

Abstract
Diabetes mellitus is one of the most potent independent risk factors for the development of diabetic cerebral vascular disease (CVD). Many evidences suggested that hyperglycemia caused excess free fatty acids, the loss of endothelium-derived nitric oxide, insulin resistance, the prothrombotic state, endothelial dysfunction, the abnormal release of endothelial vasoactivators, vascular smooth muscle dysfunction, oxidative stress, and the downregulation of miRs participated in vessel generation and recovery as well as the balance of endotheliocytes. In turn, these abnormalities, mainly via phosphatidylinositol 3 kinase, mitogen-activated protein kinase, polyol, hexosamine, protein kinase C activation, and increased generation of advanced glycosylation end products pathway, play an important role in inducing diabetic CVD complication. A deeper comprehension of pathogenesis producing diabetic CVD could offer base for developing new therapeutic ways preventing diabetic CVD complications, therefore, in the paper we mainly reviewed present information about the possible pathogenesis of diabetic CVD complication.
Keywords: Complication, Diabetes mellitus, Cerebral vascular disease, Pathway, Pathogenesis
Core tip: A better understanding pathogenesis of diabetic cerebral vascular disease (CVD) could provide the basis for developing novel therapeutic strategies against diabetic CVD complication. Our article highlights the pathogenesis as some promising options to prevent CVD complications in diabetes, including metabolic and vascular changes and main pathways are involved in diabetic CVD complication.
INTRODUCTION
Diabetic mellitus (DM) is a chronic disease leading to a fault of insulin due to pancreas dysfunction, which causes hyperglycemia with metabolic imbalances in carbohydrate, fat and protein[1]. Morbidities of DM significantly elevate in late decades, which are primarily due to alter in life style, an elevation in the incidence of obesity and longevity. Current projections estimate that the number of people with DM will nearly double by 2025[2,3].
About 100 million populations suffer from DM in the world[4], Among them, five to ten percent are type 1 DM of insulin dependence and ninety to ninety-five percent are type 2 DM (T2MD) of non insulin dependence. The current evidences demonstrated that morbidities of T2MD would increase owing to life styles leading to obesity[5]. T2DM is a very common disease, which has an asymptomatic period between the actual onset of diabetic hyperglycemia and clinical diagnosis. This stage has been evaluated to sustain at the fewest 4-7 years, and 30%-50% patients of T2DM are still unknown. This leads to the development of chronic complications of diabetes, which remain the chief problems in diabetic care, and which cause a lack of fitness to work, disability, and premature death[6,7].
Among the chronic complications of diabetes, the vasculopathy is the first serious complication. The vasculopathy related to DM was traditionally divided into two major parts. Firstly, diabetic complications associated micrangium including retina, nephridium and neural system lesion; secondly, the atherothrombotic complications related to macro-arteries like myocardial infarction, hypertension, peripheral artery lesion[8].
DM is one of the well known risk factor for cere-brovascular accident[9,10]. Prolonged untreated DM contributes to micrangium lesion, hypoxic and ischemic damages of tissues, which elevates the danger of apoplexy and aggravates cerebral lesion caused by blood insufficiency[10,11]. Its incidence in DM patients is 2-6 times more than non DM[12-14] and ultimately its complications and subsequent prevalence is higher yet. Patients demo-nstrate a progressive atherosclerosis in cerebral arteries and increased a vascular reaction to vascular constrictors, a deregulated reaction to vascular dilators and damaged automatic regulation of brain blood stream. Changed endothelium function of small arteries and a damaged vascular motor function of resistance vessel can lead to change mediation of local blood stream and deficient perfusion of tissue in diabetic patients[10,15,16].
Many studies[8,17-19] stress the strong link between the cerebral vascular disease (CVD) complications and DM and describe a close association between CVD microvascular complications and DM, suggest that excess free fatty acids (FFAs), the loss of endothelium-derived nitric oxide (NO), insulin resistance, the prothrombotic state, endothelial dysfunction, the abnormal release of endothelial vasoactivators, vascular smooth muscle (VSM) dysfunction, oxidative stress and the miRs downregulation participated in vessel generation, vessel recovery as well as endothelium balance generate diabetic CVD complications by these major mechanisms of phosphatidylinositol 3 kinase, mitogen-activated protein kinase, polyol, hexosamine, generation of advanced glycosylation end products (AGEs) and protein kinase C (PKC) pathways activation[20,21]. The aim of this review is to review the possible pathogenesis of diabetic CVD complication.
SUPERFLUOUS FREE FATTY ACIDS
DM facilitates lipolysis, reduces uptaking of skeletal muscle, which result in superfluous concentrations of FFAs. Moreover, elevates the flux of FFAs into liver causes the stimulation of triglycerides synthesis, assembly and secretion of very low-density lipoprotein (VLDL) particles. Hypertriglyceridemia and the decreased high-density lipoprotein (HDL) cholesterin as the trans-portation of cholesterin from HDL to VLDL have been determined to be strongly relative to atherosclerosis. It also is likely that FFAs generation promotes reactive oxygen species (ROS) and PKC. PKC elevation and phosphatidylinositol 3 kinase (PI3-K) downregulation may cause endothelial impairment.
Recycling concentrations of FFAs rise in DM because of the superfluous releasion derived from adipose tissue as well as the reducing uptake of skeletal muscle[22-24]. FFAs can damage endothelial function via a series of mechanisms such as elevating oxygen-mediated free radicals generation, PKC activation and dyslipidemia aggravation[25-27]. FFAs levels increase activates PKC, reduces insulin receptor substrate-1 associated PI3-K activity[25,28]. Increased triglyceride levels decreases HDL through facilitating cholesterin transportation from HDL to VLDL[29]. These disturbances alter LDL configuration, elevating the quantity of the more LDL of small density contributing to atherosclerosis[30,31]. Hypertriglyceridemia and decreased HDL are suggested to be relative to endothelial dysfunction[21,32-34].
THE LOSS OF ENDOTHELIUM-DERIVED NITRIC OXIDE
Endothelial and vascular smooth muscle cells (VSMCs) dysfunction and an inclination of thrombus formation result in atherogenesis as well as the relative complications. Because endothelial cells (ECs) mediate the vascular function and structure, they take on an important anato-mical location on interaction of circulatory blood and vascular wall. In normal conditions, ECs active substances, synthesize and release vascular activators to preserve vessel balance, to ensure a normal blood stream and nutritious transportation while avoiding thrombus formation as well as white blood cell permeation[35]. One of key molecules produced by ECs is NO, it is generated by an endothelial NO synthase (eNOS) via a 5 electrons oxidation of the guanidine nitrogen terminal of L-arginine[36].
The NO biologic availability is a vital element in vessel abnormality, which results in vascular dilation activated guanylyl cyclase in VSMCs[36]. Furthermore, NO prevents vascellum from internal lesion like atherogenesis-mediated molecule signal that stops platelet and leukocyte interacting with vessel wall and inhibits VSMCs proliferation and migration[37,38]. Contrarily, ECs reduction-mediated NO induces elevated pro-inflammatory transcription factor nuclear factor kappa B (NFκB) activity which causes a leukocyte adhesion molecules expression, chemokines as well as cytokines generation[39]. The effects facilitate mono cells and VSMCs to migrate into the internal membrane and macrophage foam cells formation, producing an early morphologic alteration of atherogenesis[39-43]. Disorder of endothelium function such as damaged endothelium dependent and NO-derived relaxation is identified in cell and animal studies of DM[21,44-47].
INSULIN RESISTANCE
Insulin resistance is another vital pathogenesis that exerts a major effect on the diabetic CVD complication. Insulin exerts effects by two pathways including PI3-K and mitogen-activated protein kinase (MAPK). Insulin signal producing by PI3-K has effects of anti-proliferative and anti-coagulant, the effects activated by MAPK have a proatherogenic function. On base of insulin resistance, although the first pathway is damaged, the second pathway maintains intact. Therefore, the decrease endothelial dependent vasodilatation as well as increase mitosis effects is a key result[48,49].
Insulin resistance also critically takes part in vascular dysfunction in patients with T2DM[50]. In fact, the reduction of PI3-K/Akt pathway causes eNOS depression, decreases NO generation[51]. Combining with decreasing NO synthesis, intracellular oxidization of stored FFAs produces ROS contributing to vascular inflammation, AGEs synthesis, inhibited PGI2 synthase activity, and PKC activation[51,52].
Rised ROS concentrations closely related with insulin resistance remove NO generation, generate peroxynitrite accompanying with a more decrease of NO biologic availability. Decreased cell concentrations of NO activate pro-inflammatory pathways promoted by increasing cytokine generation. In fact, TNF-α and IL-1 facilitate NFκB activity and adhesion molecules expression. TNF-α also induces C reactive protein expression which lowers the regulation of eNOS and elevates adhesion molecules and endothelin-1 (ET-1) generation[50,53].
