

Abstract
Chronic treatment with β2 adrenoceptor agonists is recommended as a first-line maintenance therapy for chronic obstructive pulmonary disease (COPD). However, a potential consequence of long-term treatment may be the loss of functional response (tachyphylaxis) over time. In this study, we have investigated the tendency of such agonists, with a range of efficacies, to develop functional desensitization to cAMP responses in primary human bronchial smooth muscle cells following prolonged agonist exposure. The data show that upon repeat exposure, all agonists produced functional desensitization to the same degree and rate. In addition, β2 adrenoceptor internalization and β-arrestin-2 recruitment were monitored using β2·eGFP visualization and the PathHunter™ β-arrestin-2 assay, respectively. All agonists were capable of causing robust receptor internalization and β-arrestin-2 recruitment, the rate of which was influenced by agonist efficacy, as measured in those assays. In summary, although a relationship exists between agonist efficacy and the rate of both receptor internalization and β-arrestin-2 recruitment, there is no correlation between agonist efficacy and the rate or extent of functional desensitization.
Keywords: β2 adrenoceptor, agonist efficacy, COPD, functional desensitization
Introduction
Inhaled β2 adrenoceptor agonists are widely used for the treatment of asthma and chronic obstructive pulmonary disease (COPD), providing symptomatic relief by inducing bronchodilation via relaxation of airway smooth muscle. Specifically, relaxation is brought about when these agonists activate β2 adrenoceptors expressed on airway smooth muscle cells, increasing the activity of adenylate cyclase and leading to increases in intracellular levels of cAMP. The β2 adrenoceptor agonists currently used in the management of asthma and COPD can be classified as either short-acting β2 adrenoceptor agonists (SABA), for example, salbutamol; long-acting β2 adrenoceptor agonists (LABA), for example, formoterol and salmeterol; or ultra LABA, for example, indacaterol, which is based on their duration of action after a single inhaled dose, that is, 4–6, 12, and 24 h, respectively. Short-acting agonists are recommended as first-line monotherapy for patients with mild COPD, whereas for patients with more a more severe disease state, longer acting β2 adrenoceptor agonists are recommended, either for use alone or in combination with inhaled corticosteroids (Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease [GOLD[ 2013).
Chronic treatment with β2 adrenoceptor agonists are recommended as a first-line maintenance therapy for COPD. However, a potential consequence of long-term treatment may be the loss of functional response (tachyphylaxis) over time through G protein coupled receptor (GPCR) desensitization. There have been a number of studies exploring loss of clinical responsiveness to LABA therapy in the clinic within the first 2 weeks of beginning treatment (Bhagat et al. 1995; Aziz et al. 1998; Drotar et al. 1998; Giannini et al. 2011). This loss of responsiveness usually manifests itself as a rapid desensitization to the bronchoprotection afforded by LABA therapy, possibly through tachyphylaxis of receptor signaling on inflammatory cells, in particular mast cells (Scola et al. 2004, 2009). However, the bronchodilator properties of LABA therapy appear to be much more resistant to tolerance, possibly due to the higher receptor reserve present in airway smooth muscle cells, compared to mast cells (Chong and Peachell 1999; Giembycz 2009), or the relatively lower expression of proteins involved in receptor regulation (McGraw and Liggett 1997). The amount of tachyphylaxis to chronic LABA therapy has been less comprehensively studied, and many of the results are contradictory and often complicated by differences in study design. In some cases salmeterol has been shown to produce tolerance to bronchodilation (Donohue et al. 2002, 2003) compared to long-acting muscarinic antagonist therapy. In other studies there was no evidence of tolerance to chronic treatment of either formoterol or salmeterol (FitzGerald et al. 1999; Hanania et al. 2005). In a more recent study, salmeterol and formoterol were shown to suffer from a slight loss of efficacy between 13 and 52 weeks of continuous treatment although it was not found to be significant in terms of clinically meaningful outcomes (Donohue et al. 2008).
The suggestion that chronic treatment with higher efficacy agonists may lead to more tachyphylaxis is a much debated topic. Developing a better understanding of the relationship between agonist efficacy and the functional desensitization of receptors may aid in the interpretation of observed clinical responses to agonist-based therapies, including β2 adrenoceptor agonists, and importantly, may also help illuminate best therapeutic approaches for future treatment paradigms with agonist-based therapies. Early studies examining the desensitization of the β2 adrenoceptor showed a clear relationship between agonist efficacy and receptor desensitization, with partial β2 agonists causing less phosphorylation and internalization than full agonists (January et al. 1997; Clark et al. 1999; Moore et al. 2007). However, these early experiments conducted with the β2 agonists were designed so that receptor occupancy was matched for each agonist, regardless of efficacy. This effectively negated any influence of receptor reserve (spare receptors not required to produce a maximum response in the presence of the agonist). In the clinic, the dose of agonist chosen to treat patients is determined based on therapeutic effect (e.g., bronchodilation) and not on receptor occupancy. A more recent study by Düringer et al. (2009) addressed this problem by comparing the desensitization profile of β2 agonists at equi-effective concentrations (i.e., concentrations that produce equal functional responses), rather than equal occupancy, thus more closely reflecting the clinical situation. This study showed that under conditions of continuous agonist stimulation, reduction in receptor responsiveness was proportional to the initial functional effect and similar for all agonists studied. These results were in contrast to previous studies, questioning whether high-efficacy agonists necessarily induce more desensitization than partial agonists (Charlton 2009).
In the clinic, an acute (sudden) loss in response to a drug after its administration is termed tachyphylaxis. When the same effect is observed in vitro in experiments using cell cultures or isolated tissues we use the term desensitization. However, the term desensitization is also used to describe many of the individual processes in the pathway leading to desensitization such as β-arrestin-2 recruitment, receptor phosphorylation, and internalization, and recent studies suggest that these processes do not necessarily shut down receptor-mediated signaling. Indeed, it has been shown that some internalized receptors, including the β2 adrenoceptor, are still capable of signaling from endosomes (Mullershausen et al. 2009; Calebiro et al. 2010; Irannejad et al. 2013) and that β-arrestin-2 is a signaling molecule in its own right, able to initiate non-G protein-mediated pathways upon recruitment to the receptor (Wei et al. 2003; Shenoy and Lefkowitz 2005). It is therefore important to clarify the terminology used in this manuscript. The IUPHAR guide on terms used in Quantitative Pharmacology states that tachyphylaxis and desensitization are “overlapping terms that refer to a spontaneous decline in the response to a continuous application of agonist, or to repeated applications or doses. No mechanism is implied by either term, and it is recommended that desensitization be used when the fade or tachyphylaxis is considered to be a direct consequence of receptor activation” (Neubig et al. 2003). As we feel that tachyphylaxis is a term more commonly used to describe this loss of response in in vivo or clinical settings, we have chosen desensitization as a more appropriate term for these in vitro studies and further clarified the term as “functional desensitization” to describe a loss in functional cAMP response following agonist administration. This term is intended to encompass a number of mechanisms including G protein uncoupling, receptor phosphorylation, β-arrestin recruitment, receptor internalization, and downregulation, all of which may contribute to an overall loss in the ability of the cells to respond to agonists and produce cAMP. This we believe to be the most appropriate readout of tachyphylaxis. In addition, we have used the term receptor modifications to incorporate biochemical processes such as β-arrestin-2 recruitment, receptor phosphorylation, and internalization.
