

Abstract
The CD133 cell surface protein expresses the AC133 epitope that is associated with cancer progenitor cells and resistance to traditional anticancer therapies. We report that the endoplasmic reticulum Golgi intermediate compartment residing acetyltransferases, ATase1 and ATase2, can physically interact with CD133 to acetylate the protein on three lysine residues predicted to reside on the first extracellular loop of CD133. Site-directed mutagenesis of these residues mimicking a loss of acetylation and downregulation or inhibition of ATase1/ATase2, resulted in near complete abolishment of CD133 protein expression. We also demonstrate that targeting ATase1/ATase2 results in apoptosis of CD133 expressing acute lymphoblastic leukemia cells. Taken together, we suggest that lysine acetylation on predicted extracellular residues plays a key role in expression and trafficking of CD133 protein to the cell surface and can be targeted to disrupt CD133 regulation and function.
Keywords: Membrane protein, ERGIC, trafficking, post-translation modifications, protein processing
COMMUNICATION
The AC133 epitope of the 120 kDa pentaspan transmembrane glycoprotein CD133 has been used as a cell-surface marker of stem cells and cancer stem cells (CSCs) from a broad range of tissue types (1). Cells marked by AC133 have also been shown to have increased resistance to traditional cancer treatments, including chemo- and radio-therapies, and this may explain certain cases of cancer relapse (1). Despite its utility as a widely used marker to isolate, enrich or characterize CSCs, CD133 post-translational regulation remains poorly understood. Interestingly, CD133 protein is maintained during colon CSC differentiation, whereas expression of the cell-surface AC133 epitope specifically marks CSCs, leading to the hypothesis that CD133 post-translational modifications regulate cell-surface expression of the AC133 epitope (2). Indeed, recent evidence shows that CD133 trafficking to the plasma membrane and cell-surface expression of its AC133 epitope is directly regulated by N-glycosylation (3). Importantly, loss of CD133 N-glycosylation results in intracellular retention of CD133, without significantly impacting total CD133 levels (3).
Cell surface expression of CD133 is also tightly regulated through endocytosis (4). More specifically, CD133 directly interacts with the tubulin deacetylase HDAC6, which promotes expression of CD133 at the cell surface (4). This observation provides a model for retention and turnover of CD133 at the cell surface, but does not illuminate how CD133 traffics from the endoplasmic reticulum (ER) and Golgi apparatus where it is N-glycosylated, to the cell surface. To further understand the mechanism by which CD133 traffics to the cell surface, we explored the possibility that additional post-translational modifications on CD133 contribute to its trafficking.
Here, we report that CD133 is acetylated on specific lysine residues exposed to the ER lumen and that the Nε-lysine acetylation of CD133 is mediated by ATase1 and ATase2, two acetyl-CoA:lysine acetyltransferases that are found in the ER/ER Golgi intermediate compartment (ERGIC). Importantly, inhibiting intraluminal CD133 lysine acetylation destabilizes the protein and thereby influences expression at the cell surface. Our study implicates the acetylation of lysine residues facing the lumen of the ER as an important mechanism of CD133 post-translational regulation and trafficking.
Acetylation of multiple lysine residues in the first extracellular loop of CD133
To identify proteins that physically associate with the pentaspan membrane protein CD133 (also known as PROM1), we generated an HEK293 cell line that stably expresses a versatile affinity (VA)-tagged version of CD133 (3, 5) and found that the tubulin deacetylase HDAC6 associates with CD133 by immunoprecipitation followed by tandem mass spectrometry (IP-MS) analysis (4). It was further elucidated that HDAC6 deacetylase activity is required to stabilize CD133 at the cell surface and loss of HDAC6 catalytic activity rapidly promotes endocytosis of CD133 followed by lysosomal degradation (4). The interaction between CD133 and HDAC6 lead us to hypothesize that CD133 may be directly acetylated. As a result, examination of CD133 IP-MS data identified three acetylated lysine residues, K216, K248 and K255, on CD133 (Figure 1A). These three acetylated lysine residues are positioned on the first predicted extracellular loop of CD133 (Figure 1B). Since HDAC6 is primarily localized in the cytoplasm and we could not detect any CD133 acetylated peptides predicted to reside in intracellular regions of CD133, we suggest that CD133 may not be a direct substrate of HDAC6.
Figure 1. CD133 can be lysine acetylated.

