

Abstract
Molecular targeting of drug delivery nanocarriers is expected to improve their therapeutic index while decreasing their toxicity. Here we report the identification and characterization of novel peptide ligands specific for cells present in high-risk neuroblastoma (NB), a childhood tumor mostly refractory to current therapies. To isolate such targeting moieties, we performed combined in vitro/ex-vivo phage display screenings on NB cell lines and on tumors derived from orthotopic mouse models of human NB.
By designing proper subtractive protocols, we identified phage clones specific either for the primary tumor, its metastases, or for their respective stromal components. Globally, we isolated 121 phage-displayed NB-binding peptides: 26 bound the primary tumor, 15 the metastatic mass, 57 and 23 their respective microenvironments. Of these, five phage clones were further validated for their specific binding ex-vivo to biopsies from stage IV NB patients and to NB tumors derived from mice. All five clones also targeted tumor cells and vasculature in vivo when injected into NB-bearing mice. Coupling of the corresponding targeting peptides with doxorubicin-loaded liposomes led to a significant inhibition in tumor volume and enhanced survival in preclinical NB models, thereby paving the way to their clinical development.
Keywords: Phage display screening, peptides, nanocarriers, liposomes, targeted therapy, neuroblastoma
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in children, accounting for about 8% of childhood cancers. Approximately 40% of NB tumors are classified as high-risk; their management includes combinations of chemotherapy, autologous stem cell transplantation, surgery, and radiation therapy. Despite this aggressive treatment, children with high-risk NB have very poor 5-year overall survival rate, due to relapsed and/or treatment-resistant tumors [1, 2]. A further increase in therapeutic dose intensity is not feasible, because it will lead to prohibitive short-term and long-term toxicities. New approaches with targeted therapies may improve efficacy and decrease toxicity [3].
Extensive investigation of NB biology has recently resulted in the identification of a wide range of potential druggable targets, most of which are commonly deregulated in different solid tumors, e.g., disialoganglioside GD2 [3], anaplastic lymphoma kinase (ALK) [4], aurora kinase A (AURKA) [5], epidermal growth factor receptor (EGFR) [6], insulin growth factor 1 receptor (IGF1R) [7], mammalian target of rapamycin (mTOR) [7], and vascular endothelial growth factor receptor 2 (VEGFR2) [8]. Several compounds have proved to be highly active in preclinical models, and at least 15 genes are presently in the clinical pipeline as targets for NB therapy [9]. However, small molecule inhibitors and blocking antibodies suffer from a number of limitations, including toxic side effects, induction of resistance, and low response rates. Together, reports from the literature and from clinical trials provide evidence that, unfortunately, for more than 50% of patients with high-risk NB there are substantially no curative options available.
The use of drug delivery systems allows site specific delivery of higher payload of active agents associated with lower systemic toxicities compared to the use of convetional (‘free’) drugs; the possibility of imparting selectivity to the carrier to the cancer foci through the use of a targeting moiety (e.g., a peptide or an antibody) further enhances drug efficacy and safety [10-13]. The peptide motifs, NGR [14] and CPRECES [15], home to the vasculature of different tumor types and we have recently exploited them to vehiculate chemotherapeutic agents to tumor blood vessels in preclinical models of NB [16-18]. However, NB-targeting peptides with greater selectivity, which are expected to substantially improve current therapeutic regimens, have not been identified thus far.
We report here the identification of novel peptide ligands, selected by combined in vitro/ex-vivo phage display screenings in preclinical models of human NB. Five sequences with differential specificity for either epithelial or stromal components of the tumors were validated in vitro and in vivo for their targeting specificity. The capability of peptide-targeted drug delivery systems to counteract tumor progression was investigated in biologically relevant murine models of human NB. We demonstrated that this approach allows a substantial improvement in therapeutic efficacy compared to both free drug and untargeted systems.
Materials and Methods
Cells lines and human samples
The neuroblastoma (NB) cell lines (GI-LI-N, HTLA-230 and IMR-32) were grown in complete Dulbecco's Modified Eagle Medium (DMEM) or RPMI-1640 medium, as previously described [16, 19]. Human umbilical vein endothelial cells (HUVECs) were maintained in endothelial cell basal medium-2 (Cambrex Bio Science), as described [20]. Cells were tested for mycoplasma contamination and characterized by cell proliferation, morphology evaluation, and multiplex short tandem repeat profiling test. Human samples (07-B-822, 07-B-1173 and 07-B-1312A2) derived from stroma poor, stage IV, NB patients, were provided, after informed consent, by Bio-bank, Istituto Giannina Gaslini, Genoa, Italy.
