

Abstract
Solid phase synthesis of HBS helices involving the Fukuyama–Mitsunobu reaction and triphosgene coupling is described.
α-Helices constitute the largest class of protein secondary structures, and play a major role in mediating protein–protein interactions. The organic chemistry community has focused significant attention on studying different approaches to either stabilize this conformation in peptides or mimic it with nonnatural scaffolds.1 We have developed a new strategy for constraining short peptides in stable α-helical conformations.2 This strategy, termed the hydrogen bond surrogate approach, involves replacement of one of the main chain hydrogen bonds with a carbon–carbon bond. In an α-helix, a hydrogen bond between the C=O of the ith amino acid residue and the NH of the i + 4th amino acid residue nucleates and stabilizes the helical structure (Fig. 1). To mimic the C=O–H–N hydrogen bond with a carbon–carbon bond, we utilized a ring closing metathesis (RCM) reaction between appropriately placed olefins on the peptide chain. A crucial feature of this helix design is that all side-chains are available for solvent exposed contacts with biomolecular targets. The crystal structure of a short HBS α-helix shows that the RCM-based macrocycle faithfully reproduces the conformation of a canonical α-helix.3 We have also demonstrated that hydrogen bond surrogate (HBS) α-helices can target chosen protein receptors in cell-free and cell-culture assays.4
Fig. 1.
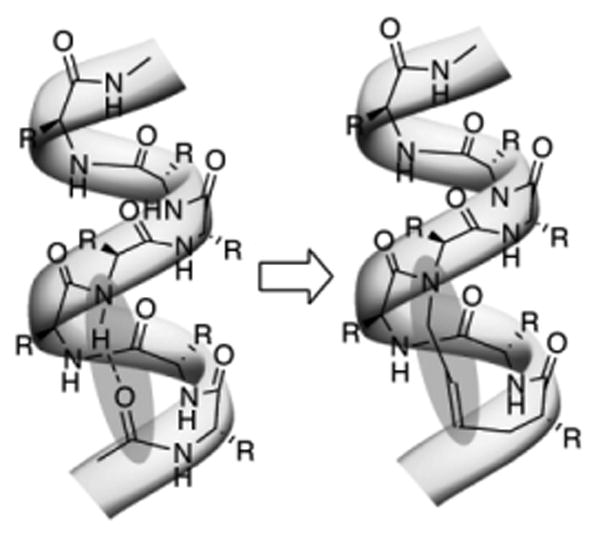
An HBS α-helix features a carbon–carbon bond in place of an N-terminal hydrogen bond. R = amino acid side chains.
The ring-closing metathesis reaction is the key step in our synthesis of HBS peptides, and we have reported optimized procedures for this step on solid-phase under oil-bath heating and microwave irradiation conditions.5 However, despite gaining access to high-yielding protocols for the difficult RCM reaction, we have been unable to prepare diverse HBS sequences. The synthesis strategy we typically utilize for the preparation of these artificial helices from bis-olefins is depicted in Scheme 1. We prepare bis-olefin 1 by standard Fmoc solid phase synthesis protocols, introduce the N-allyl functionality as a dimer, and cap the peptide chain with 4-pentenoic acid. The N-allyl dimer 4 is prepared in solution using the Fukuyama nitrobenzenesulfonamide chemistry,6 as described in a previous report.7 We prepare the N-allyl containing dimer in solution because of the inherent unreactivity of the N-allylamino acids toward activated carboxylic acids.8 Coupling to N-allyl amino acids is significantly more challenging than coupling to N-methyl amino acids. The solution phase synthesis allows us to isolate purified dimer for further elaboration of the peptide on solid phase. The analogous synthesis of the bis-olefin entirely on solid phase leads to inseparable mixtures. However, the requirement to prepare the dimer in solution limits our choice of amino acid residues to those that do not require protecting groups for the side chain functionality. Although, we have prepared such dimers as needed,4,5 the manipulation of orthogonal protecting groups for the amine, carboxylic acid and the side chain functionality restricts the sequence of dimers that can be synthesized.
