, MMP9, S100A9, STAT1, STAT5B, IL17RA |
| Interferon signaling | 0.00004 | 108 | FCGR1A, IL1B, CCL8, STAT1, STAT5B, TNFSF10, LILRB2 | |
| Death Domain receptors and caspases in apoptosis | 0.00007 | 117 | CASP5, NAIP, TNFSF10, TNFRSF10C, BAG4, CARD6, NLRP12 | |
| Nitric oxide signaling | 0.00017 | 95 | FOSL2, GUCY1B3, IL1B, MMP9, STAT1, PDE5A | |
| Vitamin, mediator, and cofactor metabolism Alpha-tocotrienol | 0.00082 | 19 | ACSL1, FOSL2, CYP4F2 | |
| Thrombopoetin signaling via JAK-STAT pathway | 0.00082 | 20 | MPL, STAT1, STAT5B | |
| IL-12, 15, 18 signaling | 0.00134 | 55 | ATF1, FOSL2, STAT1, PELI1 | |
| Growth hormone signaling via STATs and PLC/IP3 | 0.00181 | 27 | CRK, STAT1, STAT5B | |
| Intracellular pattern recognition receptors | 0.00204 | 105 | CASP5, FOSL2, IL1B, STAT1, NLRP12 | |
| Blood vessel morphogenesis | 0.00237 | 343 | CRK, NRG1, IL1B, STAT1, STAT5B, PDE5A, PDE7B, EGLN1, PROK2 | |
| IL-17 signaling pathways | 0.00280 | 68 | CXCL1, IL1B, MMP9, IL17RA | |
| Inflammasome | 0.00308 | 116 | CASP5, FOSL2, IL1B, STAT1, NLRP12 |
Genes shown in italics were upregulated upon dilmapimod treatment. All other genes were downregulated.
Only the top 12 regulated pathways are shown for the 6-h time point.
To determine the interconnection between the pathways identified and the key genes involved, IPA network analysis was performed using the list of significantly regulated genes in Table3. This produced a network showing the interactions between these genes (Fig.1) and suggests a central role for IL-1β at 2 and 6 h (1.4-fold decrease and P < 0.01 at both timepoints), and also for STAT1 at 6 h (1.4-fold decrease and P < 0.01). At 1 h, there were no clearly regulated genes at the center of the network.
Figure 1.

Network showing connections between the differentially expressed genes contained within the enriched pathways. Nodes are colored according to gene expression changes at each time point (A)1 h; (B) 2 h; (C) 6 h. Red corresponds to upregulation and green corresponds to downregulation. Solid lines indicate direct relationship; dotted lines indicate indirect relationship.
The IPA tool was also used to identify upstream transcriptional regulators and cytokines that could potentially explain the gene expression changes caused by dilmapimod; the results are shown in Table4 and Figure2. There were no significant regulators predicted at the 1 h time point, probably due to the low number of significant gene expression changes seen. Nuclear factor Kb (NF-κB), IL1β, and TNF-α were predicted to be drivers of the gene expression changes seen at 2 and 6 h. Other upstream regulators were also identified at 6 h, including IFN-γ.
Table 4.
Upstream cytokines and transcriptional regulators predicted1 to be effected by dilmapimod treatment
| Time point2 | Upstream regulator | Predicted activation state | Target genes regulated by dilmapimod3 |
|---|---|---|---|
| 2 h | NFkB (complex) | Inhibited | CARD8, CASP9, CCL8, CEBPD, CXCL1, ICAM1, IL1B |
| IL1B | Inhibited | ADM, CEBPD, CXCL1, ICAM1, IL1B, NFIL3 | |
| TNF | Inhibited | ADM, CARD8, CEBPD, CXCL1, ICAM1, IL1B, TDRD7 | |
| 6 h | IL1RN | Activated | CXCL1, IL1B, MMP9, PELI1, TNFSF10 |
| IFNG | Inhibited | ADM, BST1, CARD6, CASP5, CCL8, CFLAR, CXCL1, FCGR1A, FCGR1B, IL17RA, IL1B, MMP9, NAMPT, PELI1, S100A9, SLC11A1, STAT1, TLR1, TNFSF10, VNN3 | |
| IFN gamma (complex) | Inhibited | IL17RA, IL1B, MMP9, STAT1, TNFSF10 | |
| NFkB (complex) | Inhibited | CCL8, CFLAR, CXCL1, FCGR1A, IL1B, MMP9, NAMPT, NLRP12, SLC22A4, TNFRSF10C, TNFSF10 | |
| IRF1 | Inhibited | IL17RA, IL1B, MMP9, STAT1, TNFSF10 | |
| IL1A | Inhibited | CCL8, CXCL1, IL1B, MMP9, S100A9, VNN3 | |
| TNFSF11 | Inhibited | BST1, CCL8, IL1B, MMP9, OSCAR, STAT1 | |
| TNF | Inhibited | ACSL1, ADM, BCL6, CAV1,CD47, CFLAR, CXCL1, FOSL2, IL1B, MMP9, NAIP, NAMPT, NEFM, NLRP12, S100A9, SLC22A4, STAT1, STEAP4, TNFSF10, VNN3 | |
| IL1B | Inhibited | ADM, CFLAR, CXCL1, IL1B, MMP9, NAMPT, S100A9, SLC22A4, STAT1, TNFSF10 |
Prediction based on Ingenuity IPA upstream regulator analysis.