Animal models
Animals were purchased from Harlan Laboratories (Harlan Italy, S.Pietro al Natisone, Italy) and were housed under pathogen-free conditions; experiments were reviewed and approved by the licensing and ethical committee of IRCCS Azienda Ospedaliera Universitaria San Martino - IST Istituto Nazionale per la Ricerca sul Cancro (Genoa, Italy), and by the Italian Ministry of Health. For the orthotipic model, 5-week-old athymic (nu/nu) female mice were injected with 1×106 GI-LI-N or HTLA-230 cells in the left adrenal gland, as described [16]. No mice died as a result of the surgery. Animals were monitored at least twice a week for evidence of tumor development and quantification of tumor size, and were sacrificed by cervical dislocation after being anesthetized with xilezine (Xilor 2%, Bio98 Srl, Milan, Italy), when they showed signs of poor health, e.g. abdominal dilation, dehydration, or paraplegia. For the pseudometastatic model, 4-week-old female athymic (nu/nu) mice were injected intravenously (i.v.) in the tail vein with 4×106 HTLA-230 cells, as previously described [19]. Body weight and general physical status were recorded daily, and mice were sacrificed by cervical dislocation after the administration of xilezine, when they showed signs of poor health.
Phage display biopanning on preclinical models of human neuroblastoma
Cultured GI-LI-N and HTLA-230 cells were detached with 0.25% trypsin-ethylene diamino tetra-acetic acid (EDTA) solution (Invitrogen, Milan, Italy). Fresh tissues from orthotopically implanted GI-LI-N and HTLA-230 animal models were dissected with a scalpel in Iscove's Modified Dulbecco's Minimum Essential Medium (IMDM supplemented with 2% fetal calf serum, FCS) in bath ice.). 1010 transducing units (T.U.) of a X7 (X = any amino acid) phage library (Ph.D.™-7 Phage Display Peptide Library Kit, New England Biolabs, Ipswich, MA) was added to 5×105 target cells in binding medium, and incubated 4 h at 4°C (first round). For successive rounds, phages were first pre-adsorbed on control cells/tissues for 1 h at 4°C and were subsequently incubated with target tissues for 2 h at 4°C. After 5 washes in binding medium, bound phages were recovered and amplified by infection of K91Kan Escherichia coli bacteria in log-phase. Phage particles were purified from bacterial culture supernatants by precipitation in NaCl/poly (ethylene glycol-8000) and titrated as described [21, 22]; phage DNA was extracted and sequenced following the instructions of the Ph.D.™-7 kit (New England Biolabs).
Ex-vivo validation of phage clones targeting neuroblastoma tissues
Fifteen phage clones, chosen on the basis of the presence of repeated tripeptide motifs, were validated for their binding to NB tissues both from patients with stage IV NB and mice orthotopically implanted with GI-LI-N cells. Tumors, frozen in optimum cutting temperature (OCT) medium (Miles Chemical Co., Elkhart, IN), were cut in 5 μm -sections, fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, and histologically evaluated by staining with Mallory Trichrome (Bio-Optica, Milan, Italy). For the phage overlay binding assay, sections were washed twice in phosphate buffered saline (PBS), saturated with Protein Block Serum-Free (DAKO, Milan, Italy), and overlayed for 1 h at 4°C with 108 T.U./μl of each phage clone. After extensive washing, phages were revealed by staining with a rabbit anti-fd bacteriophage antibody (1:100, Sigma, St. Louis, MO) and DcEnVision + System HRP (DAKO, Milan Italy) as substrate. Non-epithelial tumor components were identified by staining with rabbit polyclonal anti-Factor VIII (1:100, DAKO), rat monoclonal anti-CD31 (1:100, BD Biosciences, Franklin Lakes, NJ), Cy3-conjugated anti-α-smooth muscle actin mouse monoclonal (SMA-Cy3, 1:200, Sigma), and rabbit polyclonal anti-collagen I (1:200, AbCam, Cambridge, UK). For immunofluorescence staining and confocal analyses (TCS SP2 confocal microscope, Leica Microsystems, Mannheim, Germany), AlexaFluor® secondary antibodies (Invitrogen) were used.