a) MS analysis of CD133 Flag immunoprecipitated from HEK 293/CD133-VA lysates identified CD133 to be acetylated at K216, K248 and K255. MS analysis was performed as previously described (12) with the addition that post-translational modifications for acetylation were also detected. b) Mapping of acetylated lysine sites on the predicted topology of the CD133 protein at the plasma membrane. c) Endogenous immunoprecipitation of CD133 from Caco-2 cell lysate was analyzed by Western Blot for lysine acetylation of endogenous CD133 protein. Acetylated CD133 was detected using an anti-Ac-lysine antibody (7F8, Santa Cruz Biotechnology Inc.). d) Lysates from Flag-tagged wild type CD133-F and the CD133 K-to-Q Caco-2 stable cells were Flag immunoprecipitated and analyzed by Western Blot to determine CD133 lysine acetylation. IP of CD133 was performed as previously described (12) with exception of 0.5% Triton X-100 was used for IP of CD133 for MS analysis and RIPA buffer was used for IP of CD133 for in vitro acetylation experiments in an effort to disrupt any protein-protein interactions. CD133 K-to-Q mutant was generated as previously described (3) with the following primer setsCD133(K255Q) sense 5′-CAT GGC AAC AGC GAT CCA AGA GAC CAA AGA GGC G-3′ and antisense 5′-CGC CTC TTT GGT CTC TTG GAT CGC TGT TGC CAT G-3′; CD133(K248Q) sense 5′-CCT GTT CTT GAT GAG ATT CAA TCC ATG GCA ACA GCG ATC-3′ and antisense 5′-GAT CGC TGT TGC CAT GGA TTG AAT CTC ATC AAG AAC AGG-3′; CD133(K216Q) sense 5′-CAA CAC TAC CAA GGA CCA AGC GTT CAC AGA TCT GAA C-3′ and antisense 5′-GTT CAG ATC TGT GAA CGC TTG GTC CTT GGT AGT GTT G-3′
To confirm that the CD133 polypeptide is acetylated in vivo, we immunoprecipitated CD133 from Caco-2 cell (Figure 1C) and used an antibody that detects acetylated lysine residues. Western blot analyses using anti-acetyl lysine antibodies detected a band at ~120kDa corresponding to acetylated CD133 (Figure 1C).
To further confirm that CD133 is acetylated on lysines and investigate whether additional acetylated lysine residues exist on CD133 that were not identified by IP-MS, we performed site-directed mutagenesis on CD133 and substituted lysines 216, 248 and 255 to glutamines (hereafter referred to as CD133 K-to-Q mutant). Substituting glutamine for lysine removes a positively charge amino acid, in an attempt to mimic acetylated lysine. We introduced either wild type CD133 or the CD133 K-to-Q mutant into Caco-2 cells and monitored lysine acetylation status by Flag immunoprecipitation (IP) coupled with an anti-acetyl lysine immunoblotting. A single band cross-reacting with the anti-acetyl lysine antibody was present at ~120-kDa for wild-type CD133, but not for the CD133 K-to-Q mutant (Figure 1D), suggesting that K216, K248 and K255 are the primary CD133 lysines acetylated in Caco-2 cells. Importantly, no significant differences in cell surface and total protein expression levels between wild-type CD133 and the CD133 K-to-Q mutant were observed (data not shown).
Lysine acetylation regulates CD133 expression
To investigate the role of the acetylated lysine residues, we generated a CD133 K-to-R mutant by substituting lysine residues 216, 245 and 255 to arginines, which maintains a positive charge amino acid with the inability to be acetylated. In contrast to the CD133 K-to-Q mutant, the CD133 K-to-R mutant resulted in the near complete loss of CD133 protein expression from whole cell lysates from HEK293 and Caco-2 stable cell lines (Figure 2A). Importantly, the K-to-R mutation did not noticeably affect CD133 transcript levels (Figure 2B), so we suggest that differences in CD133 protein expression between the CD133 wild-type and CD133 K-to-R constructs are not likely from to differences in transcription efficiency or mRNA stability, but from differences post-transcriptionally, such as protein expression and regulation.
Figure 2. CD133 requires lysine acetylation for its stability.