No upstream effectors were predicted at the 1-h time point.
Genes shown in italics were upregulated upon dilmapimod treatment. All other genes were downregulated.
Figure 2.

Network showing predicted upstream effectors and associated downstream changes in response to dilmapimod at (A) 2 h and (B) 6 h. Gray nodes show predicted upstream effectors of the gene expression changes. Green nodes were downregulated by dilmapimod treatment; Red nodes were upregulated by dilmapimod treatment. Solid lines indicate direct relationship; dotted lines indicate indirect relationship.
IPA network analysis was used to explore the connectivity between p38 MAPK kinase and CRP and fibrinogen. The shortest path networks connecting p38 MAPK kinase (MAPK14) to CRP via genes that were changed in response to dilmapimod at any time point or identified as potential upstream drivers of the gene expression changes in the previous analysis (Table4) were identified. The resulting network is shown in Figure3; STAT1, MMP9, CAV1, and IL-1β were identified as genes regulated by dilmapimod in this study that could also influence fibrinogen levels. IL-1β was identified as a potential regulator of CRP.
Figure 3.
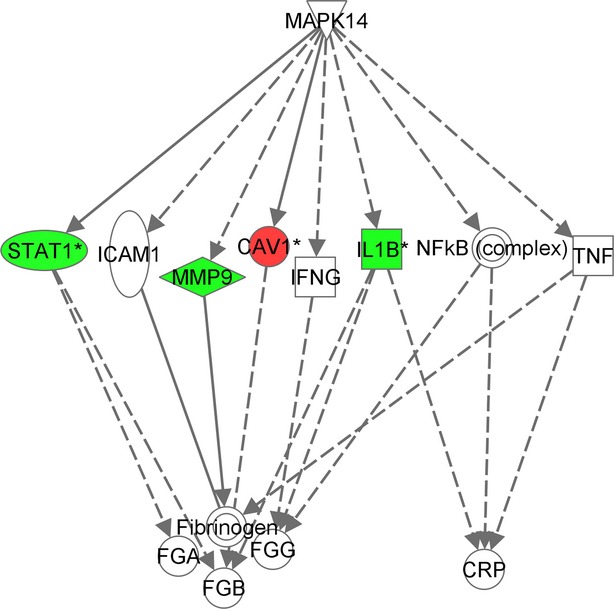
Network showing connections between p38 MAPK kinase, fibrinogen, and CRP. Nodes are colored according to gene expression changes at 6 h. Red corresponds to upregulation and green corresponds to downregulation. Solid lines indicate direct relationship; dotted lines indicate indirect relationship.
Real-time PCR analysis of gene expression changes in blood and sputum
Real-time PCR analysis showed that dilmapimod significantly reduced the gene expression levels of IL-1β (1.4-fold), IL-8 (1.4-fold), and MMP-9 (2.1-fold) in blood at 6 h, while HSP27 gene expression was significantly increased (1.2-fold) (Fig.4). There were no significant changes for CCL5, IL-6, or TNF-α. The PCR results confirmed the significant changes observed by microarray analysis for IL-1β and MMP-9.
Figure 4.