Scheme 1.
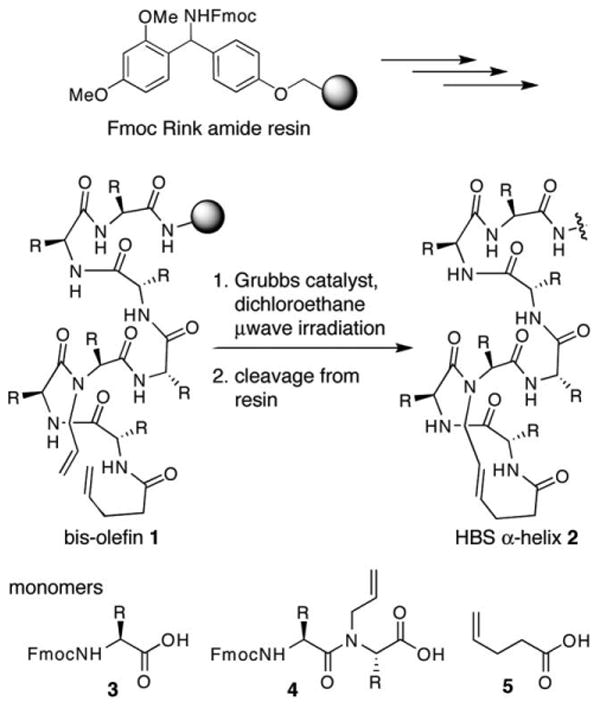
Reported synthesis of HBS α-helices.
N-Allylamino acids are significantly less nucleophilic than N-methyl amino acids for coupling with Fmoc amino acids. We found most standard coupling reagents to provide low yields of the desired products even with excess reagents, multiple coupling cycles, and microwave irradiation. After extensive investigations with common and exotic coupling reagents, we discovered that triphosgene (bis-(trichloromethyl)carbonate) delivers the coupled product in acceptable yields. The optimized solid-phase methodology is compatible with all standard side chain protecting groups and offers the option to drive the difficult coupling reaction to completion through the use of excess reagents.
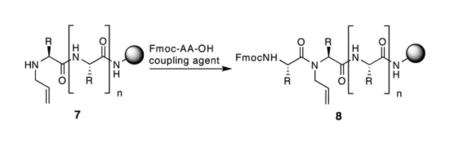
Scheme 2 illustrates the new method, which has proven to be highly effective for the preparation of a diverse set of HBS sequences on solid phase (Table 1). We utilized microwave assisted Fukuyama–Mitsunobu chemistry to prepare the N-allylamino peptide on standard Fmoc resins for the subsequent amide bond formation reaction with triphosgene. The optimized approach bypasses the need for preparation of the N-allyl dimer. The optimization studies for each step were initially performed with peptide 9, and the best conditions were then tested with the other sequences. We chose bisolefin 9 for the optimization studies because it has been a difficult sequence to access with our previous approach. Coupling of the t-butyl protected glutamic acid residue to an allylamine, in the solution phase synthesis of FmocGlu(OtBu)-N-allylAla-OH, is difficult because of the bulky side chain and the troublesome protecting group manipulations. The optimized conditions described below provide high yields of the bis-olefin peptides consisting of six to sixteen residues independent of the sequence.
Scheme 2.
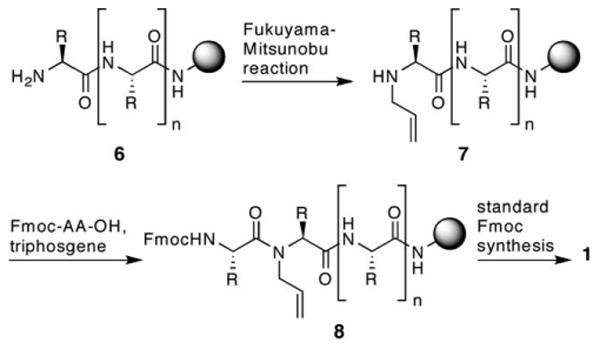
Optimized synthesis of bis-olefin 1.