a) Flag immunoblotting of lysates from Caco-2 cells and AC133 immunoblotting of lysates from HEK293 cells stably expressing either CD133-VA wild type or K-to-R mutant, or an empty vector. Immunoblotting of actin was used as loading control. CD133 K-to-R mutant was generated as previously described (3) with the following primer sets: CD133(K255R) sense 5′-GGC AAC AGC GAT CAG AGA GAC CAA AGA GGC G-3′, antisense 5′-CGC CTC TTT GGT CTC TCT GAT CGC TGT TGC C-3′; CD133(K248R) sense 5′-GTT CTT GAT GAG ATT CGG TCC ATG GCA ACA GCG-3′ and antisense 5′-CGC TGT TGC CAT GGA CCG AAT CTC ATC AAG AAC-3′; CD133(K216R) sense 5′-CAC TAC CAA GGA CCG GGC GTT CAC AGA TC-3′ and antisense 5′-GAT CTG TGA ACG CCC GGT CCT TGG TAG TG-3′;. b) Caco-2 stables were analyzed for versatile affinity tagged CD133 transcript levels by quantitative PCR with the following primers CD133-F: sense, 5′-GGA CGT GTA CGA TGA TGT TG-3′ and antisense, 5′-CAC CGT CAT GGT CTT TGT AG-3′. Primers do not recognize endogenous CD133. Transcript levels were normalized to actin and relative to CD133 VA wild type. Error bars represent the standard deviation of three independent replicates (n=3). p-value was calculated against the control using a two-tailed Student’s t-test. c) FACS analysis of HEK293 cells stably expressing either CD133-VA wild type or the K-to-R mutant, or an empty vector stained with AC133-APC. Cell staining for flow cytometry and analysis was performed as previously described (9). d) Flag (against CD133-VA) and CANX immunofluorescence of HEK293 cells stably expressing either CD133-VA wild type or the K-to-R mutant. Cells were stained with Hoechst 33342 to visualize nuclei. Immunofluorescence were performed as previously described (3). Scale bar, 25μm. e) Wild type and K-to-R mutant CD133 expressing HEK293 cells were treated with either vehicle only or with MG132 for 24 hours as previously described (4).
To pinpoint the stage at which lysine acetylation impinges on CD133 protein expression and trafficking, we conducted a series of experiments. To determine the localization pattern of the CD133 K-to-R mutant, we performed co-immunofluorescence for CD133 and for the ER chaperone calnexin (CANX). As previously demonstrated (3), wild type CD133 was found to be localized both at the ER compartment and plasma membrane (Figure 2C). CD133 K-to-R mutant expression was undetectable in the ER, as determined by the lack of co-localization with CANX, and only a relatively low level of CD133 K-to-R mutant protein was detected at the plasma membrane (Figure 2C). To quantify the cell surface expression of the AC133 epitope, a marker of primitive cells, we performed fluorescent activated cell sorting (FACS) analysis of wild type CD133 and the CD133 K-to-R mutant(Figure 2D). Similar to what was observed at the total protein level (Figure 2A) and localization pattern (Figure 2C), cell surface AC133 expression for the CD133 K-to-R mutant was drastically reduced compared to wild type CD133 (Figure 2D). It is noteworthy, that CD133 expression in extracellular particles or subcellular compartments other than the plasma membrane or the ER have not been investigated in this study.
Given that the CD133 K-to-R mutant is undetectable in the ER, but wild type CD133 is highly expressed at this early stage in trafficking, we suggest that the majority of CD133 K-to-R mutant protein is not stabilized in the ER during production and degraded prior to reaching the cell surface due to lack of acetylation on key residues resulting in improper protein folding and maturation. This is supported by the observation that the CD133 K-to-R mutant is, at least in part, degraded by a proteasome dependent pathway as treatment of the CD133 expressing HEK293 stable cell lines with the proteasome inhibitor MG132 could partially rescue CD133 K-to-R protein levels, but did not noticeably impact wild type CD133 levels (Figure 2E). This led us to hypothesize that CD133 is a substrate for an acetyltransferase, and possibly a deacetylase, that is localized in the ER lumen and/or Golgi lumen.
CD133 physically interacts with the ER residing acetyltransferases ATase1 and ATase2
A recent study has reported the identification of two ER/ERGIC residing Nε-lysine acetyltransferases, ATase1 and ATase2 (also known as NAT8B and NAT8, respectively), which can physically interact and acetylate β-site APP cleaving enzyme 1 (BACE1) in the ER lumen (6). The transient lysine acetylation of BACE1 regulates its ability to reach the Golgi apparatus and the cell surface (7). ATase1 and ATase2 share an 88% amino acid sequence similarity and are single membrane spanning proteins and both have the C-terminal catalytic domain exposed to the ER lumen (6). Given these findings, we hypothesized that ATase1 and ATase2 may acetylate the lysine residues located on the CD133 first extracellular loop.