The present study was conducted to compare the ability of a number of β2 adrenoceptor agonists with a range of intrinsic efficacies to cause functional desensitization in primary human bronchial smooth muscle cells. In an attempt to more accurately reflect the conditions in the clinic, whereby patients are on continuous treatment with the same LABA to maintain target coverage, we have used the same agonist for the subsequent rechallenge after prolonged agonist treatment. In addition, we have studied β-arrestin-2 recruitment and receptor internalization in two highly sensitive recombinant systems. Our results show all of the agonists tested were able to recruit β-arrestin-2 and promote receptor internalization with a range of intrinsic efficacies. When cAMP accumulation was studied following prolonged exposure to each agonist it was shown that all of the agonists produced the same degree of functional desensitization. We therefore conclude that there is no correlation between intrinsic efficacy for cAMP accumulation and functional desensitization of this response.
Materials and Methods
Materials
PathHunter™ CHO-K1 β2-adrencoceptor: β-arrestin-2 cells (CHO-β2:arrestin) and PathHunter™ lysis and Flash detection reagents were purchased from DiscoveRx (Birmingham, UK). Human bronchial smooth muscle cells were purchased from Lonza (Slough, UK). McCoys 5A, Ham’s F12 media supplemented with l-glutamine, CO2-independent medium, heat inactivated fetal bovine serum (FBS), geneticin, hygromycin, penicillin, streptomycin, trypsin-ethylenediaminetetraacetic acid (EDTA), Dulbecco’s phosphate-buffered saline solution, Hank’s balanced salt solution without phenol red (HBSS w/o phenol red), HEPES, and Lipofectamine2000™ were all purchased from Invitrogen (Paisley, UK). Smooth muscle cell growth medium and supplements were purchased from Promocell (Heidelberg, Germany). AlphaScreen cAMP detection kit (containing streptavidin-coated donor beads, anti-cAMP acceptor beads, biotinylated cAMP, and cAMP standard) 96-well Viewplates™ and white 384-well CulturPlates were purchased from Perkin Elmer Life Sciences (Boston, MA). Bovine serum albumin (BSA), Tween-20, isoprenaline, rolipram, were purchased from Sigma Aldrich (Poole, UK).
Cell culture
Human bronchial smooth muscle cells (hBSMc) were routinely cultured at 37°C, 5% CO2 in smooth muscle cell growth medium, supplemented with FBS (5% v/v), epidermal growth factor (recombinant human – 0.5 ng mL−1), basic fibroblast growth factor (recombinant human – 2 ng mL−1), and insulin (recombinant human – 5 μg mL−1).
U2OS cells were maintained in McCoys 5A medium supplemented with FBS (10% v/v) at 37°C, 5% CO2. A stable cell line expressing a β2 adrenoceptor eGFP construct was generated using Lipofectamine2000™ transfection according to manufacturer’s instructions, followed by clonal selection (from here on referred to as U2OS-β2·eGFP). Expression was maintained using antibiotic selection (500 μg mL−1 geneticin).
CHO-β2:arrestin cells (DiscoveRx) were maintained in Ham’s F12 nutrient mix supplemented with FBS (10% v/v), penicillin (100 IU mL−1), streptomycin (100 μg mL−1), hygromycin (200 μg mL−1), and geneticin (600 μg mL−1) at 37°C, 5% CO2.
For experiments, all of the cells detailed above were harvested using trypsin/EDTA and seeded on to multiwell plates in their subculture medium.
Measurement of cAMP in hBSMc cells using AlphaScreen technology EC50 determinations
Cells were seeded into 96-well ViewPlates™ at 20,000 cells per well in culture medium and grown at 37°C, 5% CO2 for 24 h. Spent medium was removed and cells washed 1× with assay buffer (HBSS w/o phenol red, 5 mmol/L HEPES, 5 μmol/L Rolipram and 0.1% [v/v[ BSA). A range of concentrations of agonist were added to the wells and incubated with the cells for 2 h at 37°C. This was followed by the addition of lysis buffer (dH2O, 0.3% [v/v[ Tween-20) containing 20 units mL−1 streptavidin-coated donor beads and biotinylated cAMP (preincubated for 30 min), and 20 units mL−1 anti-cAMP acceptor beads added to the buffer just before addition to the assay plate. The plate was then incubated in the dark, at room temperature for 60 min, and read on the Envision plate reader (Perkin Elmer). A cAMP standard curve was constructed in each experiment – the concentration range covered was 10,000–0.001 nmol/L. The cAMP standard was diluted in assay buffer and lysis buffer containing the bead mix was added to the standard curve at the same time it was added to the wells of the assay.
Experiments to investigate loss of receptor function following prolonged incubation with agonist
As described previously, cells were seeded into 96-well ViewPlates™ at 20,000 cells per well in culture medium and grown at 37°C, 5% CO2 for 24 h. Spent medium was removed from each well and replaced with either fresh medium or a range of concentrations of β2 adrenoceptor agonists which had been serially diluted in cell culture medium containing 0.01% (w/v) ascorbic acid to prevent compound oxidation. The cells were incubated with agonist at 37°C, 5% CO2 for between 1 and 24 h, before the medium was removed and the cells were washed 5× with assay buffer (HBSS w/o phenol red, 5 mmol/L HEPES, 5 μmol/L rolipram, and 0.1% [v/v[ BSA) to remove any exported cAMP and BackSeal was applied to the plate. A range of agonist concentrations which matched those used for pretreatment were diluted in assay buffer and transferred to the cell plate. In effect, if cells had been pretreated with a range of concentrations of isoprenaline, then the same concentration range of isoprenaline was added back to the cells following the wash step. As a control, cells which had been incubated with media alone were also exposed to the same concentration range of agonist. The cells were incubated for 2 h at 37°C, as this gave a robust signal in this cAMP accumulation assay. The incubation was terminated by the addition of lysis buffer (dH2O, 0.3% [v/v[ Tween-20) containing 20 units mL−1 streptavidin-coated donor beads and biotinylated cAMP (preincubated for 30 min) and 20 units mL−1 anti-cAMP acceptor beads, added to the buffer just before addition to the assay plate. The assay plate and standard curve plate were incubated in the dark, for at least 60 min, at room temperature. The plate was then read on the Envision plate reader (Perkin Elmer).
Internalization assay
U2OS-β2·eGFP cells were seeded overnight in black, clear bottomed 384-well ViewPlates (Greiner, UK) at 3000 cells/well. On the day of the experiment, spent medium was removed and replaced with 40 μL CO2-independent medium supplemented with FBS (10% v/v) and Hoechst nuclei stain (1 μmol/L). Cells were stimulated with 20 μL of β2 adrenoceptor agonists for between 0 and 2 h, and imaged using ImageXpress Micro automated imaging system on the IX500 (Molecular Devices, Sunnydale, CA). All experimental manipulations with viable cells were performed at 30°C to minimize assay medium evaporation during extended time courses. Images were collected using an 40× objective and a Peltier cooled CCD camera (1280 × 1024 pixel). Exposure times were 5 msec for DAPI (nuclear stain) and 200 msec for fluorescein isothiocyanate (β2·eGFP). Cellular image analysis was performed using MetaXpress software and Timelapse analysis journals embedded with the Transfluor application module to measure the appearance of vesicles (1.4–2.2 μm) containing the β2·eGFP receptor over time.