Synthesis and in vitro validation of the NB-targeting peptides
To reproduce the original molecular environment of their phage-displayed counterpart, selected NB-targeting peptides were synthesized with the addition of the YSHS and GGG sequences at their N- and C-terminal, respectively. To favor the accessibility of liposome-bound peptides and to allow their coupling to maleimido groups (see below), an additional Cys residue was inserted at their C-terminal, resulting in the following peptide sequences (the NB-binding motifs are underlined): #1: YSHSYEGLISRGGGC; #5: YSHSHSYWLRSGGGC; #8: YSHSWSWPRELGGGC; #10: YSHSALAAHKLGGGC; #14: YSHSKSFFLSHGGGC. In some experiments, the scrambled (SCR) peptide YSHSLAKALHAGGGC was used as a control. For the cellular association assays, biotin-conjugated NB-targeting peptides (concentration range: 20-200 μg/mL) were incubated with 1×106 cells for 1 h at 4°C. Samples were washed in PBS, followed by incubation for 30 min with Cy3-labelled streptavidin (Cy™3-Streptavidin, GE Healthcare). After extensive washing, Cy3-positive cells were counted by flow cytometry, using a FACScan instrument (Becton-Dickinson Immunocytometry Systems).
Preparation of neuroblastoma-targeted liposomes
Stealth™ liposomes (SL) were synthesized from HSPC:CHOL:DSPE-PEG2000, 2:1:0.1 molar ratio, and HSPC:CHOL:DSPE-PEG2000:DSPE-PEG2000-MAL, 2:1:0.08:0.02 molar ratio, as previously described [16]. The hydrated liposomes were sequentially extruded (LiposoFast-basic extruder, Avestin, Inc, Leiden, Holland) through polycarbonate filters of pore size ranging from 0.2 μm down to 0.1 μm to produce primarily unilamellar vesicles. SL size, polydispersity, and zeta-potential were analyzed by dynamic light scattering using the zeta sizer Nano-S particle sizer at a fixed angle (90°) (Malvern Instruments, Malvern, UK). Doxorubicin (DXR) was loaded into SL via an ammonium sulphate gradient, as previously reported [19]. Targeted SL were produced by mixing freshly-prepared nanocarriers with an equimolar (with respect to DSPE-PEG2000-MAL) amount of peptide for 16 h at 4°C under argon, followed by incubation in a 10-fold excess of 2-mercapethanol for 1 h to neutralize remaining maleimido groups. Uncoupled peptides were removed by passing the reaction mixture through a Sepharose CL-4B column in HEPES buffer, pH 7.4. Coupling efficiency was determined by the quantification of SL-associated peptides with the CBQCA Protein Quantification Kit (Molecular Probes Europe, Leiden, The Netherlands), as described [18]. 3H- and 14C-dual labeled targeted liposomes were synthesized as previously described [23].
Cell apoptosis and viability assays
Detection of apoptotic/hypodiploid cells was performed as described [24]. Briefly, GI-LI-N (6×105/well), HTLA-230 (6×105/well) and HUVECs (1.2×106/collagen-pre-coated well) were seeded in 6-well plates in complete medium; after 24 h, 50 μM of DXR encapsulated in either untargeted or peptide-targeted SL was added to the culture medium and cells were incubated for 1 h at 37°C. Cells were then washed twice in PBS and cultured in fresh medium for additional 47 h, before being incubated in 3.4 mM sodium citrate, 20 μg/mL propidium iodide (PI) (Sigma) and 100 μg/mL RNase A (Qiagen Italy, Milan, Italy) for 30 min at 37°C in the dark.
For proliferation and cell cycle evaluation, untargeted or peptide-targeted SL-treated cells were pulse-labeled with 10 μmol/L bromodeoxyuridine (BrdU; Sigma) for 30 min, as previously reported [20]. BrdU uptake was detected by cell staining with FITC-conjugated mouse monoclonal anti-BrdU antibody (Becton Dickinson) at a final concentration of 5 μg/mL for 30 min at room temperature. After extensive washing, cells were resuspended in PBS containing 5 μg/mL PI and the distribution of BrdU (FITC) versus DNA content (PI) was assessed by flow cytometry. In all the experiments, SCR-SL[DXR] were used to validate the peptides targeted, DXR-loaded liposomes-driven cytotoxic effects.