Our efforts to demonstrate a physical interaction between CD133 and ATase1 or ATase2 by IP followed by Western blotting or MS analyses were unsuccessful (data not shown), which is not surprising if such an interaction were transient or sensitive to detergents in lysis buffers. Therefore, we turned to the membrane yeast two-hybrid (MYTH) assay to gain evidence for an interaction between CD133 and ATase1/ATase2(8). The MYTH assay relies on the reconstitution of two complementing ubiquitin fragments that result in the recognition and cleavage by an endogenous yeast protease. When cleaved, the LexA transcription factor (T) that is fused to the C-terminus of ubiquitin (Cub) translocates into the yeast cell nucleus for activation of the MYTH reporters (ADE2/HIS3/β-galactosidase) (8). CD133 and the control CD4 transmembrane protein were fused to the signal sequence of the α-mating pheromone precursor (MFα) at the N’terminus and Cub-T fragment at the C’terminus. MFα-CD133-Cub-T and MFα-CD4-Cub-T were properly integrated into the yeast membrane as assayed by N-terminal ubiquitin (Nub) I13G/NubI controls (4, 8) (Figure 3A). The bait proteins were then co-expressed with N- or C-terminally fused NubG-ATase1 or NubG-ATase2 and assayed on selective media. Consistent with previous reports that the N-terminal domains of ATase1 and ATase2, as well as the C-terminus of CD133, are exposed to the cytoplasm (6), we only observed the N-terminal NubG fused ATase1 and ATase2 to interact with CD133-C-T (Figure 3A). Importantly, the negative control MFα-CD4-C-T could not interact with either NubG-ATase1 or NubG-ATase2.
Figure 3. ATase1 and ATase2 physically interact with CD133 in the ERGIC compartment.

a) Membrane yeast two-hybrid (MYTH) was performed as previously described (4, 8). Generation of NubG fused ATase1 or ATase2 was performed by PCR amplification from pDONR223-NAT8 and pDONR223-NAT8B (Open Biosystems Inc.), respectively, with the following primers for N’terminal tagged NubG: sense 5′-GGT GGT CCA TAC CCA TAC GAT GTT CCA GAT TAC GCT GCT CCT TGT CAC ATC CGC AAA TAC-3′ and antisense 5′-GTA AGC GTG ACA TAA CTA ATT ACA TGA CTC GAG TCA CAG ACT CCC TAC C-3′ and C’terminal tagged NubG: sense 5′ CAA TAT TTC AAG CTA TAC CAA GCA TAC AAT CAA CTC AAT GGC TCC TTG TCA CAT CCG-3′ and antisense 5′-GGA GCG TAA TCT GGA ACA TCG TAT GGG TAC ATA TCC AGA CTC CCT ACC TTA GAA G-3′. PCR products were introduced into the pPR3-N or pPR3-C MYTH prey vectors by yeast homologous recombination as previously described (8). MFα-CD133-Cub-T or the control MFα-CD4-Cub-T were expressed in the yeast strain THY.AP4 along with NubG-ATase1, NubG-ATase2, ATase1-NubG or ATase2-NubG and assayed for growth on selective media. MFα-CD133-Cub-T and MFα-CD4-Cub-T were also expressed in THY.AP4 along with positive controls Ost1-NubI and Fur4-NubI and the negative controls Ost1-NubG and Fur4-NubG to demonstrate absence of self-activation and proper membrane integration. b) CD133 (pDONR223-CD133) (3), ATase1 (pDONR223-NAT8B) and ATase2 (pDONR223-NAT8) (Open Biosystems Inc.) were introduced into Gateway-compatible protein complementation assay (PCA) vectors. Vectors were transfected into HEK293 cells and immunofluorescence was performed as previously described (4), using the anti-ERGIC-53 antibody (C-6, Santa Cruz Biotechnology Inc.). CD133-VF1 and either VF2 only, VF2-ATase1 or VF2-ATase2 were co-transfected into HEK293 cells. 48 hours post-transfection, immunofluorescence was performed to determine co-localization between ERGIC-53, CD133-VF1 and either VF2 only, VF2-ATase1 or VF2-ATase2. Cells were stained with Hoechst 33342 to visualize nuclei. Scale bar, 10μm.