Adipokines associated with vasculopathy are leptin, adipocyte fatty acid-binding protein, interleukins, lipocalin-2 and pigment epithelium- derived factor, which could produce disorders of vessel function through increasing proliferation and migration of smooth muscle cells (SMCs), eNOS depression, and NFκB signaling activation accompanied with adhesion molecule expression and atherogenesis[54].
PROTHROMBOTIC STATE
Damaged fibrinolysis, as a result of enhancing generation of PAI-1 and excessive activity of platelet result from of glycoprotein IIb/IIIa superfluous expression and excessive production of thromboxane A2 in DM. Furthermore, DM rises concentrations of VII, VIII factor as well as thrombin-antithrombin compounds[48,55-57]. Haemostasis chaos elevates the risk of cerebral thrombosis.
Abnormality of platelet function as well as elevation of both glycoprotein Ib and IIb/IIIa expression in DM augment interaction between platelet-von Willebrand factor (vWF) and platelet-fibrin[58]. The internal-cellular platelet glucose level responses the external-cellular circumstance, is relative to increasing superoxide anion (O2-) generation as well as PKC activity, reduced platelet mediated NO[59]. High blood glucose more alters hematoblastic functions through damaging calcium balance, thus changes platelet activation and aggregation such as platelet construct and mediators release[60]. In DM, plasm coagulate factors VII, thrombin and impairment-dependent coagulate elements such as tissue factor (TF) elevated, and endogenic anti-coagulate factors like thrombomodulin as well as protein C reduced[61-63]. In addition, the generation of plasminogen activator inhibitor-1 (PAI-1), a fibrinolysis inhibitor elevated[64-66]. Therefore, a inclination of hematoblastic activating and aggregating accompanied with coagulate propensity is associated with a danger of thrombus formation complicated plaque burst.
Diabetic CVD largely results from an abnormality of elements participated in activation of coagulate factors and hematoblast[67]. Insulin resistance, high blood glucose involve in the nosogenesis of the prothrombotic status[68]. Insulin resistance rises PAI-1 and fibrinogen, decreases levels of tissue plasminogen activator. Hyperinsulinemia elevates TF expression in monocytes of T2DM contri-buting to increase TF procoagulant activity and thrombin production[69]. Low level of inflammation also leads TF expression in vessel endothelial cells of DM patients, and results in atherothrombosis[68,69].
Platelet hyperreactivity is one of major relations in elements leading to DM prothrombotic status[70]. A lot of mechanisms leaded to platelet dysfunction affect the adhesive, activated and aggregative stages of hema-toblast induced thrombus formation. High blood glucose changes hematoblast calcium ion homeostasis contributing to a abnormality of cellular constructure, elevates the production of pro-aggregant elements[58]. Furthermore, the increase production of glycoproteins Ib and IIb/IIIa in DM subjects contributes to thrombosis through interacting with vWF and fibrin.
In DM, the elevation of glucose levels contributes to the activation of PKC, down-regulates the production of platelet derived NO, rises the formation of O2-[59], and also triggers the disorder of calcium homeostasis in platelets[60]. The abnormal calcium regulation may significantly lead to disordered activity, because the intraplatelet calcium mediates a shape change, secretion, aggregation and thromboxane formation of platelet. Their disorders may cause by decreasing endothelial generation of the antiaggregants NO and prostacyclin, increasing generation of fibrinogen, and increasing generation of platelet activators like thrombin and vWF[58]. In general, diabetic disorders elevate the activation of platelet and depress internal depressors of platelet activity.
DM increases a blood coagulability which makes it more likely that atherosclerotic plaque rupture or erosion will lead to the thrombotic occlusion of artery. T2DM has damaged fibrinolytic capacity owing to increasing levels of PAI-1 in atherogenetic damage and nonateromatous artery[71]. DM elevates the TF (a forceful procoagulant) expression and plasm coagulate elements like factor VII, reduces contents of internal anti-coagulate factors like antithrombin III and protein C[62,63,72]. A number of these disorders are associated with the occurrence of hyperglycemia and proinsulin split products[73]. Therefore, DM increases a tendency of coagulation accompanying with damaging fibrinolysis, facilitates the formation and persistence of thrombi.
ENDOTHELIAL DYSFUNCTION
The endothelium is an organ composing of a mono cell layer arraying the intimal surface of the vasculature, serving as a paclose between blood and tissues. The normal paracrine and autocrine functions of endothelial cells include the synthesis of a series of substances that moderate vascular relaxation, mediate local inflammation, depress leucocyte migration and affect platelet activation.
Endothelial dysfunction consists of many abnor-malities, encompassing changed vasomotor activity, VSMC dysfunction, excess generation of inflammatory cytokines and chemokines, damaged platelet function and abnormal coagulation, which contribute to elevating vasoconstriction, inflammation and thrombosis[74].
The endothelium in DM is more frail in producing atherosclerotic plaques compared with the endothelium of non DM. A series of mechanisms may lead to the elevated risk of generating atherosclerotic plaques in DM. The abnormal cluster of hyperglycemia, increased FFAs and insulin resistance in DM, targeting the endothelial cell, causing oxidative stress and endothelial dysfunction[75]. The endothelial dysfunction leads to a defective endothelium dependent vasorelaxation and vasoconstriction, a migration of monocytes, a VSMCs transport into internal membrane as well as a generation of macrophage foam cells, which trigger an atherogenesis production. As far as ET-1, besides a vascular constrictive function, which has the function of proinflammation, mitogenesis and proliferation yet[48,76].
Hyperglycemia, elevated FFAs as well as insulin resistance exert through a usual pathogenesis characterized by rising ROS generation (Especially O2-), subsequently result in damaging endothelial dependent NO induced relaxation. Elevated ET-1 generation and VSMCs proliferation also lead to endothelium dysfunction. Hy-pertriglyceridemia, consequent atherogenesis and platelet hyperactivity as well as diminished fibrinolysis and hypercoagulability are features of vessel circumstances in DM. In general, an initial and progressive atherogenesis, endothelium dysfunction as well as elevated thrombus production are extremely susceptible to generating thrombotic occlusive events at brain circle for this type of DM[20].
Mechanisms of the endothelial dysfunction encompass elevating polyol pathway flux, changed cell redox status, increasing generation of diacylglycerol, specific PKC isoforms activation, and exacerbated non-enzymatic production of AGEs. A lot of pathways promote the generation of oxidative and nitrosative mediated oxidants and free radicals like O2- and peroxynitrite, exerting a important effect in the mechanism of the DM-relative endothelial dysfunction. The cellular sources of ROS like O2- are diverse and encompass AGEs, NADH, NADPH oxidases, mitochondrial respiratory chain, xanthine oxidase, arachidonic acid cascade, and microsomal enzymes[77-82]. The oxygen and nitrogen stress mediated by hyperglycemia results in DNA-lesion as well as succedent poly (ADP-ribose) polymerase (PARP) activation[83]. The endothelial dysfunction progression was related to a concurrent NAD+ and NADPH loss in vascular systems, PARP depression inversed their alterations. Endothelium dysfunction in DM is relied on a PARP-derived, inverse cell NADPH insufficient[83,84].
A mono layer of ECs is seated in the internal mem-brane of all vascellum, which offers a metabolically active interactive spot between blood and vessels regulating blood influx, nutritious transport, coagulation and thrombosis, and leukocyte diapedesis[85]. ECs synthetize a lot of key bioactive substances, such as ROS, prostaglandin, endothelin as well as angiotensin II, which modulates vascular effect and structure. Moreover, it diminishes platelet activation, inhibits inflammation by decreasing leukocyte adhesion to endothelium and migrating into the vascular wall, and reduces VSMCs proliferation and migration[38,86,87]. In general, these characters prevent atherogenesis and protect the vascular vessel.
DM damages endothelium dependent vasodilation prior to the generation of atheroma[88,89]. Many of fundamental mechanisms lead to the lower bioavailability of vasoactivators in DM. Hyperglycemia diminishes generation of NO by inhibiting the activation of eNOS synthase and elevating the generation of ROS, particularly O2-, in endothelial and VSMCs[78].
Insulin resistance contributes to excessive release of FFAs from adipose tissue[90], activating the signal enzyme PKC, depressing PI3-K, and increasing the generation of ROS-mechanisms[27]. Generation of peroxynitrite decreases synthetizing the vessel dilatory and antiplatelet prostanoid prostacyclin[91]. The increased levels of FFAs in DM trigger the production of oxidized low-density lipoproteins (Ox-LDL), including vital initiating events for atherosclerosis. Ox-LDL can impair ECs and increase adhesion molecules expression like P-selectin[92] and chemotactic factors like monocyte chemoattractant protein-1, macrophage colony stimulating factor[93,94] and thus lead to endothelial dysfunction in DM[78,95].