Measurement of β-arrestin-2 recruitment using enzyme fragment complementation
The PathHunter™ β-arrestin-2 assay (DiscoveRx) uses enzyme fragment complementation between two portions of β-galactosidase to measure recruitment of β-arrestin-2 to a GPCR after activation. The larger portion (EA tag) is fused to β-arrestin-2 and the smaller portion (ProLink™) is fused to the C-terminus of the GPCR of choice. Activation of the receptor causes recruitment of β-arrestin-2 to the GPCR thus forming a functional β-galactosidase enzyme whose activity can be measured by the addition of a chemiluminescent substrate. CHO-β2:arrestin cells were seeded into opaque 384-well CulturPlates (PerkinElmer, UK) at 3000 cells/well and grown at 37°C, 5% CO2 for 24 h prior to agonist treatment. On the day of the assay, medium was removed and replaced with HBSS containing 0.1% BSA (w/v) and 20 mmol/L HEPES, at pH 7.4. Cells were stimulated with a range of β2 adrenoceptor agonists for between 15 min and 6 h at 37°C, 5% CO2. Following incubation, 25 μL of proprietary Flash reagent mixed with lysis buffer was added to each well, and luminescence was read after 3 min on the Leadseeker plate reader (GE Healthcare, UK).
Data analysis
The amount of cAMP detected by the AlphaScreen cAMP kit was quantified using the Envision plate reader (Perkin Elmer). To account for the interassay variation in levels of cAMP which were produced, in each experiment, the data for the cells which had been pretreated with agonist were expressed as a percentage of its respective control cells which had not been pretreated with agonist.
Analysis was performed using Prism 4.0 (GraphPad Software Inc., San Diego, CA). % maximum isoprenaline response values for each agonist concentration response curve were analyzed by nonlinear regression, sigmoidal dose response (variable slope) according to the following equation:
where Y is the % maximum isoprenaline response, Top denotes maximal asymptotic percentage response, and Bottom denotes the minimal asymptotic percentage response. This generated pEC50 values and % Emax for each agonist under all conditions. Data are summarized as mean ± SEM from n number of individual experiments.
For internalization time-course experiments, data were analyzed by nonlinear regression, polynomial: first order (straight) line according to the following equation:
where Y is the % maximum isoprenaline response. The rate of internalization was calculated using the slope of the fit between 2 and 15 min postagonist addition.
To calculate the rate of β-arrestin-2 recruitment, data were analyzed using nonlinear regression, one phase exponential association according to the following equation:
where Y is the % maximum isoprenaline response, starting at zero and ascending to Ymax with the rate constant K. The rate half-time is calculated as 0.69/K.
To determine if rates were significantly different, a one-way analysis of variance (ANOVA) was performed, followed by Tukey multiple comparison test.
Results
Investigating cAMP signaling in hBSMc following chronic agonist treatment
By monitoring β2 adrenoceptor agonist-induced cAMP signaling in the hBSMc, we demonstrated that the agonists tested showed a range of potencies and intrinsic efficacies in these cells (Fig. 1, Table 1).
Figure 1.
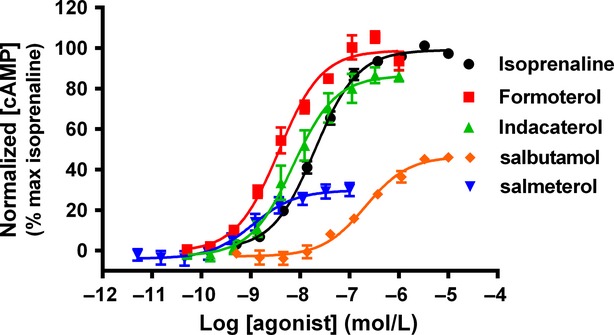
β2 adrenoceptor-mediated cAMP accumulation in hBSMc. Concentration effect curves for cAMP generation were determined in hBSMc following exposure to β2 adrenoceptor agonists for 2 h. For each individual experiment, data have been normalized to the maximum amount of cAMP produced after addition of 10 μmol/L isoprenaline, and are expressed as means ± SEM for three independent experiments.
Table 1.
Potency and efficacy of β2 adrenoceptor agonists in cAMP accumulation, β-arrestin-2 recruitment, and internalization assays.
| cAMP |
Internalization |
β-arrestin-2 recruitment |
||||
|---|---|---|---|---|---|---|
| pEC50 | % max response | pEC50 | % max response | pEC50 | % max response | |
| Isoprenaline | 7.8 ± 0.11 | 100 | 8.2 ± 0.2 | 100 | 7.8 ± 0.11 | 100 |
| Formoterol | 8.8 ± 0.19 | 98.5 ± 1.8 | 9.1 ± 0.2 | 96.6 ± 5.8 | 8.8 ± 0.12 | 108 ± 4.8 |
| Indacaterol | 8.2 ± 0.09 | 87.2 ± 3.6 | 8.6 ± 0.1 | 95.1 ± 3.6 | 7.8 ± 0.10 | 98.3 ± 6.0 |
| Salbutamol | 6.7 ± 0.03 | 48.2 ± 1.4 | 7.0 ± 0.1 | 68.8 ± 3.9 | 6.9 ± 0.07 | 56.0 ± 3.9 |
| Salmeterol | 9.0 ± 0.26 | 34.2 ± 5.8 | 8.9 ± 0.3 | 43.7 ± 4.1 | 8.4 ± 0.08 | 27.1 ± 2.5 |
Data are expressed as means ± SEM for three independent experiments (3–5 for internalization). Potency and efficacy of β2 adrenoceptor agonists in cAMP accumulation using hBSMc (AlphaScreen), β-arrestin-2 recruitment in PathHunter CHO-β2:arrestin cells, and internalization in U2OS-β2·GFP cells. pEC50 values were calculated after a 2 h (cAMP and internalization) or 4 h stimulation (β-arrestin-2 recruitment) with β2 adrenoceptor agonists. Emax was calculated as a percentage of maximal isoprenaline response.
When hBSMc were pretreated with a range of concentrations of each agonist for 3 h or more, washed to remove any exported cAMP, and then exposed to the same concentration of agonist for a further 2 h, each of the agonists tested showed a loss in maximal amount of cAMP production compared to control cells (Fig. 2). In addition, the degree of reduction in cAMP signaling correlated with the length of agonist pretreatment, with the greatest reduction observed after 24 h of agonist exposure. After this duration of agonist exposure the levels of cAMP produced by readdition of each agonist had been reduced by similar amounts, when compared to their own control responses (75.9% for isoprenaline; 76.2% for formoterol; 73.3% for indacaterol; 80.4% for salmeterol). Figure 3A shows that, additionally, the rate of loss of cAMP signaling, which plateaus between 6 and 24 h, is almost identical for each of the agonists tested. The data in Figure 2, 3A were calculated by comparing each agonist to itself in control conditions. The data in Figure 3B show the loss of cAMP signaling with time, but it is plotted as a percentage of the maximal control isoprenaline response. This highlights the differences in the initial levels of cAMP which are produced between the high–moderate and low-efficacy agonists and also shows that in the case of salmeterol the levels of cAMP after 24 h pretreatment are only approximately 5% of the maximal control isoprenaline response. In addition, it also shows that even after full desensitization (post 24 h), isoprenaline, formoterol, and indacaterol still generate approximately the same amount of cAMP in these cells as salmeterol does predesensitization (t = 0). Finally, the loss of cAMP signaling at equi-effective concentrations was studied. In order to look at this the concentration of agonist which produced 20% of the control isoprenaline response was determined from Figure 1 (4.5 nmol/L isoprenaline, 4 nmol/L indacaterol, 11.2 nmol/L salmeterol, and 1.2 nmol/L formoterol). This concentration of agonist was then used to calculate the % cAMP signal remaining at each time point (Fig. 3C). The data show that at equi-effective concentrations all of the agonists show a similar rate of loss of cAMP signaling. The % cAMP response remaining after 3 h treatment with each agonist was compared by one-way ANOVA, and shown to be significantly different between isoprenaline and salmeterol (P < 0.001), indacaterol and salmeterol (P < 0.05), and isoprenaline and formoterol (P < 0.05), suggesting that salmeterol is apparently faster to desensitize than the fuller agonists.