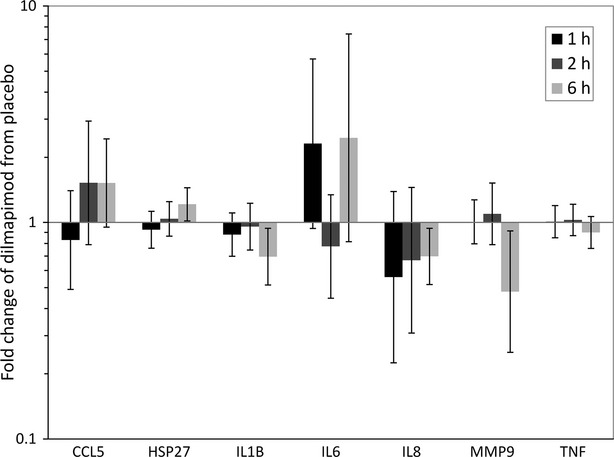
Gene expression changes in whole blood in response to dilmapimod treatment at 1, 2 or 6 h post-dose. Bars represent the ratio (fold-change) of the geometric mean of dilmapimod versus placebo groups with 95% confidence interval.
IL1-β gene expression in sputum cells, as measured by PCR, was significantly reduced (1.7-fold) at 2 h post-dose (Fig.5). There were no changes in sputum cell CCL4, CXCL1, CXCL10, HSP27, IL-6, IL-8, PPARγ, or TNF-α gene expression.
Figure 5.
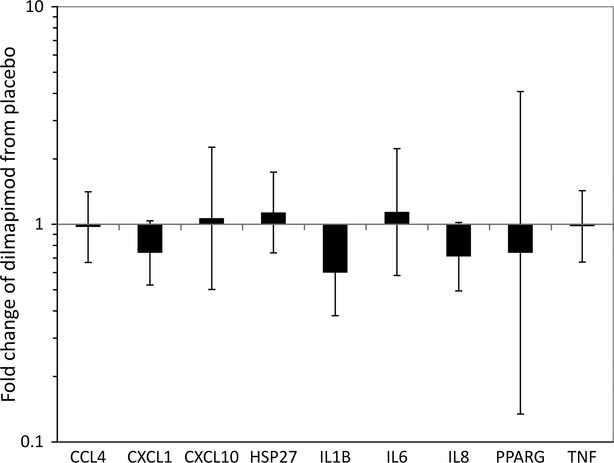
Gene expression changes in sputum in response to dilmapimod treatment at 2 h post-dose. Bars represent the ratio (fold-change) of the geometric mean of dilmapimod versus placebo group with 95% confidence interval.
Discussion
A single dose of the p38 MAPK inhibitor dilmapimod caused a range of gene expression changes in the whole blood of COPD patients. Microarray analysis identified a set of 6 genes that were downregulated at both 2 and 6 h post-dose. Of these genes, real-time PCR analysis of IL-1β confirmed the downregulation in both blood and sputum samples. Pathway and network analysis of the gene array results demonstrated central roles for IL-1β and STAT1 in the regulation of gene expression changes caused by dilmapimod. These analyses suggested IL-1β and STAT1 as potential regulators of fibrinogen levels, but only IL-1β as a potential regulator of CRP. This analysis reveals the inflammatory pathways regulated by dilmapimod, and suggests an important role for IL-1β as a p38 MAPK-regulated cytokine that can influence both fibrinogen and CRP levels.
It is known that p38 MAPK inhibitors can exert anti-inflammatory effects through regulation of transcription (Whitmarsh 2010). The novelty of this analysis was to identify specific genes and pathways in COPD patients that are regulated by a p38 MAPK inhibitor. A key finding was the reduction in IL-1β gene expresson levels in both the blood and sputum. IL-1β signals through the IL-1 receptor, activating transcription factors such as NF-κB), resulting in inflammatory cell activation and the secretion of proinflammatory cytokines and chemokines (Weber et al. 2010). IL-1β exists as an inactive precursor that is cleaved by caspase-1 to produce the biologically active form. Inflammasomes, including the Nlrp3 (NOD-like receptor family, pyrin domain containing 3) inflammasome, are able to cleave inactive procaspase-1, thus releasing active caspase-1. The sputum supernatant levels of IL-1β are increased in COPD patients compared to controls (Pauwels et al. 2011), and are further upregulated during COPD exacerbations (Bafadhel et al. 2011). The potential role of IL-1β in the pathophysiology of COPD has been investigated in cigarette smoke-induced inflammation in mice; pulmonary inflammation was dependent on IL-1β signaling (Botelho et al. 2011; Pauwels et al. 2011). In the current study, dilmapimod reduced IL-1β gene expression levels (1.4-fold in blood and 1.7-fold in sputum), and significantly regulated the expression of additional genes involved in the inflammasome pathway leading to activation of caspase. The alteration of IL-1β signaling by dilmapimod may, therefore, be an important mechanism by which this p38 MAPK inhibitor could exert therapeutic benefit.