To further validate the interaction between CD133 and ATase1/ATase2, we performed a bi-fluorescence complementation assay, which relies on complementation of the Venus fluorescent protein fragments (VF1 and VF2) due to specific protein-protein interaction pairs. This allows interactions between proteins fused with complementary Venus fragments to be visualized and localized by fluorescent microscopy. Following co-transfection of CD133-VF1 with either VF2-ATase1 or VF2-ATase2 in HEK293 cells, we observed a strong fluorescent signal within the ERGIC compartment as determined by ERGIC-53 co-staining (Figure 3B). Importantly, we did not observe any fluorescence when the VF2 alone control was expressed with CD133-VF1, which supports the specificity of the interaction between CD133 and ATase1/ATase2.
ATase1 and ATase2 acetylate CD133
ATase1 and ATase2 were previously shown to acetylate BACE1 both in vitro and in vivo (6). To assess whether the Nε-lysine acetylation of CD133 can be mediated by ATase1 or ATase2, we performed an in vitro acetylation assay where purified wild-type CD133 or the CD133 K-to-Q were incubated with either purified ATase1 or ATase2 in the presence of radiolabeled acetyl-CoA, and quantified the amount of radiolabel remaining after washing. We found that either ATase1 or ATase2 could acetylate wild type CD133 in vitro, but not the control CD133 K-to-Q mutant (Figure 4A).
Figure 4. ATase1 and ATase2 regulate CD133 stability.

a) An in vitro acetylation assay was performed as previously described (6). Briefly, affinity-purified wild-type or K-to-Q mutant CD133 was incubated with [3H]acetyl-CoA (American Radiolabeled Chemicals) in the presence of the acetyl-CoA:lysine acetyltransferase, ATase1 or ATase2, purified using the ProFound kit (Pierce-Thermo Scientific). The reaction was performed in 200 μl of acetylation buffer (50 mM Tris-HCl (pH 8.0), 0.1 mM EDTA, 1 mM dithiothreitol, 10% glycerol, 20 μM acetyl-CoA) for 1 h at 30°C. The reaction was stopped by adding an equal volume of ice-cold acetylation buffer and immediate immersion in ice. Following the reaction, CD133 was immunoprecipitated using anti-FLAG magnetic beads (Sigma-Aldrich) and then counted on a liquid scintillation counter. As a control, CD133 was incubated with [3H]acetyl-CoA in the absence of the enzyme (ATase1 or ATase2). Error bars represent the standard deviation of three independent replicates (n=3). b) HEK293/CD133-F and Caco-2 cells were treated with two independent shRNAs targeting either ATase1 (shATase1-1: TRCN0000035450, ATase1-2:TRCN0000035451, Sigma-Aldrich Inc.) or ATase2 (shATase2-1: TRCN000035564 and shATase2-2: TRCN0000035568 Sigma-Aldrich Inc). Lentiviral production and infection was performed as previously described (3). Lysates were analyzed by immunoblotting for CD133 using the anti-AC133 antibody. ATase1 and ATase2 knockdown at the protein level was monitored by immunoblotting using the anti-NAT8 (Ap4967c, Abgent Inc.) and anti-NAT8B (ab97885, Abcam Inc.) antibodies. Immunoblotting of actin was used as loading control. c) ATase1, ATase2, and CD133 transcript levels were monitored by quantiative PCR in the presence of shATase1 and shATase2 knockdown using primers for ATase1 or ATase2 (6). Transcript levels were normalized to actin and are relative to the control shRNA. Error bars represent the standard deviation of four independent replicates (n=4). *p<0.05, #p<0.01. p-value was calculated against the control using a two-tailed Student’s t-test. d) FACS analysis of Caco-2 cells transduced with either a control shRNA, shATase1-1 or shATase2-1. Cell staining for flow cytometry and analysis was performed as previously described (9).
To study the effects of ATase1 and ATase2 on CD133 expression in vivo, we knocked down either ATase1 or ATase2 using two lentiviral-based short-hairpin RNAs (shRNAs) targeting independent sites of the ATase1 or ATase2 transcripts. Western blot and quantitative PCR analyses demonstrated that ATase1 and ATase2 shRNAs were able to efficiently knockdown their targets at the protein and mRNA levels, respectively (Figure 4B and 4C). Notably, knockdown of either ATase1 or ATase2 resulted in a downregulation in total CD133 protein levels (Figure 4B), but did not significantly alter CD133 transcript levels (Figure 4C). Consistent with the downregulation of total CD133, cell surface expression of the AC133 epitope was noticeably decreased suggesting that ATase1 or ATase2 knockdown most likely impacts plasma membrane localized CD133. Taken together, this suggests that CD133 protein expression is positively regulated by ATase1/ATase2 mediated lysine acetylation.