Figure 2.
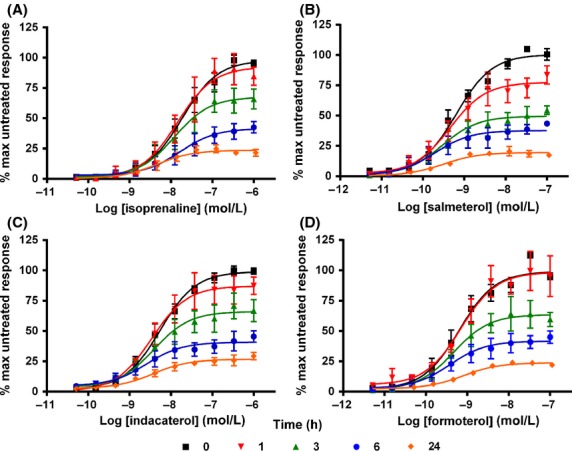
Loss of cAMP signaling in hBSMc following chronic β2 adrenoceptor agonist exposure. hBSMc were pretreated with indicated concentrations of isoprenaline (A), salmeterol (B), indacaterol (C), or formoterol (D) for between 0 and 24 h, washed, and then exposed the same concentration of agonist for a further 2 h. For each individual experiment, data have been normalized to the maximum amount of cAMP produced by agonist in cells which have not been previously exposed to agonist. Data shown are expressed as means ± SEM for three independent experiments.
Figure 3.
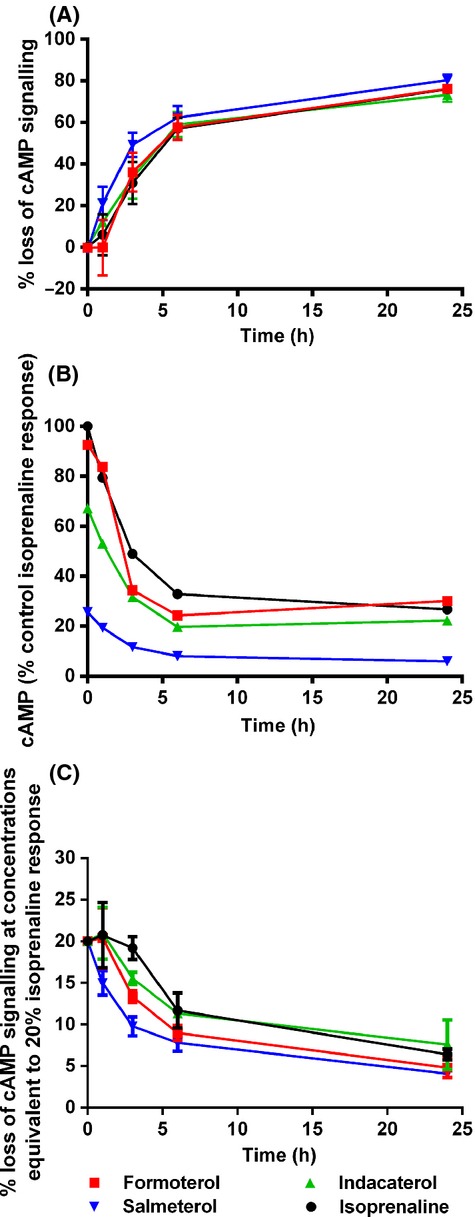
Rate of loss of cAMP signaling in hBSMc following chronic β2 adrenoceptor agonist exposure. (A) Rate of loss of cAMP signaling in hBSMc following pretreatment with β2 adrenoceptor agonists. Data shown are expressed as means ± SEM for three independent experiments. (B) Relative levels of cAMP (% response compared to isoprenaline) produced in human bronchial smooth muscle cells following treatment with different β2 adrenoceptor agonists over a 24 h time period. Data shown are single determinations from a single experiment, which is representative of three independent experiments. (C) Rate of loss of cAMP signaling in human bronchial smooth muscle cells following pretreatment with a concentration of β2 adrenoceptor agonist which produces an amount of cAMP equivalent to 20% of the isoprenaline response. Specifically, these concentrations were 4.5 nmol/L isoprenaline, 4 nmol/L indacaterol, 11.2 nmol/L salmeterol, and 1.2 nmol/L formoterol. These concentrations were taken from the data generated in Figure 2 and normalized to 20% isoprenaline. Data shown are expressed as means ± SEM for three independent experiments.
Monitoring β2 adrenoceptor internalization over time
After demonstrating that all the ligands we tested resulted in functional desensitization of the cAMP response in hBSMc, we then aimed to determine if these agonists were capable of causing receptor internalization. Unfortunately there are, as yet, no reagents suitable for monitoring endogenous β2 adrenoceptors using immunocytochemical (ICC) or immunofluorescent (IFC) approaches. In addition, the level of receptor expression in hBSMc was insufficient to obtain specific binding of fluorescently labeled agonists or antagonists (data not shown). The only radioligand currently available with sufficiently high specific activity to do detect the low levels of receptors expressed in primary cells is [I125[-cynopindolol, which is a highly lipophilic ligand that demonstrates significant cell penetration and membrane partitioning. For this reason we were unable to use radioligand binding to measure receptor internalization due to significant contamination from internalized receptors.
In order to monitor receptor internalization, we therefore used U2OS cells expressing endogenous β-arrestin-2, which were stably transfected with an eGFP-tagged β2 adrenoceptor. This assay was used to measure the kinetics and extent of receptor internalization in response to agonist treatment. To this end, a time course of β2 adrenoceptor internalization was constructed for each agonist over a range of concentrations (10,000–0.1 nmol/L).
For all of the agonists tested internalization of the β2 adrenoceptor was observed, as shown in Figure 4. In addition, this was shown to be concentration-dependent (Fig. 5A) and pEC50 values could be determined, as shown in Table 1. The extent to which receptors internalized varied between agonists, with isoprenaline causing the greatest amount of internalization, followed by formoterol, indacaterol, and salbutamol with salmeterol causing the least amount of internalization. From these data, EC80 concentrations were derived and the rate of internalization was compared for each agonist (Fig. 5B, Table 2). The rates were shown to be significantly different when compared by one-way ANOVA (P < 0.0001). Isoprenaline had the fastest onset of receptor internalization, followed by formoterol, indacaterol, salbutamol, and salmeterol.