IPA network analysis demonstrated likely central roles for IL-1β and STAT1 (Fig.1). Further IPA analysis of upstream transcriptional regulators and cytokines demonstrated that NFkB, IL-1β, and TNF-α were the most strongly predicted drivers of gene expression regulation at 2 and 6 h (Fig.2). These analyses suggest complex anti-inflammatory effects of dilmapimod impacting more than one inflammatory pathway. Inhibition of p38 can lower mRNA levels of IL-1β (or TNF-α, Kumar 2003) through modulation of several transcription factors including CEBP (CCAAT/enhancer-binding protein; Baldasarre et al. 1999), which appears to be one key anti-inflammatory mechanism as detailed above. There is also evidence for a role of p38 in activation of NFkB under various conditions (Berghe et al. 1998; Madrid et al. 2001; Jijon et al. 2004) which would represent another key transcriptional target and set of pathways. An interesting finding was the central role of STAT1 in mediating dilmapimod effects. STAT1 is known to regulate the transcription of a large number of inflammatory genes (Ramana et al. 2000), and so inhibition of STAT1 may result in a broad profile of anti-inflammatory effects.
The real-time PCR measurements also showed significant suppression of MMP-9 by dilapimod at 6 h, confirming the microarray findings. MMP-9 is a protease that is believed to play an important role in the destruction of lung tissue that occurs in emphysema (Elkington and Friedland 2006). The genes selected for real-time PCR analysis were decided “a priori”, before the results of the microarray data. We selected genes that are implicated in the pathophysiology of COPD and/or the p38 MAPK pathway. IL-8 gene expression was also reduced by dilmapimod, although this was not observed in the microarray experiments. Real-time PCR analysis is prone to less variability, and hence is usually interpreted as a more accurate result than microarray.
Clinical trials of p38 MAPK inhibitors have demonstrated a reduction in CRP levels in patients with rheumatoid arthritis and COPD (Cohen et al. 2009; Lomas et al. 2012; Macnee et al. 2013). However, this effect in rheumatoid arthritis has not been sustained over time (Genovese 2009; Genovese et al. 2011), leading to the theory that compensation occurs that eventually overcomes p38 MAPK inhibition of CRP (Genovese 2009; Singh 2013). In COPD clinical trials, there is evidence that both CRP and fibrinogen are suppressed by p38 MAPK inhibitors, with sustained suppression observed for CRP at 6 weeks (Macnee et al. 2013) and for fibrinogen but not CRP at 12 weeks studies (Lomas et al. 2012). A 24-week study in COPD also showed suppression of CRP and fibrinogen; at 24 weeks this effect was not statistically significant, which could be attributed to decreases in CRP and fibrinogen levels in the placebo group, although at 12 weeks data were consistent with previous studies (Watz et al. 2014).
The regulation of CRP and fibrinogen is complex, and controlled by different factors (Fish and Neerman-Arbez 2012; Agassandian et al. 2014). CRP is known to be produced by the liver, but can also be synthesized by other cells included respiratory epithelia and circulating monocytes (Agassandian et al. 2014). We used network analysis to model the mechanisms by which p38 MAPK inhibitors could act to regulate CRP and fibrinogen; this could occur indirectly through p38 MAPK-mediated inhibition of inflammatory pathways such as IL-1β signaling. IL-1β was a potential regulator of both CRP and fibrinogen, further confirming the other analyses conducted indicating a prominent role for IL-1β in the coordination of the anti-inflammatory effects of dilmapimod. Based on this analysis, fibrinogen appears to also be regulated by several processes that are modulated by dilmapimod, including STAT1, and therefore fibrinogen inhibition by dilmapimod may be less liable to be overcome by compensatory pathways such as those that impact CRP.
The potential limitations of this study are that only a single dose and short time points were studied, and different changes may be observed at longer time points or after a longer duration of treatment. This is an important consideration, as the effects of anti-inflammatory treatments for COPD should be considered over the long term. This is particularly important for p38 MAPK inhibitors where the effects may reduce over time (Singh 2013). Also, the use of whole blood for microarray analysis is well known to lead to modest gene expression changes compared to isolated cell types. However, in the context of a clinical trial with multiple samples requiring standardization, the use of whole blood for this purpose was a practical solution.