Targeting CD133 using an ATase1/ATase2 inhibitor
It was recently demonstrated that both ATase1 and ATase2 can be specifically targeted using a non-competitive small molecule inhibitor termed compound 19 (10). Consistent with shRNA knockdown of ATase1 or ATase2 (Figure 4B), treatment of Caco-2 cells with compound 19 resulted in downregulation of total CD133 levels (Figure 5A). To demonstrate that compound 19 may have potential in treating certain cancers, we employed the acute lymphoblastic leukemia (ALL) cell line SEM-K2, which we previously demonstrated that CD133 was required for its survival (9). We showed that CD133 loss and apoptosis in SEM-K2 cells can be triggered using tubacin, a small molecule inhibitor of HDAC6 deacetylase activity, but not niltubacin (Figure 5B) (4). To determine whether loss of CD133 mediated through ATase1 and ATase2 inhibition has a similar effect on SEM-K2 cells, we treated cells with compound 19 (10). Consistent with our findings that targeting ATase1/ATase2 results in CD133 downregulation (Figure 4B), downregulation of CD133 occured withcompound 19 treatment (Figure 5B). Importantly, inhibiting ATase1/ATase2 with compound 19 resulted in SEM-K2 apoptosis, suggesting that targeting CD133 lysine acetylation, and possibly other ATase targets, has a survival impact in the ALL cell line SEM-K2.
Figure 5. Targeting ATase1/ATase2 results in CD133 downregulation and apoptosis.
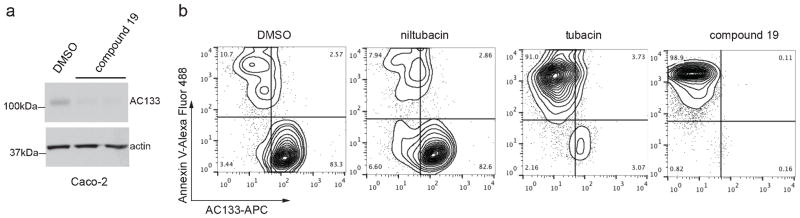
a) Caco-2 cells were treated with either DMSO or 10μM of compound 19 for 24 hours. Lysates were analyzed by immunoblotting for CD133 using the anti-AC133 antibody. b) SEM-K2 cells were treated for 24 hours with either DMSO or 10μM compound 19. Cells were co-stained with AC133-APC and Annexin-488 and analyzed by flow cytometry as previously described (9).
Conclusion
The results described above implicate extracellular acetylation of CD133 as an important regulatory mechanism for CD133 expression. We show that acetylation of three lysine residues in CD133 predicted to be in the first extracellular loop of the protein, which face the lumen of the ER and Golgi apparatus during translation of the nascent protein and trafficking to the cell surface, are important regulatory sites for proper expression of the protein. The enzymes responsible for the Nε-lysine acetylation of CD133 are ATase1 and ATase2, two ER/ERGIC-based acetyl-CoA:lysine acetyltransferases.
A similar mechanism of post-translational regulation by ATase1 and ATase2 was originally described for BACE1, a type I membrane protein that is involved with the pathogenesis of Alzheimer’s disease (6, 7). In contrast to BACE1(6), we did not observe an increase in CD133 levels when the cells were treated with ceramide (data not shown), a lipid second messenger known to increase both the influx of Acetyl-CoA into the ER lumen (7, 11) and the expression of ATase1 and ATase2 (6). This is consistent with our observations that a CD133 mutant mimicking constitutive lysine acetylation (CD133 K-to-Q) as well as the overexpression of ATase1/ATase2 did not increase CD133 levels (data not shown). Given this, it is conceivable that the regulation of CD133 by Nε-lysine acetylation differs from BACE1 in the sense that it displays saturation kinetics, with CD133 being the rate-limiting molecule in the enzymatic reaction.
We demonstrate that CD133 expression is positively regulated by ATase1/ATase2, and that perturbing CD133 acetylation may be a viable approach to indirectly targeting CD133 regulation and function. This is particularly attractive as we, and others, have demonstrated that CD133 has a functional role in suppressing cancer cell differentiation and, in some scenarios, a role in cancer cell survival (9). Future studies that examine CD133 expression due to acetylation could help shed light on the role of extracellular acetylation, as well as help define deacetylases that participate in this process.