ABNORMAL RELEASE OF ENDOTHELIAL VASOACTIVATORS
Hyperglycemia rises the cyclooxygenase-2 mRNA expression in DM. Endothelin could be especially associated with the pathophysiology of vasculopathy in DM, because endothelin triggers inflammatory reaction, results in VSMCs contraction and growth[95]. The abnormalities of endothelium associated factors or vasoactivators generation consisting of vascular oxidative stress[96], inflammatory factors, NO, prostanoids (prostacyclin), ET-1, angiotensin II (ANG-II), tissue-type plasminogen activator (t-PA), PAI-1, vWF, adhesion molecules such as vascular cell adhesion molecule (VCAM) leukocyte adhesion molecules, intercellular adhesion molecule (ICAM) as well as cytokines[97]. These vascular activated factors contribute to elevating vascular tone, resulting in microvascular and macrovascular impairment and apoptosis of microvascular cells, consequently contributing to DM associated vascular complications[97]. In lots of pathological conditions, the abnormal balance of these regulatory mediators causes the onset and process of vascular endothelial dysfunction[98]. Vascular endothelial dysfunction elevates effects of leukocyte, smooth muscle proliferation, vascular constriction, damaged coagulating, vessel inflammation, thrombus generation, and athero-genesis, these mechanisms are the base of later DM complications like retinopathy, nephropathy, vasculopathy as well as neuropathy[99]. ET-1 is a forceful vasoconstrictor generated by ECs. The generation and the level of ET-1 in plasma elevated in DM patients, and it is reported a positive correlation between plasm ET-1 concentrations and the micro-vessel lesion of DM. Therefore, ET-1 could exert a possible key effect on endothelial dysfunction by a disorder between ECs mediated vascular dilator and vascular constrictor factors on mechanisms of the vessel complication in DM[100]. Besides its direct vasoconstrictor functions, elevated levels of ET-1 may lead to endothelial dysfunction via generating a series of vascular active substances consisting of ROS, NO and inflammatory factors[101-103].
CGRP is a key mediator of ET-1 vasoconstriction. The elevation of CGRP expression leads an abnormal balance in the CGRP/ET-1 ratio, inducing abnormal vascular constriction to result in topical endothelial dysfunction as well as vessel impairment[104,105]. VCAM-1 is one of key ECs receptors, mediating leukocyte adhesion to the vascular ECs. Current studied results have highly proposed that VCAM-1 might exert a key effect on mechanisms of atherosclerosis on account of VCAM-1 effects on leukocyte adhesion and transmigration is key as well as its expression is upregulated in the initial phases of neogenetic atheroma plaques[106]. Moreover, VCAM-1expression is upregulated by pro-inflammatory stimuli like TNF-α and IL-1β, is least partially mediated by NFκB[107]. The expression of VCAM-1 is upregulated in vessel stress circumstances like insulin resistance and chronic high blood glucose yet. In the circumstance, the elevation in the activity of VCAM-1 expression is found yet[108,109]. Besides binding leukocytes, VCAM-1 engagement leads to leukocyte transendothelial migration (TEM) via inducing gap formation between cells in the endothelial monolayer, facilitating TEM[110]. The gap formation is moderated by VCAM-1[111,112]. VCAM-1 accumulation elevates the internal cellular free calcium level yet[111]. Therefore, the expression of VCAM-1 rises the permeability of vascular endothelium as well as the TEM of leukocyte, leads to the impairment of vessel and abnormal endothelial function, as well as mediates atherogenesis production. The ICAM-1 expression in ECs is risen in atherogenesis and in the animal model of atherogenesis[113,114]. In normal conditions, ICAM-1 is presented in low concentrations in ECs, however ICAM-1 is significantly elevated while stimulating by pro-inflammatory factors such as the pro-inflammatory cytokines TNF-α, IL-1β as well as interferon-γ[114]. Soluble ICAM-1 may mediate leukocyte adhesion, migration. Promoting leukocyte to attach to the ECs surface isn’t the only effect of ICAM-1. Effect of ICAM-1 mediates signal in ECs, rises the IL-8 generation, facilitates the ICAM-1 and c-fos expression. It is likely that pro-inflammatory IL-8 and c-fos activated by ICAM-1 facilitates a positive feedback loop, contributing to excessive ICAM-1 and VCAM-1 expression and thus initiating the persistent recruitment of leukocytes to regions of atherosclerosis, facilitating the progress of atherogenesis through producing pro-inflammatory stimuli through indirect or direct endothelial dysfunction[115-117].
P-selectin is mediated by pro-inflammatory stimuli stored in internal cellular vesicles of ECs combined with the plasm membrane being responsible to many stimuli like ischemia and chronic high blood glucose yet[118]. On some conditions, P-selectin translocation to the endothelial cell surface is modulated by a ROS-dependent mechanism. P-selectin accumulation rises cytosol free Ca2+, mediates changes of cell morphology, facilitating endothelial dysfunction to ultimately generate vessel impairment[119-121].
VCAM-1, ICAM-1, P-selectin exert a key effect on vascular integrity and permeability through endothelial dysfunction[122]. Increased concentrations of soluble EAM could be one of the common causes for the pathogenesis of between atherogenetic CVD and endothelial dys-function[122]. The adhesive molecule expression in ECs facilitates the adhesion and transportation of leukocytes into the sub-endothelial space, consequently contributing to abnormal endothelial function and sub-endothelial structure alteration[123]. Elevated vessel permeability owing to structure changes can then decrease insulin transportation to insulin sensitive peripheral tissues, ultimately form insulin resistance. In addition, insulin resistance may directly contribute to endothelial dysfunction[124]. The investigation in non-diabetic subjects have proposed that lightly damaged glucose tolerance in the normal glycemic range may facilitate the process of endothelial dysfunction through side effects of oxidative stress, generation of AGEs, and increased concentrations of FFAs[125].
Vascular endothelial dysfunction may be prior to the development of insulin resistance, which results from a decrease of insulin sensitivity, generates a vicious cycle[126-128]. Our studied results provided the further evidences that endothelial dysfunction exerts a causal role in the pathogenesis of CVD in T2DM, and also highlights new insights into the possible clinical value of endothelial function in CVD of T2DM. The pathophysiologic mechanisms of CVD in T2DM could be relative to an abnormal expressive balance of ET-1, CGRP, VCAM-1, ICAM-1 and P-selectin, causing endothelial dysfunction via a series of chemical factors like ROS, NO and inflammatory factors. Alternatively, we speculated that emotion, cerebral splanchno-motor and neuroendocrine center could participate in the mechanisms of CVD in T2MD through changes of ET-1, CGRP, VCAM-1, ICAM-1 and P-selectin expression, but further researches need to be warranted[129].
VASCULAR SMOOTH MUSCLE DYSFUNCTION
The changes in vascular homeostasis owing to the dysfunction of ECs and SMCs are the primary characters of diabetic vascular diseases, which are favor of a pro-inflammatory or thrombotic status, finally contributing to artery thrombus formation. The diabetic affect on vascular function isn’t confined to ECs. The abnormal regulation of VSM function is accelerated through damaging the sympathetic nervous system function[130]. DM rises PKC activity, NFκB and oxygen free radicals generation in VSM, which is similar to the effects in ECs[131]. Furthermore, DM elevates the migration of VSMCs into early atherosclerotic impairment, replicating and generating external cellular matrix key process in later impairment production[132]. VSMCs apoptosis during atherogenetic impairment is rised yet, so that DM patients are apt to have fewer VSMCs in impairment, increasing the tendency of plaque rupture. In DM, the cytokine generation reduces VSM synthesis of collagen and elevates generation of matrix metalloproteinases, producing an elevated propensity for plaque destabilization and rupture[133].
DM promotes the VSMCs atherogenic activity. Hyperglycemia stimulates PKC, receptor for AGEs and NFκB in VSMCs, which is similar in ECs. Promotion of these systems increases generation of O2-, which leading to the oxidant gathering circumstances[27]. VSMCs are indispensable in the progression of atherosclerosis. In case the formation of the macrophage abundant fatty streak, VSMCs in the middle layer of the arteries migrate into the early intimal impairment, replicate and generate a complicate extracellular matrix vital process in the development formed atherosclerotic plaque. VSMCs heighten the atheroma by way of the collagen source, which makes it less possibly to rupture and results in thrombosis. In fact, the impairments that have disrupted and resulted in fatal thrombosis are inclined to have few VSMCs[134]. Hyperglycemic lipid modifications of LDL may partially modulate the risen migration and the following apoptosis of VSMCs in atherosclerosis impairment. LDL that has suffered nonenzymatic glycation promotes VSMCs migration, while oxidized glycated LDL can promote apoptosis of VSMCs[135]. Therefore, DM changes VSMCs function through facilitating atherosclerotic lesion formation, plaque instability[68].