Table 1. Bis-olefin peptides prepared by the optimized method.
| bisolefin | sequencea | % yieldb |
|---|---|---|
| 9 | XFEA*IYRLELLKAEEAN-NH2 | 84 |
| 10 | XFEG*IYRLELLKAEEAN-NH2 | 91 |
| 11 | XEFL*LRLWLKAibEEAibN-NH2 | 55 |
| 12 | XRKA*YKRLAMK-NH2 | 60 |
X denotes pentenoic acid residue, and A*, G* and L* refer to N-allyl A, G and L, respectively.
Refers to the amount of the desired peptide in crude mixture, after cleavage, as judged by HPLC (see Supplementary Information).
The Fukuyama–Mitsunobu reaction has emerged as the best method for N-alkylation of amino acids and peptides.9 We utilized this approach to prepare N-allylpeptide 7 on solid-phase under microwave irradiation. Table 2 shows the results of the Fukuyama and the Mitsunobu steps for allylation of the alanine residue needed for the synthesis of bis-olefin 9. Both reactions provide the desired products in high yields at room temperature and with microwave irradiation.10 To the best of our knowledge, this is the first report of a microwave-assisted Fukuyama–Mitsunobu reaction. Palladium catalyzed allylation of nitrobenzenesulfonamides also proceeds in high yields.8
Table 2. Optimized Fukuyama–Mitsunobu reaction conditions for N-allylation of amino acid residues on Rink amide Resina.
| |||
|---|---|---|---|
|
| |||
| entry | T/°C | time/h | % conversionb |
| o-nitrobenzenesulfonyl protection | |||
| 1 | 25 | 12 | quantitative |
| 2 | 100c | 0.25 | quantitative |
| Mitsunobu reaction | |||
| 3 | 25 | 6 | 70% |
| 4 | 25 | 12 | 90% |
| 5 | 100c | 0.16 | quantitative |
| o-nitrobenzenesulfonyl deprotection | |||
| 6 | 25 | 2 | quantitative |
| 7 | 50c | 0.08 | quantitative |
Optimization studies were performed with peptide 9 sequence; results of N-allylation of alanine are shown.
Determined from analytical HPLC areas.
With microwave irradiation.
The key reason limiting our access to diverse sets of HBS helices has been the inefficient coupling of an N-allylamine to the next Fmoc amino acid.8 Over the past several years, we have tested a variety of coupling agents for this reaction. Table 3 describes the effectiveness of different coupling reagents for the amide bond formation between of FmocGlu(tBu)-OH to an N(allyl)Ala peptide (for the synthesis of peptide 9) on Rink amide resin. Coupling of the t-butyl protected glutamic acid residue to an allylamine is difficult because of the bulky side chain. Traditional reagents such as HBTU, PyBOP and TBTU give only traces of the amide product.11 Coupling with pre-synthesized nosyl-amino acid chlorides8 yields a complex mixture with traces of desired product. In situ generated amino acid fluoride with DAST12 failed to provide any product. Reagents recommended for difficult couplings, such as, HATU/HOAt13 TFFH/HOAt12 and PyBrOP/DMAP14 provided low yields. DIC/HOAt afforded a 34% conversion. Significantly, bis-(trichloromethyl)carbonate,15 commonly known as triphosgene, provided greater than 90% conversion of the allylamine to the desired amide. No racemization was observed from the triphosgene mediated coupling as detected by HPLC, which agrees with the observation by Falb et al.15c Fig. 2 depicts crude HPLC traces for the amide bond formation between FmocGlu(tBu)-OH to N-allylalanine residue in 9, after cleavage of the peptide from resin. The effectiveness of all coupling reagents was tested at room temperature (data not shown) and with microwave heating. Table 3 shows optimized conditions for triphosgene mediated coupling reaction. Percent conversions reported in Table 3 are results after three different treatments (“triple coupling”) with the Fmoc amino acid and coupling agent. It is interesting to note that amino acid chlorides generated by SOCl2 are less effective than those generated with triphosgene. This result likely reflects the stability of acid chlorides. In the triphosgene method, the acid chloride is generated and coupled to the amine in situ; whereas in the thionyl chloride procedure, the acid chloride must be isolated prior to the coupling step.