Acknowledgments
We thank G. I. Chen for assistance with MS analyses, and Y. Fedyshyn and B. Fedyshyn for technical assistance. This work was supported by funds from the Stem Cell Network (to J.M.), the Canadian Institutes of Health Research (to J.M.), the Ontario Institute for Cancer Research (to J.M.) and the NIH/NIA (AG028569; to L.P.).
Abbreviations
- CSC
Cancer Stem Cells
- Cub
C’terminal ubiquitin
- ER
Endoplasmic Reticulum
- ERGIC
Endoplasmic Reticulum Golgi Intermediate Compartment
- IP-MS
immunoprecipitation coupled with tandem mass spectrometry
- MFα
α-mating pheromone precursor
- MYTH
Membrane Yeast Two-Hybrid
- Nub
N’terminal ubiquitin
- T
Transcription factor
- shRNA
short-hairpin RNA
- VF
Venus fluorescent protein fragment
Footnotes
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
- 1.Ferrandina G, Petrillo M, Bonanno G, Scambia G. Targeting CD133 antigen in cancer. Expert Opin Ther Targets. 2009;13:823–837. doi: 10.1517/14728220903005616. [DOI] [PubMed] [Google Scholar]
- 2.Kemper K, Sprick MR, de BM, Scopelliti A, Vermeulen L, Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, Medema JP. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719–729. doi: 10.1158/0008-5472.CAN-09-1820. [DOI] [PubMed] [Google Scholar]
- 3.Mak AB, Blakely KM, Williams RA, Penttila PA, Shukalyuk AI, Osman KT, Kasimer D, Ketela T, Moffat J. CD133 N-glycosylation processing contributes to cell-surface recognition of the primitive cell marker AC133. J Biol Chem. 2011;286:41046–41056. doi: 10.1074/jbc.M111.261545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, Gingras AC, Mazitschek R, Neel BG, Stagljar I, Moffat J. Regulation of CD133 by HDAC6 Promotes beta-Catenin Signaling to Suppress Cancer Cell Differentiation. Cell Rep. 2012;2:951–963. doi: 10.1016/j.celrep.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mak AB, Ni Z, Hewel JA, Chen GI, Zhong G, Karamboulas K, Blakely K, Smiley S, Marcon E, Roudeva D, Li J, Olsen JB, Wan C, Punna T, Isserlin R, Chetyrkin S, Gingras AC, Emili A, Greenblatt J, Moffat J. A lentiviral functional proteomics approach identifies chromatin remodeling complexes important for the induction of pluripotency. Mol Cell Proteomics. 2010;9:811–823. doi: 10.1074/mcp.M000002-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ko MH, Puglielli L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J Biol Chem. 2009;284:2482–2492. doi: 10.1074/jbc.M804901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 2007;407:383–395. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gisler SM, Kittanakom S, Fuster D, Wong V, Bertic M, Radanovic T, Hall RA, Murer H, Biber J, Markovich D, Moe OW, Stagljar I. Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol Cell Proteomics. 2008;7:1362–1377. doi: 10.1074/mcp.M800079-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mak AB, Nixon AM, Moffat J. The mixed lineage leukemia (MLL) fusion-associated gene AF4 promotes CD133 transcription. Cancer Res. 2012;72:1929–1934. doi: 10.1158/0008-5472.CAN-11-3589. [DOI] [PubMed] [Google Scholar]
- 10.Ding Y, Ko MH, Pehar M, Kotch F, Peters NR, Luo Y, Salamat SM, Puglielli L. Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces beta-secretase (BACE1) levels and Abeta generation. J Biol Chem. 2012;287:8424–8433. doi: 10.1074/jbc.M111.310136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonas MC, Pehar M, Puglielli L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J Cell Sci. 2010;123:3378–3388. doi: 10.1242/jcs.068841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen GI, Tisayakorn S, Jorgensen C, D’Ambrosio LM, Goudreault M, Gingras AC. PP4R4/KIAA1622 forms a novel stable cytosolic complex with phosphoprotein phosphatase 4. J Biol Chem. 2008;283:29273–29284. doi: 10.1074/jbc.M803443200. [DOI] [PMC free article] [PubMed] [Google Scholar]