Figure 4.
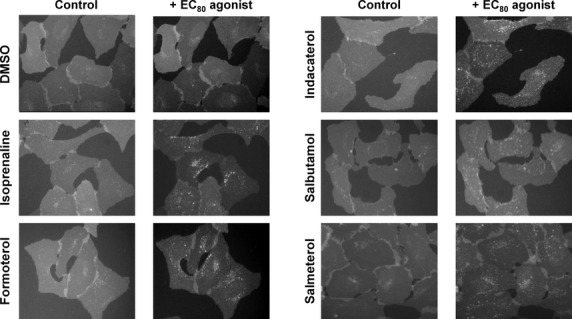
β2 adrenoceptor agonist-mediated receptor internalization. Agonist-dependent formation of β2·eGFP containing vesicles following stimulation of U2OS-β2·eGFP cells with an EC50 concentration of agonist (derived from Fig. 5) for 60 or 90 min in the case of salmeterol. Images were performed using ImageXpress Micro automated imaging system on the IX500 (Molecular Devices). Images were collected using a 40× objective and a Peltier cooled CCD camera. β2·eGFP receptors were detected using fluorescein isothiocyanate filter and a 200 msec exposure time.
Figure 5.
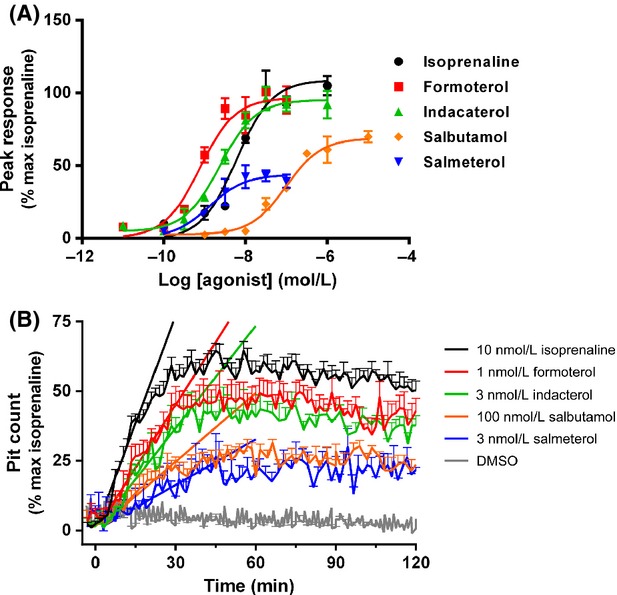
Concentration- and time dependency of receptor-mediated β2-adrenoceptor internalization in U2OS-β2·GFP cells. (A) Concentration-dependent increases in β2 adrenoceptor internalization were assessed between 0 and 2 h stimulation with the indicated concentrations of β2 adrenoceptor agonists. (B) Time-dependent increases in β2 adrenoceptor internalization for between 0 and 2 h after stimulation of cells with approximately EC80 concentrations of agonist (as determined from A). Slope factors (K) determined for initial rate of β2 adrenoceptor internalization (between 0 and 45 min) from polynomial first order (straight) line fit. For each individual experiment, data have been normalized to the maximum amount of β2 adrenoceptor internalization detected after addition of 10 μmol/L isoprenaline, and are expressed as mean ± SEM for 3–5 independent experiments.
Table 2.
Rate of β2 adrenoceptor-mediated β-arrestin-2 recruitment and internalization.
| K value (% max isoprenaline response per minute) |
||
|---|---|---|
| Internalization | β-arrestin-2 recruitment | |
| Isoprenaline | 2.58 ± 0.15 | 0.032 ± 0.002 |
| Formoterol | 1.50 ± 0.17 | 0.024 ± 0.001 |
| Indacaterol | 1.22 ± 0.10 | 0.016 ± 0.001 |
| Salbutamol | 0.82 ± 0.15 | 0.013 ± 0.001 |
| Salmeterol | 0.55 ± 0.16 | 0.008 ± 0.002 |
Data are expressed as means ± SEM for three independent experiments (3–5 for internalization). Rate of receptor-mediated internalization in U2OS-β2·GFP cells and β-arrestin-2 recruitment in PathHunter CHO-β2:arrestin cells, generated using an EC80 concentration of agonist. Slope factors (K) were determined for rate of β-arrestin-2 recruitment by fitting data to a monoexponetial curve, or initial rate of β2 adrenoceptor internalization (between 0 and 45 min) from polynomial first order (straight) line fit.
β-arrestin-2 recruitment
Using the highly sensitive U2OS-β2·eGFP internalization assay, we were able to demonstrate that all agonists tested were capable of causing β2 adrenoceptor internalization. We then aimed to determine if all agonists were also capable of causing β-arrestin-2 recruitment. The PathHunter™ β-arrestin-2 assay was used to measure β-arrestin-2 recruitment to the β2 adrenoceptor as a consequence of agonist stimulation of the receptor. Full concentration response curves were performed for each agonist for between 15 min and 6 h (Fig. 6A). The potency was lower at 15 min than 2 h for all of the agonists tested, although the time taken to reach full potency varied for each agonist. Despite this, rank order of potency did not change with increasing stimulation time, the mean pEC50 values for each agonist following a 2 h stimulation are shown in Table 1. A time course of β-arrestin-2 recruitment was then constructed for EC80 concentrations of each agonist (Fig. 6B).
Figure 6.
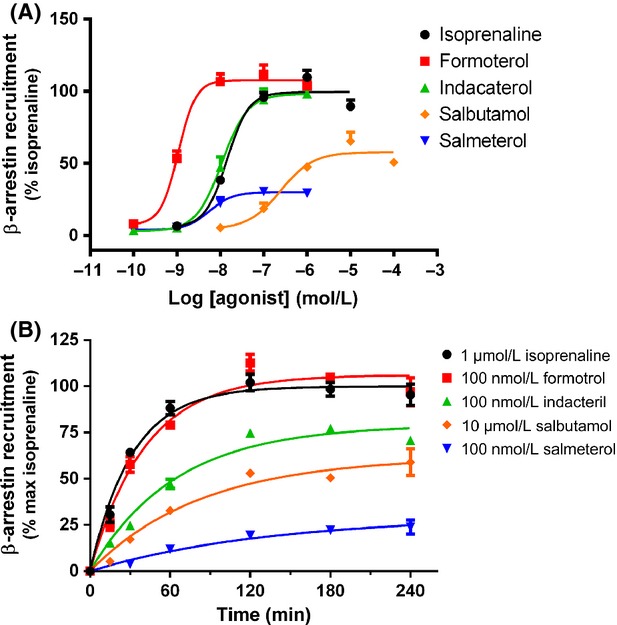
Concentration- and time dependency of receptor-mediated β-arrestin-2 recruitment in PathHunter™ CHO-β2:arrestin cells. (A) Concentration-dependent increases in β-arrestin recruitment were assessed between 0 and 4 h stimulation with the indicated concentrations of β2 adrenoceptor agonists. (B) Time-dependent increases in β-arrestin-2 recruitment for between 0 and 4 h after stimulation of cells with approximately EC80 concentrations of agonist (as determined from part A). Data were fit by nonlinear regression to a one phase exponential association to determine half-life of β-arrestin-2 recruitment. For each individual experiment, data have been normalized to the β-arrestin-2 recruitment detected after addition of 10 μmol/L isoprenaline for 4 h, and are expressed as mean ± SEM for three independent experiments.