In conclusion, we have demonstrated that a single dose of dilmapimod results in the regulation of a number of inflammatory genes in whole blood of COPD patients. A consistent finding was the reduction in IL-1β expression levels in whole blood and induced sputum, suggesting a prominent role for IL-1β in the coordination of the anti-inflammatory effects of p38 MAPK inhibitors in COPD. Network analysis revealed different mechanisms capable of regulating CRP and fibrinogen levels; while IL-1β was involved in the regulation of both of these proteins, other pathways including STAT1 signaling were found to be involved in fibrinogen regulation only. This analysis suggests signaling pathways by which p38 MAPK inhibitors may have differential effects on CRP and fibrinogen levels in COPD.
Acknowledgments
We wish to thank Ted Cook for his contribution to this study.
Glossary
Abbreviations
- ANOVA
anaylsis of variance
- COPD
Chronic obstructive pulmonary disease
- CRP
C-reactive protein
- DTT
dithiothreitol
- FEV1
forced expiratory volume in 1 sec
- IPA
ingenuity pathway analysis
- MAD
median absolute deviation
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-κB
- PCA
principal component analysis
- RMA
robust multichip average
- TLR
toll-like receptor
- TNF-α
tumor necrosis factor-alpha
Disclosures
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
. Primer sequences for real-time PCR studies.
Table S2. Probesests differentially regulated by dilmapimod 25 mg compared to placebo at 1 h post-dose (P < 0.01, FC > 1.3).
Table S3. Probesets differentially regulated by dilmapimod 25 mg compared to placebo at 2 h post-dose (P < 0.01, FC > 1.3).
Table S4. Probesets differentially regulated by dilmapimod 25 mg compared to placebo at 6 h post-dose (P < 0.01, FC > 1.3).
References
- Agassandian M, Shurin GV, Ma Y, Shurin MR. C-reactive protein and lung diseases. Int J Biochem Cell Biol. 2014;53C:77–88. doi: 10.1016/j.biocel.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Armstrong J, Harbron C, Lea S, Booth G, Cadden P, Wreggett KA, et al. Synergistic effects of p38 mitogen-activated protein kinase inhibition with a corticosteroid in alveolar macrophages from patients with chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2011;338:732–740. doi: 10.1124/jpet.111.180737. [DOI] [PubMed] [Google Scholar]
- Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184:662–671. doi: 10.1164/rccm.201104-0597OC. [DOI] [PubMed] [Google Scholar]
- Baldasarre JJ, Yanhua B, Bellone CJ. The role of p38 mitogen-activated protein kinase in IL-1beta transcription. J Immunol. 1999;162:5367–5373. [PubMed] [Google Scholar]
- Berghe WV, Plaisance S, Boone E, De Bosscher K, Schmitz ML, Fiers W, et al. p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J Biol Chem. 1998;273:3285–3290. doi: 10.1074/jbc.273.6.3285. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;22:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Botelho FM, Bauer CMT, Finch D, Nikota JK, Zavitz CCJ, Kelly A, et al. IL-1α/IL-1R1 expression in chronic obstructive pulmonary disease and mechanistic Relevance to smoke-induced neutrophilia in mice. PLoS ONE. 2011;6:e28457. doi: 10.1371/journal.pone.0028457. doi: 10.1371/journal.pone.0028457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung FE. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest. 2011;139:1470–1479. doi: 10.1378/chest.10-1914. [DOI] [PubMed] [Google Scholar]
- Cohen SB, Cheng TT, Chindlore C, Damianov N. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthrits Rheum. 2009;60:335–344. doi: 10.1002/art.24266. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Elkington PTG, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax. 2006;61:259–266. doi: 10.1136/thx.2005.051979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faner R, Tal-Singer R, Riley JH, Celli B, Vestbo J, Macnee W, et al. on behalf of the ECLIPSE Study Investigators. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax. 2013;69:666–672. doi: 10.1136/thoraxjnl-2013-204778. [DOI] [PubMed] [Google Scholar]
- Fish RJ, Neerman-Arbez M. Fibrinogen gene regulation. Thromb Haemost. 2012;108:419–426. doi: 10.1160/TH12-04-0273. [DOI] [PubMed] [Google Scholar]