In vivo homing of neuroblastoma-targeted phage clones and of neuroblastoma-targeted liposomes
For the phage homing studies, mice bearing orthotopic implants of GI-LI-N cells were inoculated i.v. into the tail vein with 1×109 T.U. of each NB-targeting phage clone. Animals were perfused with complete medium at 10 min and 24 h after inoculation before being euthanized; tumors and healthy organs were collected and fixed in Bouin solution. Four micrometer tissue slices were stained with the anti-fd bacteriophage and anti-SMA-Cy3 antibodies using standard protocols. Fluorescence was examined with a Laser Scanning Spectral confocal microscope (TCS SP2, Leica MicroSystems). Staining was quantified on 3-5 confocal images/experimental point by using the Image Processing and Analysis software in Java (ImageJ). Furthermore, digital images were processed with Adobe Photoshop CS2 (Adobe Systems, San Jose, CA) to adjust contrast and assemble the final plates. For the tumor accumulation studies, 35 days after orthotopic tumor cell implantation (mean volume ∼250 mm3), mice received a single injection of untargeted or peptides-targeted SL containing-encapsulated DXR (5 mg/kg/mouse). Twenty-four hours after the injection, mice were sacrificed, tumors were collected, and DXR accumulation was visualized by fluorescence microscopy of cryopreserved tissue sections.
Pharmacokinetic and biodistribution experiments
For pharmacokinetic studies, mice were injected via the tail vein with either a single dose or two doses (7 days interval) of peptides-targeted, dual-labeled liposomes, as previously described [18, 23]. At selected time points (2-48 hours) post-injection, mice (three mice/group) were anesthetized and sacrified by cervical dislocation. A blood sample (100 μl) was collected by heart puncture and counted for the [3H]- and [14C]-labels in a Packard beta-counter. Blood correction factors were applied to all samples as reported [23]. Biodistribution was performed in mice orthotopically implanted with NB GI-LI-N cells, as previously reported [23].
In vivo therapeutic studies and tumor imaging
Mice bearing the orthotopic model were treated starting from day 21 after NB cell implant; mice with the pseudo-metastatic model received the first treatment 4 h after NB cell injection. These therapeutic schedules were designed to test the effects of our targeted formulations against both established and pseudo-metastatic preclinical models of human NB, as described [16, 19]. Animals were treated i.v. once a week for 3 weeks with untargeted (SL[DXR]) or peptides-targeted SL[DXR] (5mg/kg). Scrambled peptide-functionalized liposomess were used as a control, and in every experiment a group of control mice received HEPES-buffered saline. Survival times were used as the main criterion for determining treatment efficacy. In the orthotopic model, time-dependent anti-tumor activity was also evaluated by bioluminescence imaging (BLI) and X-ray analyses. For this purpose, the GI-LI-N cell line was infected with a retrovirus expressing the firefly luciferase gene, as previously reported [17]; luciferase activity of retrovirally-transduced cells was visualized in vivo by BLI (IVIS Caliper Life Sciences, Hopkinton, MA) after a 10 min incubation with 150 μg/mL of D-luciferin (Caliper Life Sciences), as described [17]. X-ray analysis was superimposed to the luminescence for a better visualization of the tumors.
Statistics
Results are expressed as mean ± Confidence Intervals (95% CI). All the analyses were performed with Prism 5 software (GraphPad, La Jolla, CA): either Student's t test or two-way analysis of variance (ANOVA) followed by Bonferroni's post-test were used to evaluate differences within treatments; survival curves were drawn as Kaplan-Meier Cumulative Proportion Surviving graphs, and corresponding p-values were calculated by the use of the log-rank (Chi square) test. Asterisks indicate the following p-value ranges: §,* = p<0.05, §§,** = p<0.01, §§§,*** = p<0.001.