The requirement for complementation to occur between the two portions of β-galactosidase following β-arrestin-2 recruitment and the rate of the subsequent enzyme reaction that produces the luminescent signal appears to cause a lag in the kinetics observed with this assay. It has been demonstrated previously using FRET measurements that the kinetics of β-arrestin-2 associations with the β2 adrenoceptor occur with a t1/2 of 19.6 sec (Krasel et al. 2004), although these experiments were performed in the presence of cotransfected GRKs which may also contribute to the faster rates observed. In contrast, measurement of the same associations with the β-galactosidase fragmentation complementation method used here can take 1–4 h to reach maximal activation (Fig. 6B). This is similar to previous work using complementation assays, where a treatment time of 1 h was required to produce a maximal response (Carter and Hill 2005). This implies that while the assay is useful in measuring comparative rates, allowing us to rank agonists against each other, it does not represent the true rate of β-arrestin-2 recruitment that would be observed in a physiological system.
Despite this limitation, we were able to measure differences in the onset of action for each agonist. Isoprenaline had the fastest onset of β-arrestin-2 recruitment followed by formoterol, indacaterol, salbutamol, and salmeterol, K values are shown in Table 2. Rates of β-arrestin-2 recruitment determined from EC80 concentrations of agonist were compared by one-way ANOVA, and shown to be significantly different with P < 0.0001. In addition, when comparing individual agonists, the high-efficacy ligands isoprenaline and formoterol were shown to have significantly faster rates of β-arrestin-2 recruitment than the lower efficacy ligands (all with P < 0.05).
Discussion
β2 adrenoceptor agonists have been used extensively in the treatment of pulmonary disorders such as asthma and COPD. As these disorders are chronic in nature they require long-term treatment with the β2 adrenoceptor agonists which may lead to a loss of responsiveness through tachyphylaxis. There have been a number of clinical studies that have investigated the long-term β2 adrenoceptor therapy (20–24 weeks), but very few so far that have investigated tachyphylaxis for periods of greater than 6 months. Those that have been completed so far have yielded conflicting results with some studies indicating tachyphylaxis does occur, whereas others suggest little or no loss of efficacy upon long-term treatment (Steffensen et al. 1995; FitzGerald et al. 1999; Donohue et al. 2002, 2003; Hanania et al. 2005). Here we have attempted to model some of the processes involved in tachyphylaxis in order to understand these discrepancies more fully. To this end, we have investigated a number of β2 adrenoceptor agonists for their ability to cause in vitro functional desensitization of cAMP responses in hBSMc after repeat exposure to the same agonist. In addition, we have investigated their ability to cause receptor internalization and the recruitment of β-arrestin-2.
Early studies examining receptor desensitization of the β2 adrenoceptor showed a clear relationship between agonist efficacy and desensitization, with partial β2 agonists causing less phosphorylation and internalization than full agonists (January et al. 1997; Clark et al. 1999). However, these early experiments conducted with the β2 agonists were designed so that receptor occupancy was matched for each agonist, regardless of efficacy. This effectively negated any influence of receptor reserve and meant that agonists were tested at concentrations that gave markedly different functional effects. Importantly, clinical doses are chosen based on functional effect not receptor occupancy, so these early studies are not useful for understanding the tendency of particular agonists to induce tachyphylaxis in the clinic. One way to compensate for this is to compare the functional desensitization elicited by agonists at concentrations which generate the same functional response, termed an equi-effective concentration. To date, only Düringer et al. (2009) and more recently Cooper et al. (2011) have used equi-effective concentrations of agonists while investigating functional desensitization at the β2 adrenoceptor. In both of these studies, chronic (12–24 h) stimulation of small airways or isolated human airway smooth muscle cells resulted in similar loss of responsiveness to a subsequent challenge by a high-efficacy agonist (isoprenaline or formoterol). However, use of a full agonist after chronic challenge may underestimate the amount of functional desensitization that would be observed to a partial agonist after chronic treatment with the same partial agonist. In contrast, the use of a partial agonist to challenge cells after chronic treatment with a full agonist may result in greater levels of functional desensitization. As discussed in Charlton (2009), partial agonists are required to activate a higher number of receptors to produce the same response as a full agonist and so are more susceptible to loss of functional receptors than the full agonists. In the clinic, patients are on continuous treatment with the same LABA to maintain target coverage. In an attempt to more accurately reflect these conditions in our in vitro studies, we have used the same agonist for the subsequent rechallenge after prolonged agonist treatment. In addition, we chose to run full concentration response curves for each agonist.
This protocol revealed that all of the agonists tested produced functional desensitization to the cAMP response in hBSMc to the same extent. In addition, the rate of loss of cAMP signaling was shown to be similar for all of the agonists tested when considering either the maximum loss in signaling or equi-effective concentrations. Consequently, from this study our data indicate that neither the extent nor rate of functional desensitization are correlated with agonist efficacy. The time course we have observed in our functional desensitization studies is somewhat slower than might be expected if these responses were solely due to receptor phosphorylation, arrestin binding to the receptor or receptor internalization. We started to see functional desensitization after 3 h of treatment (1 h for salmeterol) that successively increased over time. This suggests that functional desensitization is a combination of many factors and is a much more complex process. This most likely includes specific receptor modifications such as phosphorylation, β-arrestin recruitment, and internalization at the shorter time points, followed by receptor downregulation at the later time points. In addition, we now know that processes such as these do not necessarily lead to complete shutdown of receptor signaling but may activate alternative signaling pathways (Shenoy and Lefkowitz 2005).
This lack of correlation may be explained by revisiting the relationship between efficacy and receptor occupancy. Although lower efficacy agonists cause less receptor modification such as β-arrestin-2 recruitment and internalization, they require a greater number of receptors to be occupied to generate a subsequent response and will therefore be sensitive to even a small reduction in receptor number. In contrast, although higher efficacy agonists cause more receptor modification they require fewer receptors to be activated to give a subsequent response, so will be more resistant to loss of receptors. Interestingly, as has been shown in this study, in both situations you would expect to observe some functional desensitization, this is discussed in more detail by Charlton (2009). In these studies, we observed rapid functional desensitization to each agonist within the first 6 h of treatment, which plateaued between 6 and 24 h.
It is also worth considering the relative levels of cAMP that are produced for each of the agonists following prolonged agonist exposure and functional desensitization, highlighted by the data shown in Figure 3B. Initially the amount of cAMP produced by salmeterol was just over 20% of that produced by isoprenaline. After 24 h exposure, the response was reduced by about 75–80% for all of the agonists tested. This means that salmeterol only generates 4% of the original isoprenaline response, compared to 20% for isoprenaline and formoterol and approximately 15% for indacaterol. So, despite the fact that each agonist shows the same percentage of functional desensitization of the cAMP response, isoprenaline, formoterol, and indacaterol generate a similar absolute amount of cAMP after chronic exposure as salmeterol does before desensitization. This suggests that very low-efficacy agonists may be more sensitive to loss of functional response upon repeated exposure and might not maintain a sufficient cAMP signal to sustain full 24 h bronchodilation.