- Gaffey K, Reynolds S, Plumb J, Kaur M, Singh D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42:28–41. doi: 10.1183/09031936.00170711. [DOI] [PubMed] [Google Scholar]
- Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009;60:317–320. doi: 10.1002/art.24264. [DOI] [PubMed] [Google Scholar]
- Genovese MC, Cohen SB, Wofsy D. A 24-week, randomized double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with rheumatoid arthritis. J Rheumatol. 2011;38:846–854. doi: 10.3899/jrheum.100602. [DOI] [PubMed] [Google Scholar]
- Jijon H, Allard B, Jobin C. NFkB inducing kinase activates NFkB transcriptional activity independently of IkB kinase gamma through a p38 MAPK-dependent RelA phosphorylation pathway. Cell Signal. 2004;16:1023–1032. doi: 10.1016/j.cellsig.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signaling molecules as therapeutic targets for inflammatory diseases. Nature Rev Drug Disc. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- Lomas DA, Lipson DA, Miller BE, Willits L, Keene O, Barnacle H, et al. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012;52:416–424. doi: 10.1177/0091270010397050. [DOI] [PubMed] [Google Scholar]
- Macnee W, Allan RJ, Jones I, De Salvo MC, Tan LF. Efficacy and Safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: a randomized clinical trial. Thorax. 2013;68:738–745. doi: 10.1136/thoraxjnl-2012-202744. [DOI] [PubMed] [Google Scholar]
- Madrid LV, May MW, Reuther JY, Baldwin AS., Jr Oncoprotein suppression of tumor necrosis factor-induced NFκB activation is independent of raf-controlled pathways. J Biol Chem. 2001;276:18934–18940. [Google Scholar]
- Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S. Vanden Berghe T, (2011) Role of IL-1α and the Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38:1019–1028. doi: 10.1183/09031936.00158110. [DOI] [PubMed] [Google Scholar]
- Pizzichini E, Pizzichini MM, Efthimiadis A, Evans S, Morris MM, Squillace D, et al. Indices of airway inflammation in induced sputum: reproducibility and validity of cell and fluid phase measurements. Am J Resp Crit Care Med. 1996;154:308–317. doi: 10.1164/ajrccm.154.2.8756799. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. Complex roles of Stat1 in regulating gene expression. Oncogene. 2000;19:2619–2627. doi: 10.1038/sj.onc.1203525. [DOI] [PubMed] [Google Scholar]
- Renda T, Baraldo S, Pelaia G, Bazzan E, Turato G, Papi A, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31:62–69. doi: 10.1183/09031936.00036707. [DOI] [PubMed] [Google Scholar]
- Singh D. p38 inhibition in COPD; cautious optimism. Thorax. 2013;68:705–706. doi: 10.1136/thoraxjnl-2013-203498. [DOI] [PubMed] [Google Scholar]
- Singh D, Smyth L, Borrill Z, Sweeney L, Tal-Singer R. A randomized, placebo-controlled study of the effect of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. J Clin Pharm. 2010;50:94–100. doi: 10.1177/0091270009347873. [DOI] [PubMed] [Google Scholar]
- Smith SJ, Finney-Hayward TK, Mayer RJ, Barnette MS, Barnes PJ, Donnelly LE, et al. Differences in MAP-kinase dependent cytokine responses in cells of the monocytic lineage. J Pharm Exp Therap. 2008;324:306. doi: 10.1124/jpet.107.127670. [DOI] [PubMed] [Google Scholar]
- Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2:63–72. doi: 10.1016/S2213-2600(13)70200-5. [DOI] [PubMed] [Google Scholar]
- Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010;3:cm1. doi: 10.1126/scisignal.3105cm1. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ. A central role for p38 MAPK in the early transcriptional response to stress. BMC Biol. 2010;2010:47. doi: 10.1186/1741-7007-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
. Primer sequences for real-time PCR studies.
Table S2. Probesests differentially regulated by dilmapimod 25 mg compared to placebo at 1 h post-dose (P < 0.01, FC > 1.3).
Table S3. Probesets differentially regulated by dilmapimod 25 mg compared to placebo at 2 h post-dose (P < 0.01, FC > 1.3).
Table S4. Probesets differentially regulated by dilmapimod 25 mg compared to placebo at 6 h post-dose (P < 0.01, FC > 1.3).