OXIDATIVE STRESS
The generation of ROS is severely controlled in normal cells, but excessive generation at the condition of metabolized disorder contributes to cell lesions. O2-. and NO are correspondingly nonvalent, however, while the both are combined they produce highly active peroxynitrites that impairs and diminishes protein and lipid. Moreover, O2-. and NO can impair the iron sulfur center of enzyme and other protein, releasing iron atom and thus depressing enzyme and protein activity. There are a number of key proteins that are highly sensitive to the type of inhibition such as complexes I-III in the electron transfer chain, aconitase in the trichloroacetic acid cycle as well as biotin synthase[136,137].
The production of lipid, protein and nucleic acid com-pounds participates in lots of complicated chain reactions in ways of a series of biological substrates containing reactive methylene groups. Intermediate productions in these chain reactions can have very strongly oxidative effects, thus cell lesion can be comprehensive[138,139]. Lipids locate in plasm, mitochondria and endoplasmic reticulum membranes are main attacked objects of ROS and peroxidation. Terminal productions of lipid peroxidation like lipid peroxides can be toxic to cells, require to be resolved by glutathione. In the same way, proteins and nucleic acids can be suffered by peroxidation and nitrosylation. The terminal products aren’t commonly directly toxic to cells, the gather of inactive proteins can excessively increase the ability of cells to recycle them, DNA impairment is known to promote the pathogenesis of apoptosis.
Diabetic CVD complications are majorly owing to a prolonged exposure of hyperglycemia[140]. The early trigger high glucose levels change vessel function is the disorder between NO bio-availability and gather of ROS, contributing to endothelial dysfunction. In fact, high blood glucose mediated production of O2- inactivates NO to generate peroxynitrite (ONOO-), a forceful oxidant, it easily pass through phospholipid membranes, causes substrate nitration[21]. Hyperglycemia mediated ROS generation promotes a series of cellular mechanisms such as polyol and hexosamine flux, AGEs, PKC activation and NFκB induced vascular inflammation[52,141]. One of the major resources of ROS in the condition of high blood glucose is represented by PKC and its downstream subjects. The circumstance of high blood glucose promotes a chronic increase of diacyglycerol concentrations in ECs accompanying the following membrane translocation of conventional and nonconventional PKC isoforms. In case activated, PKC would be responsible for different structure and functions alterations in vascular systems such as changes in permeability, inflammation, angiogenesis, growth, external cell matrix expansion and apoptosis of cells[142]. A key result of PKC activation is ROS production. Hyperglycemia mediated the PKC activation rises superoxide generation by NADPH oxidase in vascular ECs[27]. PKC also contributes to elevated generation of ET-1, which is favor of vasoconstricting and platelet aggregating[142].
In the vascular wall, the generation of PKC-depended ROS takes part in the atherogenetic progression via promoting vessel inflammation yet[52,143]. ROS contributes to upregulation and nuclear translocation of NFκB subunit p65, thus, the transcription of pro-inflammatory genes encodes for monocyte chemoattractant protein-1 (MCP-1), selectins, VCAM-1, and ICAM-1. The latter event promotes the adhesion of monocytes to vascular endothelial cells, rolls and exudes in the subendothelium accompanying the following production of foam cells. The production of IL-1 and TNF-α derived from a active macrophage keeps elevation of adhesion molecule through augmenting NFκB signal in the endothelial cell and also stimulates SMCs growth and proliferation[144].
Endothelial dysfunction in DM is the subsequence of damaged NO availability and the risen synthesis of vascular constrictor and prostanoid[144]. The up-regulation of PKC induced cyclooxygenase-2 (COX-2) is related to an elevation of thromboxane A2 as well as a downregulation of prostacyclin (PGI2) release[145]. The data speculate that PKC is upstream signaling molecules which affect vessel balance at the condition of hyperglycemia[145]. Production of AGEs contributes to cell disorders by triggering of a AGEs receptor (RAGE) activation[146,147]. AGE-RAGE signal conversely promotes ROS-sensitive biochemical pathways like the hexosamine flux[52]. At the circumstance of hyperglycemia, an elevated flux of fructose-6-phosphate promotes a series of reactions leading different glycosilated patterns being responsible for down-regulation of enzymes involved in vessel balance. Especially, OglcNAcylation at the Akt site of eNOS protein contributes to decreasing eNOS activities and ECs disorders[52,148]. Furthermore, transcription factors glycosylation results in upregulation of inflammation (TGFα, TGFβ1) and prothrombus genes (PAI-1)[148,149]. Hyperglycemia mediated ROS generation promotes the polyol pathway flux participated in vessel redox stress yet[141,150]. Therefore, hyperactivation of the pathway is relative to elevated atherogenetic damages in a DM mouse[151,152].
ABNORMALITY OF MIRS PARTICIPATE IN ANGIOGENESIS, VASCULAR REPAIR AND ENDOTHELIAL HOMEOSTASIS
MicroRNAs (miRs) are a currently found one type of small no coding RNAs known as important effects on mechanisms of high blood glucose mediated vessel lesion[153,154]. The small no coded RNAs mediate many aspects of DM vasculopathy through moderating gene expression at the posttranscriptional time. DM shows a obvious abnormality of miRs participated in angiogenesis, vascular repair and endothelial homeostasis[155]. When ECs are exposed to prolonged hyperglycemia, miR-320 is largely expressed and triggers a series of angiogenic factors and their receptors such as vascular endothelial growth factor and insulin like growth factor-1 (IGF-1). Increased expression of the miR is relative to decreasing cellular proliferation and migration. When the miR decrease recoveries the characters and elevates IGF-1 expression, facilitating angiogenesis and vascular repair[156].
High blood glucose also elevates the miR-221 expression, a mediator of angiogenesis for c-kit receptor is associated with migrating as well as homing of endothelial progenitor cells (EPCs)[157]. miR-221 and 222 were identified to induced AGE mediated vessel lesion yet[157]. In fact, the decrease of miR-222 expression in human ECs exposed to hyperglycemia and in a DM mouse results in AGE associated ECs disorders through targeted, cyclin depended kinase proteins participated in cellular cycle depression (P27KIP1 and P57KIP2)[157]. A current research revealed that miR-503 severely participated in high blood glucose mediated endothelial dysfunction in a DM mouse, is elevated in muscles of ischemia limbs in DM patients[158]. The pernicious function of miR-503 at the condition of DM has been identified by the interaction with CCNE and cdc25A, which are key mediators of cell cycle process influencing the migration and proliferation of ECs. It is interesting that miR-503 depression can actualize the normalization of post-ischemic novel vascularization and blood stream repairs in a DM mouse. The studied results offer a base to predict protective effects on regulating miR-503 expression against DM vasculopathy.
Assay of plasm miR demonstrated a largely decrease of miR-126 in a cohort of DM patients[155]. Current studied results propose that down-regulation of miR-126 expression is in part responsible for damaging vascular recovery capacities in DM[159,160]. The expression of miR-126 decreases in EPCs isolated from DM and transfection using anti-miR-126 diminished the proliferation and migration of EPCs[159,160]. By comparison, the recovered expression of miR-126 facilitated EPCs associated a recovery capacity and depressed apoptosis. The miR-126 effect in EPCs is regulated by Spred-1, an inhibitor of Ras/ERK signal pathway, a key mediator of cellular cycles. In general, the findings provide further evidences that miRs promotes a series of complicated signaling network through triggering the genes expression participated in the differentiation, migration as well as survival of cell[161].
In summary, many factors affect the diabetic CVD complication, which are summaried in the Figure 1 of diagrammatic sketch. Such factors will promote optimal understanding of the pathogenesis of the diabetic CVD complication and lead to the identification of the specific preventive therapy. Ultimately, the knowledge gained from these previous studies can be used to obtain the potential drug for preventing the diabetic CVD complication.
Figure 1.
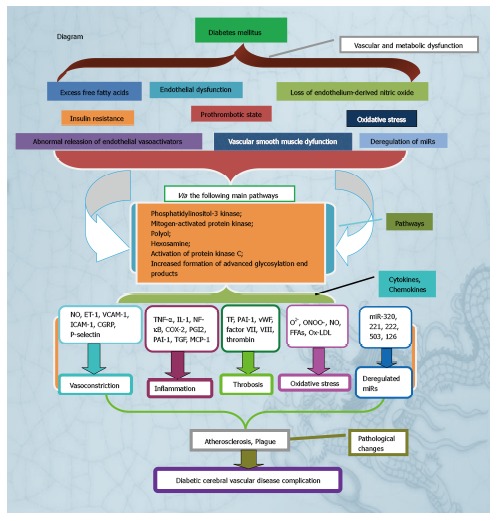
Diagram of pathogenesis of diabetic cerebral vascular disease complication. NO: Endothelium-derived nitric oxide; ET-1: Endothelin-1; VCAM: Vascular cell adhesion molecule; ICAM: Intercellular adhesion molecule; TNF: Tumor necrosis factor; IL: Interleukin; NF-κB: Nuclear factor-κB; COX-2: Cyclooxygenase-2; PGI2: Prostacyclin: PAI-1: Prothrombus genes; TGF-β: Transforming growth factor beta; MCP-1: Monocyte chemoattractant protein-1; TF: Tissue factor; Ox-LDL: Oxidized low-density lipoproteins; FFAs: Free fatty acids.