Table 3. Effectiveness of different coupling reagentsa.
| ||||
|---|---|---|---|---|
|
| ||||
| entry | coupling reagentb, c | T/°C | time/h | % conversiond, e |
| 1 | triphosgene | 45 | 0.5 | 94% |
| 2 | DIC/HOAt | 45 | 0.5 | 34% |
| 3 | HATU/HOAt | 45 | 0.5 | 11% |
| 4 | TFFH/HOAt | 45 | 0.5 | 9% |
| 5 | PyBrOP/DMAP | 45 | 0.5 | trace |
| 6 | HBTU | 25 | 12 | trace |
| 7 | TBTU | 25 | 12 | trace |
| 8 | PyBOP | 25 | 12 | trace |
| 9 | amino acid chloridef | 25 | 2 | trace |
| 10 | DAST | 25 | 2 | trace |
Optimization studies were performed with peptide 9 sequence; results of amide bond formation between an N-allylalanine residue and FmocGlu(tBu)-OH on Rink amide resin are shown.
With microwave irradiation; see the experimental section for details.
DMF was used as a solvent with all coupling agents except for THF with triphosgene.
Determined from analytical HPLC.
Results after three different treatments with the Fmoc amino acid and coupling agent.
Pre-synthesized nosyl amino acid chloride.8
Fig. 2.
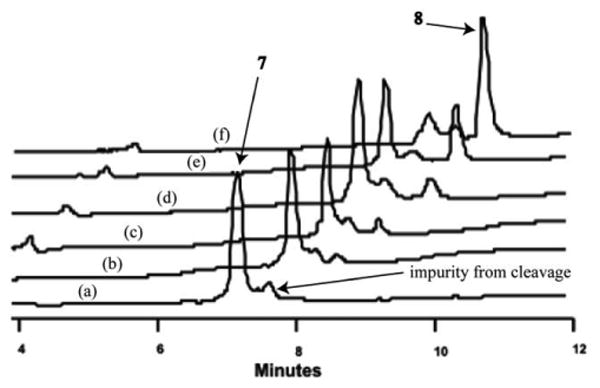
Representative crude HPLC traces for conversion of N-allylamine 7 to amide 8 with different coupling agents as described in Table 3. Peptide was deprotected and cleaved from Rink amide resin with TFA, and monitored at 220 nm. (a) N-allyl peptide, (b) PyBrOP/DMAP, (c) TFFH/HOAt, (d) HATU/HOAt, (e) DIC/HOAt, (f) triphosgene.
We find triphosgene to be a superior coupling reagent compared to every other combination tested for a variety of sequences (Table 1), except when the allyl bearing amino acid is a glycine. N-Allylglycines may be coupled to other Fmoc amino acids in high yields with the standard DIC/HOAt combination. N-Allylglycines containing sequences can be prepared either with the outlined Fukuyama–Mitsunobu method or through substitution of bromoacetamide with allylamine (Scheme 3). This strategy is analogous to that used in the synthesis of peptoids (N-alkylglycine oligomers),16 and begins with coupling of bromoacetic acid to a peptide chain followed by addition of allylamine to afford 15. Coupling of unhindered allylglycines proceeds in high yields with standard coupling agents. This simplified strategy is attractive for N-allylglycine containing HBS helices.
Scheme 3.
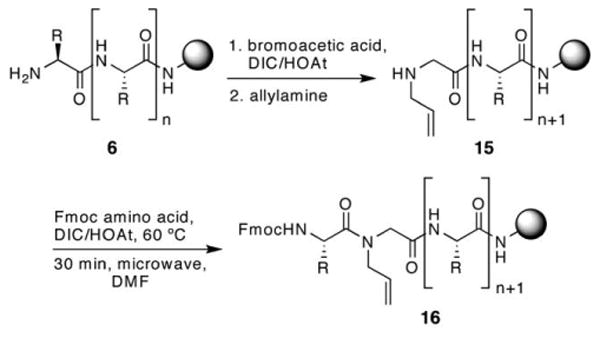
Synthesis of N-allylglycine containing peptides.