We have reason to speculate that the levels of cAMP required to cause bronchodilation may be quite low given that in the presence of substantial functional desensitization in vitro, in the clinic 24 h bronchodilation is observed with LABAs. Additionally, in a number of studies (Steffensen et al. 1995; FitzGerald et al. 1999; Donohue et al. 2002, 2003; Hanania et al. 2005) loss of efficacy is not observed following long-term treatment with β2 adrenoceptor agonists. In this study we have measured global cAMP production which could be a potential limitation as contraction/relaxation could be driven by compartmentalization of high concentrations of cAMP into areas localized with contractile machinery. It has also been demonstrated that there is a higher receptor reserve present in airway smooth muscle cells compared to other cells, such as mast cells, that express the β2 adrenoceptor agonists. This may help to explain why the airway is more resistant to substantial receptor internalization and β-arrestin-2 recruitment, to maintain bronchodilation when other cell types rapidly desensitize immunomodulatory effects such as histamine release (Chong and Peachell 1999).
One of the major steps involved in clathrin-dependent GPCR internalization is the recruitment of β-arrestin-2 following agonist occupation of the receptor and GRK-site phosphorylation ultimately leading to receptor internalization. We have investigated this pathway at two points, to determine whether the β2 adrenoceptor agonists are capable of activating these processes. First, we have used an eGFP-tagged β2 adrenoceptor and endogenous β-arrestin-2 to monitor real-time changes in surface receptor expression using fluorescence imaging, and second the PathHunter™ assay to measure recruitment of β-arrestin-2 to activated receptor using a complementation assay.
To monitor receptor internalization, we have used a system that utilizes endogenous β-arrestin but a recombinant eGFP-tagged receptor, which allowed us to develop a highly sensitive assay. In low expression systems, although receptors may be causing internalization, if there are not enough receptors in the same locale no fluorescent signal will be detected. This may lead to incorrect assumptions regarding partial agonists, and may explain why some studies demonstrate internalization to salmeterol (January et al. 1998; Kallal et al. 1998; Cooper et al. 2011), and others do not, despite showing GRK-site phosphorylation (Moore et al. 2007). We have shown that all of the agonists tested were capable of inducing receptor internalization with different intrinsic efficacies. In addition, we observed a relationship between the degree and the rate at which internalization occurred, such that agonists that were more fully efficacious in this assay had faster internalization rates than the agonists which were partial. This is in contrast to work by Moore et al. (2007), who concluded that the partial agonist salmeterol was incapable of causing significant endocytosis of the β2 adrenoceptor. The initial endocytosis experiments in their study were performed after 15 min, in which time we do not observe endocytosis in response to salmeterol either. Using quantitative Enzyme-linked immunosorbent assay (ELISA), they then demonstrated that over a 60 min incubation, salmeterol was a weak partial agonist, causing only 5% internalization compared to 60% for formoterol. However, when overexpressing eGFP-labeled β-arrestin-2, salmeterol was capable of causing significant receptor internalization after 15 min treatment (Moore et al. 2007). In agreement with our results, Cooper et al. (2011) demonstrated using adenoviral expression of Ad5-CMV-human β2 adrenoceptor-YFP in human lung slices (airway epithelial cells) that salmeterol is capable of internalizing the β2 adrenoceptor, but at a much reduced level compared to formoterol.
Using the complementation assay, we also demonstrated that all ligands were capable of recruiting β-arrestin-2, and that similar to the internalization assay the agonists did so with a range of intrinsic efficacies and rates. Again, this is in contrast to the work by Moore and colleagues, who did not demonstrate any recruitment of β-arrestin-2 to the β2 adrenoceptor after salmeterol challenge. A possible explanation for these different observations is that 2 min was not sufficient time to observe arrestin recruitment in response to salmeterol. Here we have demonstrated that the time-course of β-arrestin-2 recruitment is dependent on agonist intrinsic efficacy as measured in the same assay, so it is likely that the 2 min treatment used by Moore et al. (2007) was insufficient for salmeterol, despite being appropriate for higher efficacy ligands. An alternative explanation could be that the highly sensitive assay systems we have utilized in these studies are more adept at measuring the small responses to partial agonists such as salmeterol and salbutamol.
In summary, in agreement with many other studies performed to date, we find that high-efficacy agonists cause more β-arrestin-2 recruitment and receptor internalization. Historically, a direct relationship has been assumed between the data generated from receptor modification studies such as these, and functional desensitization. However, in our study where we have investigated cAMP responses in hBMSC we have shown that the higher efficacy agonists do not produce a higher degree of functional desensitization. In fact, under conditions that are more akin to the clinical situation where cells are chronically stimulated and then rechallenged with the same agonist, all of the agonists, regardless of their efficacy, produce the same degree of functional desensitization. With this in mind, higher efficacy agonists may provide a more sustained effect as the residual levels of cAMP produced following chronic agonist treatment may still be sufficient to maintain a bronchodilatory effect, even after 24 h. In agreement with these preclinical observations, no loss of bronchodilatory effect is observed in COPD patients following prolonged administration of the high-efficacy agonist indacaterol (Chapman et al. 2011). Finally, these data may also have clinical relevance outside the field of LABA therapy where GPCR agonists are used chronically for the treatment of disease.
Disclosures
None declared.