Footnotes
P- Reviewer: Das UN, Slomiany BL S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Supported by National Nature Science foundation of China (NSFC), No. 30560042, No. 81160161 and No. 81360198; by the Education Department of Jiangxi province (EDJX), No. GJJ10303.
Conflict-of-interest: The author declares that he has no competing interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 2, 2014
First decision: September 18, 2014
Article in press: December 10, 2014
References
- 1.Bucala R. Diabetes, aging, and their tissue complications. J Clin Invest. 2014;124:1887–1888. doi: 10.1172/JCI75224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arinzon Z, Shabat S, Shuval I, Peisakh A, Berner Y. Prevalence of diabetes mellitus in elderly patients received enteral nutrition long-term care service. Arch Gerontol Geriatr. 2008;47:383–393. doi: 10.1016/j.archger.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Bethel MA, Sloan FA, Belsky D, Feinglos MN. Longitudinal incidence and prevalence of adverse outcomes of diabetes mellitus in elderly patients. Arch Intern Med. 2007;167:921–927. doi: 10.1001/archinte.167.9.921. [DOI] [PubMed] [Google Scholar]
- 4.Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med. 1997;14 Suppl 5:S1–85. [PubMed] [Google Scholar]
- 5.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 6.Piechowski-Jozwiak B, Maulaz A, Bogousslavsky J. Secondary prevention of stroke with antiplatelet agents in patients with diabetes mellitus. Cerebrovasc Dis. 2005;20 Suppl 1:15–23. doi: 10.1159/000088233. [DOI] [PubMed] [Google Scholar]
- 7.Spijkerman AM, Dekker JM, Nijpels G, Adriaanse MC, Kostense PJ, Ruwaard D, Stehouwer CD, Bouter LM, Heine RJ. Microvascular complications at time of diagnosis of type 2 diabetes are similar among diabetic patients detected by targeted screening and patients newly diagnosed in general practice: the hoorn screening study. Diabetes Care. 2003;26:2604–2608. doi: 10.2337/diacare.26.9.2604. [DOI] [PubMed] [Google Scholar]
- 8.Krentz AJ, Clough G, Byrne CD. Interactions between microvascular and macrovascular disease in diabetes: pathophysiology and therapeutic implications. Diabetes Obes Metab. 2007;9:781–791. doi: 10.1111/j.1463-1326.2007.00670.x. [DOI] [PubMed] [Google Scholar]
- 9.Harada S, Fujita-Hamabe W, Tokuyama S. Ischemic stroke and glucose intolerance: a review of the evidence and exploration of novel therapeutic targets. J Pharmacol Sci. 2012;118:1–13. doi: 10.1254/jphs.11r04cr. [DOI] [PubMed] [Google Scholar]
- 10.Fülesdi B, Limburg M, Bereczki D, Michels RP, Neuwirth G, Legemate D, Valikovics A, Csiba L. Impairment of cerebrovascular reactivity in long-term type 1 diabetes. Diabetes. 1997;46:1840–1845. doi: 10.2337/diab.46.11.1840. [DOI] [PubMed] [Google Scholar]
- 11.Unfirer S, Kibel A, Drenjancevic-Peric I. The effect of hyperbaric oxygen therapy on blood vessel function in diabetes mellitus. Med Hypotheses. 2008;71:776–780. doi: 10.1016/j.mehy.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Kleiser B, Widder B. Course of carotid artery occlusions with impaired cerebrovascular reactivity. Stroke. 1992;23:171–174. doi: 10.1161/01.str.23.2.171. [DOI] [PubMed] [Google Scholar]
- 13.Larsen FS, Olsen KS, Hansen BA, Paulson OB, Knudsen GM. Transcranial Doppler is valid for determination of the lower limit of cerebral blood flow autoregulation. Stroke. 1994;25:1985–1988. doi: 10.1161/01.str.25.10.1985. [DOI] [PubMed] [Google Scholar]
- 14.Lipsitz LA, Mukai S, Hamner J, Gagnon M, Babikian V. Dynamic regulation of middle cerebral artery blood flow velocity in aging and hypertension. Stroke. 2000;31:1897–1903. doi: 10.1161/01.str.31.8.1897. [DOI] [PubMed] [Google Scholar]
- 15.Sena CM, Pereira AM, Seiça R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim Biophys Acta. 2013;1832:2216–2231. doi: 10.1016/j.bbadis.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Moghaddasi M, Mamarabadi M, Habibi AH. A comparison of cerebral vasomotor reactivity in diabetic and nondiabetic Iranian patients. J Res Med Sci. 2010;15:50–53. [PMC free article] [PubMed] [Google Scholar]
- 17.Konig M, Lamos EM, Stein SA, Davis SN. An insight into the recent diabetes trials: what is the best approach to prevent macrovascular and microvascular complications? Curr Diabetes Rev. 2013;9:371–381. doi: 10.2174/15733998113099990077. [DOI] [PubMed] [Google Scholar]
- 18.Tandon N, Ali MK, Narayan KM. Pharmacologic prevention of microvascular and macrovascular complications in diabetes mellitus: implications of the results of recent clinical trials in type 2 diabetes. Am J Cardiovasc Drugs. 2012;12:7–22. doi: 10.2165/11594650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 19.Sosale A, Prasanna Kumar KM, Sadikot SM, Nigam A, Bajaj S, Zargar AH, Singh SK. Chronic complications in newly diagnosed patients with Type 2 diabetes mellitus in India. Indian J Endocrinol Metab. 2014;18:355–360. doi: 10.4103/2230-8210.131184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muntean C, Mitrea A, Mota M, Tudorica V. Type 2 diabetes and its implications in cerebrovascular disease. Rom J Diabetes Nutr Metab Dis. 2012;19:81–88. [Google Scholar]
- 21.Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527–1532. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 22.Boden G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Physicians. 1999;111:241–248. doi: 10.1046/j.1525-1381.1999.99220.x. [DOI] [PubMed] [Google Scholar]
- 23.Fujimoto WY. The importance of insulin resistance in the pathogenesis of type 2 diabetes mellitus. Am J Med. 2000;108 Suppl 6a:9S–14S. doi: 10.1016/s0002-9343(00)00337-5. [DOI] [PubMed] [Google Scholar]
- 24.Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest. 1994;94:2349–2356. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dichtl W, Nilsson L, Goncalves I, Ares MP, Banfi C, Calara F, Hamsten A, Eriksson P, Nilsson J. Very low-density lipoprotein activates nuclear factor-kappaB in endothelial cells. Circ Res. 1999;84:1085–1094. doi: 10.1161/01.res.84.9.1085. [DOI] [PubMed] [Google Scholar]
- 27.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 28.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 29.Sniderman AD, Scantlebury T, Cianflone K. Hyper-triglyceridemic hyperapob: the unappreciated atherogenic dyslipoproteinemia in type 2 diabetes mellitus. Ann Intern Med. 2001;135:447–459. doi: 10.7326/0003-4819-135-6-200109180-00014. [DOI] [PubMed] [Google Scholar]
- 30.Sniderman A, Thomas D, Marpole D, Teng B. Low density lipoprotein. A metabolic pathway for return of cholesterol to the splanchnic bed. J Clin Invest. 1978;61:867–873. doi: 10.1172/JCI109012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimitriadis E, Griffin M, Owens D, Johnson A, Collins P, Tomkin GH. Oxidation of low-density lipoprotein in NIDDM: its relationship to fatty acid composition. Diabetologia. 1995;38:1300–1306. doi: 10.1007/BF00401762. [DOI] [PubMed] [Google Scholar]
- 32.de Man FH, Weverling-Rijnsburger AW, van der Laarse A, Smelt AH, Jukema JW, Blauw GJ. Not acute but chronic hypertriglyceridemia is associated with impaired endothelium-dependent vasodilation: reversal after lipid-lowering therapy by atorvastatin. Arterioscler Thromb Vasc Biol. 2000;20:744–750. doi: 10.1161/01.atv.20.3.744. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn FE, Mohler ER, Satler LF, Reagan K, Lu DY, Rackley CE. Effects of high-density lipoprotein on acetylcholine-induced coronary vasoreactivity. Am J Cardiol. 1991;68:1425–1430. doi: 10.1016/0002-9149(91)90274-o. [DOI] [PubMed] [Google Scholar]
- 34.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 35.Kinlay S, Libby P, Ganz P. Endothelial function and coronary artery disease. Curr Opin Lipidol. 2001;12:383–389. doi: 10.1097/00041433-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 37.Radomski MW, Palmer RM, Moncada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Commun. 1987;148:1482–1489. doi: 10.1016/s0006-291x(87)80299-1. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitric oxide reversibly inhibits the migration of cultured vascular smooth muscle cells. Circ Res. 1996;78:225–230. doi: 10.1161/01.res.78.2.225. [DOI] [PubMed] [Google Scholar]