In conclusion, we have described a highly effective approach to solid phase synthesis of hydrogen bond surrogate helices. The modified approach utilizes triphosgene as the coupling agent for the amide bond forming reaction between unreactive N-allylamino acid residues and Fmoc amino acids. Triphosgene emerged as the most efficient coupling agent from a variety of traditional and contemporary reagents. The optimized solid phase microwave-based protocols utilize the Fukuyama–Mitsunobu synthesis of N-allylamines and provide an efficient method for synthesizing HBS sequences.
Experimental section
Representative optimized synthesis of HBS α-helices (Tables 2 and 3)
The desired free amine peptide 6 is synthesized on Rink amide resin using standard procedures. A solution of 2-nitrobenzenesulfonylchloride (3 eq) in DCM and 2,4,6-collidine (5 eq) are added to the pre-swelled resin, and the mixture is irradiated under microwaves (CEM discover) for 15 min at 100 °C. The resin is washed with DMF (¥3) and DCM (¥3), and dried under vacuum. Anhydrous THF and triphenylphosphine (5 eq) are added to the resin (containing 13) under nitrogen atmosphere, and the mixture is cooled in an ice bath. Diisopropyl azodicarboxylate (10 eq) and allyl alcohol (10 eq) are added and the mixture is subjected to microwave irradiation at 100 °C for 10 min. The resin is washed with DMF (¥3) and DCM (¥3), and dried under vacuum. DBU (5 eq) and 2-mercaptoethanol (10 eq) are added to pre-swelled resin (containing 14) in DMF under nitrogen atmosphere. The reaction mixture is then subjected to microwave irradiation at 50 °C for 5 min. The resin (containing 7) is washed with DMF (¥3) and DCM (¥3), dried under vacuum. In a separate flask Fmoc amino acid (10 eq), triphosgene (3.3 eq) and enough THF are mixed to maintain a triphosgene concentration of 0.15 M; 2,4,6-collidine (28 eq) is added, and the resulting solution is introduced to the resin (containing 7). The mixture is irradiated under microwaves at 45 °C for 30 min. The coupling step is repeated twice. The resin (containing 8) is washed with DCM (¥5), and the Fmoc group is removed with 20% piperidine in DMF. The desired Fmoc amino acid residue and 4-pentenoic acid are coupled using standard peptide synthesis methodology as described in the Supplementary Information. Ring-closing metathesis on bis-olefin is performed with Hoveyda–Grubbs II catalyst (20 mol%) in dichloroethane under microwave irradiation at 120 °C for 10 min as described.5
Representative synthesis of N-allylglycine peptides (Scheme 3)
A solution of bromoacetic acid (20 eq) with DIC (20 eq) and HOAt (10 eq) in DMF is added to pre-swelled resin, and shaken for 2 h. The resin is washed sequentially with DMF (¥3), DCM (¥3) and DMF (¥3). Resin is re-suspended in 1 M allylamine (20 eq) in DMF and shaken for 20 min. The resin (containing 15) is washed with DMF (¥3), methanol (¥3) and DCM (¥3), and treated with the desired Fmoc amino acid (20 eq), DIC (20 eq) and HOAt (10 eq) in DMF under microwave irradiation for 30 min at 60 °C. The resin is washed with DMF (¥3), DCM (¥3), DMF (¥3), and coupled with the desired Fmoc amino acid residue and 4-pentenoic acid using standard peptide synthesis methodology as described in the Supplementary Information. Ring-closing metathesis on bis-olefin 10 is performed with Hoveyda–Grubbs II catalyst (20 mol%) in dichloroethane under microwave irradiation at 120 °C for 10 min as described.5
Supplementary Material
Acknowledgments
We are grateful for financial support from the NIH (GM073943). We thank the NSF for equipment Grants MRI-0116222 and CHE-0234863, and the NCRR/NIH for Research Facilities Improvement Grant C06 RR-16572.