Glossary
Abbreviations
- ANOVA
analysis of variance
- BSA
bovine serum albumin
- COPD
chronic obstructive pulmonary disease
- FBS
fetal bovine serum
- GOLD
Global Initiative for Chronic Obstructive Lung Disease
- GPCR
G protein coupled receptor
- hBSMc
human bronchial smooth muscle cells
- HBSS
Hank’s balanced salt solution
- ICC
immunocytochemical
- IFC
immunofluorescent
- LABA
long-acting β2 adrenoceptor agonists
- SABA
short-acting β2 adrenoceptor agonists
References
- Aziz I, Tan KS, Hall IP, Devlin MM, Lipworth BJ. Subsensitivity to bronchoprotection against adenosine monophosphate challenge following regular once-daily formoterol. Eur Respir J. 1998;12:580–584. doi: 10.1183/09031936.98.12030580. [DOI] [PubMed] [Google Scholar]
- Bhagat R, Kalra S, Swystun VA, Cockroft DW. Rapid onset of tolerance to the bronchoprotective effect of salmeterol. Chest. 1995;108:1235–1239. doi: 10.1378/chest.108.5.1235. [DOI] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Persani L, Lohse MJ. Signaling by internalized G-protein-coupled receptors. Trends Pharmacol Sci. 2010;31:221–228. doi: 10.1016/j.tips.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Carter AA, Hill SJ. Characterization of isoprenaline- and salmeterol-stimulated interactions between beta 2-adrenoceptors and beta-arrestin 2 using beta-galactosidase complementation in C2C12 cells. J Pharmacol Exp Ther. 2005;315:839–848. doi: 10.1124/jpet.105.088914. [DOI] [PubMed] [Google Scholar]
- Chapman KR, Rennard SI, Dogra A, Owen R, Lassen C, Kramer B, et al. Long-term safety and efficacy of indacaterol, a long-acting β2-agonist, in subjects with COPD: a randomized, placebo-controlled study. Chest. 2011;140:68–75. doi: 10.1378/chest.10-1830. [DOI] [PubMed] [Google Scholar]
- Charlton SJ. Agonist efficacy and receptor desensitization: from partial truths to a fuller picture. Br J Pharmacol. 2009;158:165–168. doi: 10.1111/j.1476-5381.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong LK, Peachell PT. Beta-adrenoceptor reserve in human lung: a comparison between airway smooth muscle and mast cells. Eur J Pharmacol. 1999;378:115–122. doi: 10.1016/s0014-2999(99)00425-2. [DOI] [PubMed] [Google Scholar]
- Clark RB, Knoll BJ, Barber R. Partial agonists and G protein-coupled receptor desensitization. Trends Pharmacol Sci. 1999;20:279–286. doi: 10.1016/s0165-6147(99)01351-6. [DOI] [PubMed] [Google Scholar]
- Cooper PR, Kurten RC, Zhang J, Nicholls DJ, Dainty IA, Panettieri RA. Formoterol and salmeterol induce a similar degree of β2-adrenoceptor tolerance in human small airways but via different mechanisms. Br J Pharmacol. 2011;163:521–532. doi: 10.1111/j.1476-5381.2011.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue JF, Van Noord JA, Bateman ED, Langley SJ, Lee A, Witek TJ, Jr, et al. A 6-month, placebo-controlled study comparing lung function and health status changes in COPD patients treated with tiotropium or salmeterol. Chest. 2002;122:47–55. doi: 10.1378/chest.122.1.47. [DOI] [PubMed] [Google Scholar]
- Donohue JF, Menjoge S, Kesten S. Tolerance to bronchodilating effects of salmeterol in COPD. Respir Med. 2003;97:1014–1020. doi: 10.1016/s0954-6111(03)00131-8. [DOI] [PubMed] [Google Scholar]
- Donohue JF, Hanania NA, Sciarappa KA, Goodwin E, Grogan DR, Baumgartner RA, et al. Arformoterol and salmeterol in the treatment of chronic obstructive pulmonary disease: a one year evaluation of safety and tolerance. Ther Adv Respir Dis. 2008;2:37–48. doi: 10.1177/1753465808089455. [DOI] [PubMed] [Google Scholar]
- Drotar DE, Davis EE, Cockroft DW. Tolerance to the bronchoprotective effect of salmeterol 12 h after starting twice daily treatment. Ann Allergy Asthma Immunol. 1998;80:31–34. doi: 10.1016/S1081-1206(10)62935-3. [DOI] [PubMed] [Google Scholar]
- Düringer C, Grundström G, Gürcan E, Dainty IA, Lawson M, Korn SH, et al. Agonist-specific patterns of beta(2)-adrenoceptor responses in human airway cells during prolonged exposure. Br J Pharmacol. 2009;158:169–179. doi: 10.1111/j.1476-5381.2009.00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzGerald JM, Chapman KR, Cioppa GD, Stubbing D, Fairbarn MS, Till MD, et al. Sustained bronchoprotection, bronchodilatation, and symptom control during regular formoterol use in asthma of moderate or greater severity. The Canadian FO/OD1 Study Group. J Allergy Clin Immunol. 1999;103:427–435. doi: 10.1016/s0091-6749(99)70467-7. [DOI] [PubMed] [Google Scholar]
- Giannini D, Di FA, Bacci E, Dente FL, Vagaggini B, Taccola M, et al. Tolerance to the protective effect of salmeterol on allergen challenge can be partially restored by the withdrawal of salmeterol regular treatment. Chest. 2011;119:1671–1675. doi: 10.1378/chest.119.6.1671. [DOI] [PubMed] [Google Scholar]
- Giembycz MA. An estimation of β2-adrenoceptor reserve on human bronchial smooth muscle for some sympathomimetic bronchodilators. Br J Pharmacol. 2009;158:287–299. doi: 10.1111/j.1476-5381.2009.00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2013. . Available at http://www.goldcopd.org/ (Last accessed on 9 October 2014)
- Hanania NA, Kalberg C, Yates J, Emmett A, Horstman D, Knobil K. The bronchodilator response to salmeterol is maintained with regular, long-term use in patients with COPD. Pulm Pharmacol Ther. 2005;18:19–22. doi: 10.1016/j.pupt.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, et al. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495:534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- January B, Seibold A, Whaley B, Hipkin RW, Lin D, Schonbrunn A, et al. beta(2)-adrenergic receptor desensitization, internalization, and phosphorylation in response to full and partial agonists. J Biol Chem. 1997;272:23871–23879. doi: 10.1074/jbc.272.38.23871. [DOI] [PubMed] [Google Scholar]
- January B, Seibold A, Allal C, Whaley BS, Knoll BJ, Moore RH, et al. Salmeterol-induced desensitization, internalization and phosphorylation of the human beta(2)-adrenoceptor. doi: 10.1038/sj.bjp.0701658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallal L, Gagnon AW, Penn RB, Benovic JL. Visualization of agonist-induced sequestration and down-regulation of a green fluorescent protein-tagged beta2-adrenergic receptor. J Biol Chem. 1998;273:322–328. doi: 10.1074/jbc.273.1.322. [DOI] [PubMed] [Google Scholar]
- Krasel C, Vilardaga JP, Bünemann M, Lohse MJ. Kinetics of G-protein-coupled receptor signalling and desensitization. Biochem Soc Trans. 2004;32:1029–1031. doi: 10.1042/BST0321029. [DOI] [PubMed] [Google Scholar]
- McGraw DW, Liggett SB. Heterogeneity in beta-adrenergic receptor kinase expression in the lung accounts for cell-specific desensitization of the beta2-adrenergic receptor. J Biol Chem. 1997;272:7338–7344. doi: 10.1074/jbc.272.11.7338. [DOI] [PubMed] [Google Scholar]
- Moore RH, Millman EE, Godines V, Hanania NA, Tran TM, Peng H, et al. Salmeterol Stimulation Dissociates beta2-Adrenergic Receptor Phosphorylation and Internalization. Am J Respir Cell Mol Biol. 2007;36:254–261. doi: 10.1165/rcmb.2006-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K. Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol. 2009;5:428–434. doi: 10.1038/nchembio.173. [DOI] [PubMed] [Google Scholar]
- Neubig RR, Spedding M, Kenakin T, Christopoulos A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol Rev. 2003;55:597–606. doi: 10.1124/pr.55.4.4. [DOI] [PubMed] [Google Scholar]
- Scola AM, Chong LK, Suvarna SK, Chess-Williams R, Peachell PT. Desensitization of mast cell beta2-adrenoceptor-mediated responses by salmeterol and formoterol. Br J Pharmacol. 2004;141:163–171. doi: 10.1038/sj.bjp.0705599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scola AM, Loxham M, Charlton SJ, Peachell PT. The long-acting beta2-adrenoceptor agonist, indacaterol, inhibits IgE-dependent responses of human lung mast cells. Br J Pharmacol. 2009;158:267–276. doi: 10.1111/j.1476-5381.2009.00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Seven-transmembrane receptor signaling through beta-arrestin. Science STKE. 2005;2005:Cm10. doi: 10.1126/stke.2005/308/cm10. [DOI] [PubMed] [Google Scholar]
- Steffensen I, Faurschou P, Riska H, Rostrup J, Wegener T. Inhaled Formoterol Dry Powder in the Treatment of Patients With Reversible Obstructive Airway Disease. Allergy. 1995;50:657–663. doi: 10.1111/j.1398-9995.1995.tb02582.x. [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]