- 39.Zeiher AM, Fisslthaler B, Schray-Utz B, Busse R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circ Res. 1995;76:980–986. doi: 10.1161/01.res.76.6.980. [DOI] [PubMed] [Google Scholar]
- 40.Libby P. Changing concepts of atherogenesis. J Intern Med. 2000;247:349–358. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 41.Nomura S, Shouzu A, Omoto S, Nishikawa M, Fukuhara S. Significance of chemokines and activated platelets in patients with diabetes. Clin Exp Immunol. 2000;121:437–443. doi: 10.1046/j.1365-2249.2000.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohamed AK, Bierhaus A, Schiekofer S, Tritschler H, Ziegler R, Nawroth PP. The role of oxidative stress and NF-kappaB activation in late diabetic complications. Biofactors. 1999;10:157–167. doi: 10.1002/biof.5520100211. [DOI] [PubMed] [Google Scholar]
- 43.Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tesfamariam B, Brown ML, Deykin D, Cohen RA. Elevated glucose promotes generation of endothelium-derived vasoconstrictor prostanoids in rabbit aorta. J Clin Invest. 1990;85:929–932. doi: 10.1172/JCI114521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bohlen HG, Lash JM. Topical hyperglycemia rapidly suppresses EDRF-mediated vasodilation of normal rat arterioles. Am J Physiol. 1993;265:H219–H225. doi: 10.1152/ajpheart.1993.265.1.H219. [DOI] [PubMed] [Google Scholar]
- 46.Meraji S, Jayakody L, Senaratne MP, Thomson AB, Kappagoda T. Endothelium-dependent relaxation in aorta of BB rat. Diabetes. 1987;36:978–981. doi: 10.2337/diab.36.8.978. [DOI] [PubMed] [Google Scholar]
- 47.Pieper GM, Meier DA, Hager SR. Endothelial dysfunction in a model of hyperglycemia and hyperinsulinemia. Am J Physiol. 1995;269:H845–H850. doi: 10.1152/ajpheart.1995.269.3.H845. [DOI] [PubMed] [Google Scholar]
- 48.Holt R, Cockram C, Flyvbjerg A, Goldstein B J. Textbook of Diabetes. 4th ed. USA: Wiley-Blackwell; 2010. [Google Scholar]
- 49.DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. 2010;53:1270–1287. doi: 10.1007/s00125-010-1684-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 51.Du X, Edelstein D, Obici S, Higham N, Zou MH, Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116:1071–1080. doi: 10.1172/JCI23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cardillo C, Campia U, Bryant MB, Panza JA. Increased activity of endogenous endothelin in patients with type II diabetes mellitus. Circulation. 2002;106:1783–1787. doi: 10.1161/01.cir.0000032260.01569.64. [DOI] [PubMed] [Google Scholar]
- 54.Li ZY, Wang P, Miao CY. Adipokines in inflammation, insulin resistance and cardiovascular disease. Clin Exp Pharmacol Physiol. 2011;38:888–896. doi: 10.1111/j.1440-1681.2011.05602.x. [DOI] [PubMed] [Google Scholar]
- 55.Collinson DJ, Rea R, Donnelly R. Vascular risk: diabetes. Vasc Med. 2004;9:307–310. doi: 10.1191/1358863x04vm550xx. [DOI] [PubMed] [Google Scholar]
- 56.Novak V, Zhao P, Manor B, Sejdic E, Alsop D, Abduljalil A, Roberson PK, Munshi M, Novak P. Adhesion molecules, altered vasoreactivity, and brain atrophy in type 2 diabetes. Diabetes Care. 2011;34:2438–2441. doi: 10.2337/dc11-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reagan LP. Diabetes as a chronic metabolic stressor: causes, consequences and clinical complications. Exp Neurol. 2012;233:68–78. doi: 10.1016/j.expneurol.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vinik AI, Erbas T, Park TS, Nolan R, Pittenger GL. Platelet dysfunction in type 2 diabetes. Diabetes Care. 2001;24:1476–1485. doi: 10.2337/diacare.24.8.1476. [DOI] [PubMed] [Google Scholar]
- 59.Assert R, Scherk G, Bumbure A, Pirags V, Schatz H, Pfeiffer AF. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia. 2001;44:188–195. doi: 10.1007/s001250051598. [DOI] [PubMed] [Google Scholar]
- 60.Li Y, Woo V, Bose R. Platelet hyperactivity and abnormal Ca(2+) homeostasis in diabetes mellitus. Am J Physiol Heart Circ Physiol. 2001;280:H1480–H1489. doi: 10.1152/ajpheart.2001.280.4.H1480. [DOI] [PubMed] [Google Scholar]
- 61.Hafer-Macko CE, Ivey FM, Gyure KA, Sorkin JD, Macko RF. Thrombomodulin deficiency in human diabetic nerve microvasculature. Diabetes. 2002;51:1957–1963. doi: 10.2337/diabetes.51.6.1957. [DOI] [PubMed] [Google Scholar]
- 62.Ceriello A, Giacomello R, Stel G, Motz E, Taboga C, Tonutti L, Pirisi M, Falleti E, Bartoli E. Hyperglycemia-induced thrombin formation in diabetes. The possible role of oxidative stress. Diabetes. 1995;44:924–928. doi: 10.2337/diab.44.8.924. [DOI] [PubMed] [Google Scholar]
- 63.Ceriello A, Giugliano D, Quatraro A, Marchi E, Barbanti M, Lefèbvre P. Evidence for a hyperglycaemia-dependent decrease of antithrombin III-thrombin complex formation in humans. Diabetologia. 1990;33:163–167. doi: 10.1007/BF00404044. [DOI] [PubMed] [Google Scholar]
- 64.Ren S, Lee H, Hu L, Lu L, Shen GX. Impact of diabetes-associated lipoproteins on generation of fibrinolytic regulators from vascular endothelial cells. J Clin Endocrinol Metab. 2002;87:286–291. doi: 10.1210/jcem.87.1.8175. [DOI] [PubMed] [Google Scholar]
- 65.Kario K, Matsuo T, Kobayashi H, Matsuo M, Sakata T, Miyata T. Activation of tissue factor-induced coagulation and endothelial cell dysfunction in non-insulin-dependent diabetic patients with microalbuminuria. Arterioscler Thromb Vasc Biol. 1995;15:1114–1120. doi: 10.1161/01.atv.15.8.1114. [DOI] [PubMed] [Google Scholar]
- 66.Pandolfi A, Cetrullo D, Polishuck R, Alberta MM, Calafiore A, Pellegrini G, Vitacolonna E, Capani F, Consoli A. Plasminogen activator inhibitor type 1 is increased in the arterial wall of type II diabetic subjects. Arterioscler Thromb Vasc Biol. 2001;21:1378–1382. doi: 10.1161/hq0801.093667. [DOI] [PubMed] [Google Scholar]
- 67.Vazzana N, Ranalli P, Cuccurullo C, Davì G. Diabetes mellitus and thrombosis. Thromb Res. 2012;129:371–377. doi: 10.1016/j.thromres.2011.11.052. [DOI] [PubMed] [Google Scholar]
- 68.Beckman JA, Creager MA, Libby P. Diabetes and athero-sclerosis: epidemiology, pathophysiology, and management. JAMA. 2002;287:2570–2581. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- 69.Boden G, Rao AK. Effects of hyperglycemia and hyperin-sulinemia on the tissue factor pathway of blood coagulation. Curr Diab Rep. 2007;7:223–227. doi: 10.1007/s11892-007-0035-1. [DOI] [PubMed] [Google Scholar]
- 70.Linden MD, Tran H, Woods R, Tonkin A. High platelet reactivity and antiplatelet therapy resistance. Semin Thromb Hemost. 2012;38:200–212. doi: 10.1055/s-0032-1301417. [DOI] [PubMed] [Google Scholar]
- 71.Carr ME. Diabetes mellitus: a hypercoagulable state. J Diabetes Complications. 2001;15:44–54. doi: 10.1016/s1056-8727(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 72.Ceriello A, Giugliano D, Quatraro A, Dello Russo P, Torella R. Blood glucose may condition factor VII levels in diabetic and normal subjects. Diabetologia. 1988;31:889–891. doi: 10.1007/BF00265372. [DOI] [PubMed] [Google Scholar]
- 73.Nordt TK, Bode C. Impaired endogenous fibrinolysis in diabetes mellitus: mechanisms and therapeutic approaches. Semin Thromb Hemost. 2000;26:495–501. doi: 10.1055/s-2000-13205. [DOI] [PubMed] [Google Scholar]
- 74.Snell-Bergeon JK, Wadwa RP. Hypoglycemia, diabetes, and cardiovascular disease. Diabetes Technol Ther. 2012;14 Suppl 1:S51–S58. doi: 10.1089/dia.2012.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aljada A. Endothelium, inflammation, and diabetes. Metab Syndr Relat Disord. 2003;1:3–21. doi: 10.1089/154041903321648225. [DOI] [PubMed] [Google Scholar]