Footnotes
Electronic supplementary information (ESI) available: Synthesis and characterization of HBS-helices. See DOI: 10.1039/c000905a
References
- 1.(a) Henchey LK, Jochim AL, Arora PS. Curr Opin Chem Biol. 2008;12:692–697. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davis JM, Tsou LK, Hamilton AD. Chem Soc Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]; (c) Garner J, Harding MM. Org Biomol Chem. 2007;5:3577–3585. doi: 10.1039/b710425a. [DOI] [PubMed] [Google Scholar]
- 2.Patgiri A, Jochim AL, Arora PS. Acc Chem Res. 2008;41:1289–1300. doi: 10.1021/ar700264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J, Wang D, Zheng Q, Lu M, Arora PS. J Am Chem Soc. 2008;130:4334–4337. doi: 10.1021/ja077704u. [DOI] [PubMed] [Google Scholar]
- 4.(a) Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. J Am Chem Soc. 2010;132:941–943. doi: 10.1021/ja9082864. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang D, Lu M, Arora PS. Angew Chem, Int Ed. 2008;47:1879–1882. doi: 10.1002/anie.200704227. [DOI] [PubMed] [Google Scholar]; (c) Wang D, Liao W, Arora PS. Angew Chem, Int Ed. 2005;44:6525–6529. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Chapman RN, Arora PS. Org Lett. 2006;8:5825–5828. doi: 10.1021/ol062443z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dimartino G, Wang D, Chapman RN, Arora PS. Org Lett. 2005;7:2389–2392. doi: 10.1021/ol0506516. [DOI] [PubMed] [Google Scholar]
- 6.Kan T, Fukuyama T. Chem Commun. 2004:353–359. doi: 10.1039/b311203a. [DOI] [PubMed] [Google Scholar]
- 7.Chapman RN, Dimartino G, Arora PS. J Am Chem Soc. 2004;126:12252–12253. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]
- 8.Miller SC, Scanlan TS. J Am Chem Soc. 1998;120:2690–2691. [Google Scholar]
- 9.Fukuyama T, Jow CK, Cheung M. Tetrahedron Lett. 1995;36:6373–6374. [Google Scholar]
- 10.The microwave reactions were performed in the CEM Discover single-mode reactor with controlled power, temperature, and time settings.
- 11.Abbreviations used: DAST, (diethylamino)sulfur trifluoride; DIC, diisopropylcarbodiimide; HBTU, (2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate); HOAt, N-hydroxy-7-azabenzotriazole; PyBOP, Benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate; PyBrOP, bromotripyrrolidino-phosphonium hexafluorophosphate; TBTU, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate; TFFH, tetra-methylfluoroformamidinium hexafluorophosphate.
- 12.Carpino LA, Beyermann M, Wenschuh H, Bienert M. Acc Chem Res. 1996;29:268–274. [Google Scholar]
- 13.Carpino LA. J Am Chem Soc. 1993;115:4397–4398. [Google Scholar]
- 14.Frerot E, Coste J, Pantaloni A, Dufour MN, Jouin P. Tetrahedron. 1991;47:259–270. [Google Scholar]
- 15.(a) Su W, Gray SJ, Dondi R, Burley GA. Org Lett. 2009;11:3910–3913. doi: 10.1021/ol9015139. [DOI] [PubMed] [Google Scholar]; (b) Thern B, Rudolph J, Jung G. Angew Chem, Int Ed. 2002;41:2307–2309. doi: 10.1002/1521-3773(20020703)41:13<2307::AID-ANIE2307>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; (c) Falb E, Yechezkel T, Salitra Y, Gilon C. J Pept Res. 1999;53:507–517. doi: 10.1034/j.1399-3011.1999.00049.x. [DOI] [PubMed] [Google Scholar]
- 16.Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. Methods Enzymol. 1996;267:437–447. doi: 10.1016/s0076-6879(96)67027-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.