- 76.Avogaro A, Albiero M, Menegazzo L, de Kreutzenberg S, Fadini GP. Endothelial dysfunction in diabetes: the role of reparatory mechanisms. Diabetes Care. 2011;34 Suppl 2:S285–S290. doi: 10.2337/dc11-s239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 78.De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int Suppl. 2000;77:S26–S30. doi: 10.1046/j.1523-1755.2000.07705.x. [DOI] [PubMed] [Google Scholar]
- 80.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 81.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 82.Ceriello A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care. 2003;26:1589–1596. doi: 10.2337/diacare.26.5.1589. [DOI] [PubMed] [Google Scholar]
- 83.Garcia Soriano F L, Jagtap P, Szabó E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 84.Soriano FG, Pacher P, Mabley J, Liaudet L, Szabó C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ Res. 2001;89:684–691. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- 85.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- 86.Verma S, Anderson TJ. The ten most commonly asked questions about endothelial function in cardiology. Cardiol Rev. 2001;9:250–252. doi: 10.1097/00045415-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 87.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996;27:567–574. doi: 10.1016/0735-1097(95)00522-6. [DOI] [PubMed] [Google Scholar]
- 89.Johnstone MT, Creager SJ, Scales KM, Cusco JA, Lee BK, Creager MA. Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation. 1993;88:2510–2516. doi: 10.1161/01.cir.88.6.2510. [DOI] [PubMed] [Google Scholar]
- 90.Hennes MM, O’Shaughnessy IM, Kelly TM, LaBelle P, Egan BM, Kissebah AH. Insulin-resistant lipolysis in abdominally obese hypertensive individuals. Role of the renin-angiotensin system. Hypertension. 1996;28:120–126. doi: 10.1161/01.hyp.28.1.120. [DOI] [PubMed] [Google Scholar]
- 91.Zou M, Yesilkaya A, Ullrich V. Peroxynitrite inactivates prostacyclin synthase by heme-thiolate-catalyzed tyrosine nitration. Drug Metab Rev. 1999;31:343–349. doi: 10.1081/dmr-100101922. [DOI] [PubMed] [Google Scholar]
- 92.Vora DK, Fang ZT, Liva SM, Tyner TR, Parhami F, Watson AD, Drake TA, Territo MC, Berliner JA. Induction of P-selectin by oxidized lipoproteins. Separate effects on synthesis and surface expression. Circ Res. 1997;80:810–818. doi: 10.1161/01.res.80.6.810. [DOI] [PubMed] [Google Scholar]
- 93.Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ, Fogelman AM. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci USA. 1990;87:5134–5138. doi: 10.1073/pnas.87.13.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rajavashisth TB, Andalibi A, Territo MC, Berliner JA, Navab M, Fogelman AM, Lusis AJ. Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature. 1990;344:254–257. doi: 10.1038/344254a0. [DOI] [PubMed] [Google Scholar]
- 95.Hopfner RL, Gopalakrishnan V. Endothelin: emerging role in diabetic vascular complications. Diabetologia. 1999;42:1383–1394. doi: 10.1007/s001250051308. [DOI] [PubMed] [Google Scholar]
- 96.Cohen RA, Tong X. Vascular oxidative stress: the common link in hypertensive and diabetic vascular disease. J Cardiovasc Pharmacol. 2010;55:308–316. doi: 10.1097/fjc.0b013e3181d89670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van den Oever IA, Raterman HG, Nurmohamed MT, Simsek S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators Inflamm. 2010;2010:792393. doi: 10.1155/2010/792393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tan KC, Chow WS, Ai VH, Lam KS. Effects of angiotensin II receptor antagonist on endothelial vasomotor function and urinary albumin excretion in type 2 diabetic patients with microalbuminuria. Diabetes Metab Res Rev. 2002;18:71–76. doi: 10.1002/dmrr.255. [DOI] [PubMed] [Google Scholar]
- 99.Verma S, Anderson TJ. Fundamentals of endothelial function for the clinical cardiologist. Circulation. 2002;105:546–549. doi: 10.1161/hc0502.104540. [DOI] [PubMed] [Google Scholar]
- 100.Wedgwood S, McMullan DM, Bekker JM, Fineman JR, Black SM. Role for endothelin-1-induced superoxide and peroxynitrite production in rebound pulmonary hypertension associated with inhaled nitric oxide therapy. Circ Res. 2001;89:357–364. doi: 10.1161/hh1601.094983. [DOI] [PubMed] [Google Scholar]
- 101.Romero M, Jiménez R, Sánchez M, López-Sepúlveda R, Zarzuelo MJ, O’Valle F, Zarzuelo A, Pérez-Vizcaíno F, Duarte J. Quercetin inhibits vascular superoxide production induced by endothelin-1: Role of NADPH oxidase, uncoupled eNOS and PKC. Atherosclerosis. 2009;202:58–67. doi: 10.1016/j.atherosclerosis.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 102.Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- 103.McCulloch J, Uddman R, Kingman TA, Edvinsson L. Calc-itonin gene-related peptide: functional role in cerebrovascular regulation. Proc Natl Acad Sci USA. 1986;83:5731–5735. doi: 10.1073/pnas.83.15.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iademarco MF, McQuillan JJ, Rosen GD, Dean DC. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1) J Biol Chem. 1992;267:16323–16329. [PubMed] [Google Scholar]
- 106.van Buul JD, Voermans C, van den Berg V, Anthony EC, Mul FP, van Wetering S, van der Schoot CE, Hordijk PL. Migration of human hematopoietic progenitor cells across bone marrow endothelium is regulated by vascular endothelial cadherin. J Immunol. 2002;168:588–596. doi: 10.4049/jimmunol.168.2.588. [DOI] [PubMed] [Google Scholar]
- 107.van Wetering S, van den Berk N, van Buul JD, Mul FP, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ, Hordijk PL. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol. 2003;285:C343–C352. doi: 10.1152/ajpcell.00048.2003. [DOI] [PubMed] [Google Scholar]
- 108.Madonna R, Pandolfi A, Massaro M, Consoli A, De Caterina R. Insulin enhances vascular cell adhesion molecule-1 expression in human cultured endothelial cells through a pro-atherogenic pathway mediated by p38 mitogen-activated protein-kinase. Diabetologia. 2004;47:532–536. doi: 10.1007/s00125-004-1330-x. [DOI] [PubMed] [Google Scholar]
- 109.Okouchi M, Okayama N, Shimizu M, Omi H, Fukutomi T, Itoh M. High insulin exacerbates neutrophil-endothelial cell adhesion through endothelial surface expression of intercellular adhesion molecule-1 via activation of protein kinase C and mitogen-activated protein kinase. Diabetologia. 2002;45:556–559. doi: 10.1007/s00125-001-0773-6. [DOI] [PubMed] [Google Scholar]
- 110.Rahman A, Fazal F. Hug tightly and say goodbye: role of endothelial ICAM-1 in leukocyte transmigration. Antioxid Redox Signal. 2009;11:823–839. doi: 10.1089/ars.2008.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–819. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Galkina E, Ley K. Vascular adhesion molecules in ather-osclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–2301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 113.Pi X, Lockyer P, Dyer LA, Schisler JC, Russell B, Carey S, Sweet DT, Chen Z, Tzima E, Willis MS, et al. Bmper inhibits endothelial expression of inflammatory adhesion molecules and protects against atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2214–2222. doi: 10.1161/ATVBAHA.112.252015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep. 2009;61:22–32. doi: 10.1016/s1734-1140(09)70004-0. [DOI] [PubMed] [Google Scholar]
- 115.Deem TL, Cook-Mills JM. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood. 2004;104:2385–2393. doi: 10.1182/blood-2004-02-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Setiadi H, McEver RP. Signal-dependent distribution of cell surface P-selectin in clathrin-coated pits affects leukocyte rolling under flow. J Cell Biol. 2003;163:1385–1395. doi: 10.1083/jcb.200307178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang SX, Yan J, Deshpande SS, Irani K, Lowenstein CJ. Rac1 regulates the release of Weibel-Palade Bodies in human aortic endothelial cells. Chin Med J (Engl) 2004;117:1143–1150. [PubMed] [Google Scholar]
- 118.Panés J, Kurose I, Rodriguez-Vaca D, Anderson DC, Miyasaka M, Tso P, Granger DN. Diabetes exacerbates inflammatory responses to ischemia-reperfusion. Circulation. 1996;93:161–167. doi: 10.1161/01.cir.93.1.161. [DOI] [PubMed] [Google Scholar]
- 119.Kaplanski G, Farnarier C, Benoliel AM, Foa C, Kaplanski S, Bongrand P. A novel role for E- and P-selectins: shape control of endothelial cell monolayers. J Cell Sci. 1994;107(Pt 9):2449–2457. doi: 10.1242/jcs.107.9.2449. [DOI] [PubMed] [Google Scholar]
- 120.Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102:2165–2168. doi: 10.1161/01.cir.102.18.2165. [DOI] [PubMed] [Google Scholar]
- 121.Rodriguez CJ, Miyake Y, Grahame-Clarke C, Di Tullio MR, Sciacca RR, Boden-Albala B, Sacco RL, Homma S. Relation of plasma glucose and endothelial function in a population-based multiethnic sample of subjects without diabetes mellitus. Am J Cardiol. 2005;96:1273–1277. doi: 10.1016/j.amjcard.2005.06.070. [DOI] [PubMed] [Google Scholar]
- 122.Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170:191–203. doi: 10.1016/s0021-9150(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 123.Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:266–274. doi: 10.1161/01.ATV.0000253884.13901.e4. [DOI] [PubMed] [Google Scholar]
- 124.Raghavan VA. Insulin resistance and atherosclerosis. Heart Fail Clin. 2012;8:575–587. doi: 10.1016/j.hfc.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 125.Rask-Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- 126.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 127.Perez del Villar C, Garcia Alonso CJ, Feldstein CA, Juncos LA, Romero JC. Role of endothelin in the pathogenesis of hypertension. Mayo Clin Proc. 2005;80:84–96. [PubMed] [Google Scholar]
- 128.Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1058–L1067. doi: 10.1152/ajplung.2001.281.5.L1058. [DOI] [PubMed] [Google Scholar]
- 129.Xu R, Yang R, Hu H, Xi Q, Wan H, Wu Y. Diabetes alters the expression of partial vasoactivators in cerebral vascular disease susceptible regions of the diabetic rat. Diabetol Metab Syndr. 2013;5:63. doi: 10.1186/1758-5996-5-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McDaid EA, Monaghan B, Parker AI, Hayes JR, Allen JA. Peripheral autonomic impairment in patients newly diagnosed with type II diabetes. Diabetes Care. 1994;17:1422–1427. doi: 10.2337/diacare.17.12.1422. [DOI] [PubMed] [Google Scholar]
- 131.Hattori Y, Hattori S, Sato N, Kasai K. High-glucose-induced nuclear factor kappaB activation in vascular smooth muscle cells. Cardiovasc Res. 2000;46:188–197. doi: 10.1016/s0008-6363(99)00425-3. [DOI] [PubMed] [Google Scholar]
- 132.Suzuki LA, Poot M, Gerrity RG, Bornfeldt KE. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: lack of direct growth-promoting effects of high glucose levels. Diabetes. 2001;50:851–860. doi: 10.2337/diabetes.50.4.851. [DOI] [PubMed] [Google Scholar]
- 133.Fukumoto H, Naito Z, Asano G, Aramaki T. Immun-ohistochemical and morphometric evaluations of coronary atherosclerotic plaques associated with myocardial infarction and diabetes mellitus. J Atheroscler Thromb. 1998;5:29–35. doi: 10.5551/jat1994.5.29. [DOI] [PubMed] [Google Scholar]
- 134.Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365–372. doi: 10.1161/01.cir.104.3.365. [DOI] [PubMed] [Google Scholar]
- 135.Taguchi S, Oinuma T, Yamada T. A comparative study of cultured smooth muscle cell proliferation and injury, utilizing glycated low density lipoproteins with slight oxidation, auto-oxidation, or extensive oxidation. J Atheroscler Thromb. 2000;7:132–137. doi: 10.5551/jat1994.7.132. [DOI] [PubMed] [Google Scholar]
- 136.Brown GC, Borutaite V. Nitric oxide, cytochrome c and mitochondria. Biochem Soc Symp. 1999;66:17–25. doi: 10.1042/bss0660017. [DOI] [PubMed] [Google Scholar]
- 137.Andersson U, Leighton B, Young ME, Blomstrand E, Newsholme EA. Inactivation of aconitase and oxoglutarate dehydrogenase in skeletal muscle in vitro by superoxide anions and/or nitric oxide. Biochem Biophys Res Commun. 1998;249:512–516. doi: 10.1006/bbrc.1998.9171. [DOI] [PubMed] [Google Scholar]
- 138.Beckman KB, Ames BN. Endogenous oxidative damage of mtDNA. Mutat Res. 1999;424:51–58. doi: 10.1016/s0027-5107(99)00007-x. [DOI] [PubMed] [Google Scholar]
- 139.Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Thorpe SR. Lipoxidation products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions. Nephrol Dial Transplant. 1996;11 Suppl 5:48–53. doi: 10.1093/ndt/11.supp5.48. [DOI] [PubMed] [Google Scholar]
- 140.DeFronzo RA, Ferrannini E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- 141.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 142.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kouroedov A, Eto M, Joch H, Volpe M, Lüscher TF, Cosentino F. Selective inhibition of protein kinase Cbeta2 prevents acute effects of high glucose on vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 2004;110:91–96. doi: 10.1161/01.CIR.0000133384.38551.A8. [DOI] [PubMed] [Google Scholar]
- 144.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 145.Cosentino F, Eto M, De Paolis P, van der Loo B, Bachschmid M, Ullrich V, Kouroedov A, Delli Gatti C, Joch H, Volpe M, et al. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation. 2003;107:1017–1023. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- 146.Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 2005;83:876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 148.Fülöp N, Marchase RB, Chatham JC. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res. 2007;73:288–297. doi: 10.1016/j.cardiores.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Buse MG. Hexosamines, insulin resistance, and the complications of diabetes: current status. Am J Physiol Endocrinol Metab. 2006;290:E1–E8. doi: 10.1152/ajpendo.00329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999;13:23–30. doi: 10.1096/fasebj.13.1.23. [DOI] [PubMed] [Google Scholar]
- 151.Vikramadithyan RK, Hu Y, Noh HL, Liang CP, Hallam K, Tall AR, Ramasamy R, Goldberg IJ. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J Clin Invest. 2005;115:2434–2443. doi: 10.1172/JCI24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev. 2004;25:612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- 153.Shantikumar S, Caporali A, Emanueli C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res. 2012;93:583–593. doi: 10.1093/cvr/cvr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zampetaki A, Mayr M. MicroRNAs in vascular and metabolic disease. Circ Res. 2012;110:508–522. doi: 10.1161/CIRCRESAHA.111.247445. [DOI] [PubMed] [Google Scholar]
- 155.Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, Mayr A, Weger S, Oberhollenzer F, Bonora E, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. 2010;107:810–817. doi: 10.1161/CIRCRESAHA.110.226357. [DOI] [PubMed] [Google Scholar]
- 156.Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2009;36:181–188. doi: 10.1111/j.1440-1681.2008.05057.x. [DOI] [PubMed] [Google Scholar]
- 157.Togliatto G, Trombetta A, Dentelli P, Rosso A, Brizzi MF. MIR221/MIR222-driven post-transcriptional regulation of P27KIP1 and P57KIP2 is crucial for high-glucose- and AGE-mediated vascular cell damage. Diabetologia. 2011;54:1930–1940. doi: 10.1007/s00125-011-2125-5. [DOI] [PubMed] [Google Scholar]
- 158.Caporali A, Meloni M, Völlenkle C, Bonci D, Sala-Newby GB, Addis R, Spinetti G, Losa S, Masson R, Baker AH, et al. Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation. 2011;123:282–291. doi: 10.1161/CIRCULATIONAHA.110.952325. [DOI] [PubMed] [Google Scholar]
- 159.Meng S, Cao JT, Zhang B, Zhou Q, Shen CX, Wang CQ. Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J Mol Cell Cardiol. 2012;53:64–72. doi: 10.1016/j.yjmcc.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 160.Wang DE. MicroRNA Regulation and its Biological Significance in Personalized Medicine and Aging. Curr Genomics. 2009;10:143. doi: 10.2174/138920209788185216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J. 2013;34:2436–2443. doi: 10.1093/eurheartj/eht149. [DOI] [PMC free article] [PubMed] [Google Scholar]