Results
Identification of neuroblastoma-targeting peptide ligands by combined in vitro/ex-vivo phage display screenings
We rationalized combinations of in vitro (neuroblastoma (NB) cell lines: GI-LI-N and HTLA-230) and ex-vivo (tissues from healthy animals and from orthotopic NB models) phage display screenings to identify peptide ligands specific for: (i) whole tumor masses [Experiment #1: negative pre-selection on kidney and adrenal gland from healthy mouse, target selection on GI-LI-N-derived primary tumor; Experiment #2: pre-selection on GI-LI-N-derived primary tumor, selection on corresponding pancreatic metastasis] or (ii) tumor microenvironments [Experiment #3: pre-selection on HTLA-230 cells, selection on HTLA-230-derived primary tumor; Experiments #4 and 5: pre-selection on GI-LI-N cells, selection on GI-LI-N-derived primary tumor from a different mouse; Experiment #6: pre-selection on GI-LI-N cells, selection on GI-LI-N-derived metastasis]. We obtained enrichments of target phage binding over the controls in the following selection rounds (values reported in parenthesis): Experiment #1: Round II (3.57x); Experiment #2: Round II (1.85x); Experiment #3: Round III (3.90x); Experiment #4: Round IV (3.47x); Experiment #5: Round IV (1.79x); Experiment #6: Round IV (1.32x). We globally purified 300 NB-binding phage clones, displaying 121 single peptide sequences; of these, 26 were selected for binding to the primary tumor mass (Experiment #1), 15 to the metastatic mass (Experiment #2), 57 (Experiments #3-5) and 23 (Experiment #6) to their respective microenvironments (the corresponding experimental flowchart is reported in Figure 1).
Figure 1. Flowchart of the phage display experiments.
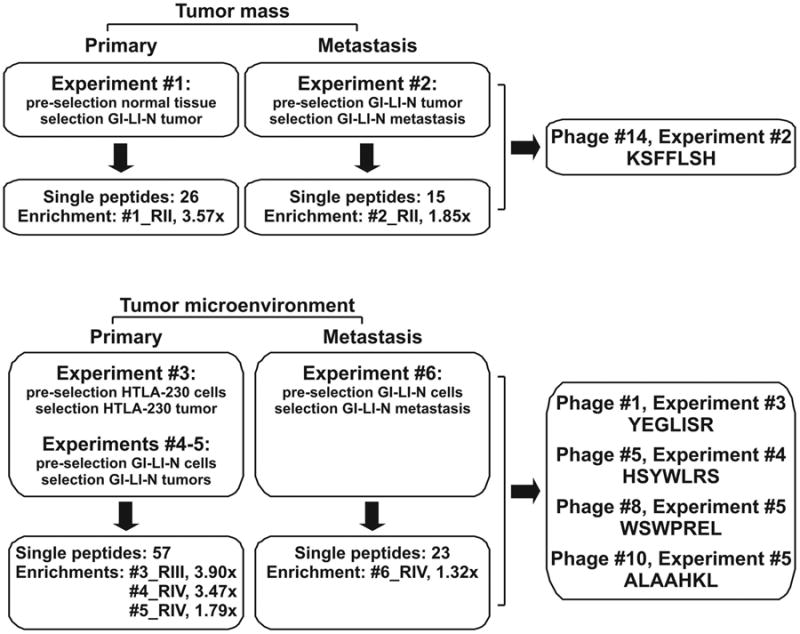
Combinations of in vitro (cell lines) and ex-vivo (tumor tissues) negative/target selection screenings were rationally designed to isolate peptide sequences specific for the whole tumoral mass or for its stromal components.
Ex vivo and in vivo validation of neuroblastoma-targeted phage-displayed peptides
We performed a first validation on a panel of phage clones selected in the different experiments (n=15) by ex-vivo overlay binding assays on tumor biopsies from NB patients. Ten out of the 15 selected phage clones showed weak or no binding (data not shown). However, five phages displayed peptides capable of high specific binding, compared to the background and to the control insertless phage (Fd-Tet), proving to be specific ligands for human NB, namely, KSFFLSH (Phage #14, Experiment #2), YEGLISR (Phage #1, Experiment #3), HSYWLSR (Phage #5, Experiment #4), WSWPREL (Phage #8, Experiment #5), ALAAHKL (Phage #10, Experiment #5) (Figures 1 and 2A). We observed that Phage #1, #5, #8, and #10 preferentially bind to non-epithelial tumor components, while Phage #14 widely recognizes the whole NB masses, as suggested by comparative histological analyses (Mallory Trichrome staining) performed on sections from the same human NB samples. We further validated the binding of these phage clones on tissue sections from mice bearing orthotopic implants of GI-LI-N cells. In this case, the stromal components of tumor masses were further characterized by staining for different specific markers, i.e., von Willebrand factor (FVIII), CD31, α-smooth muscle actin (SMA), and Collagen I (Figure 2B). These assays revealed that mouse (stromal) components are also well-targeted, as supported by a comparison between phage targeting of selected tumor components and staining for the stromal markers, supporting the rationale for in vivo preclinical applications.
Figure 2. Selected phage-displayed peptides target human neuroblastoma from patients and from orthotopically implanted mouse model ex-vivo.
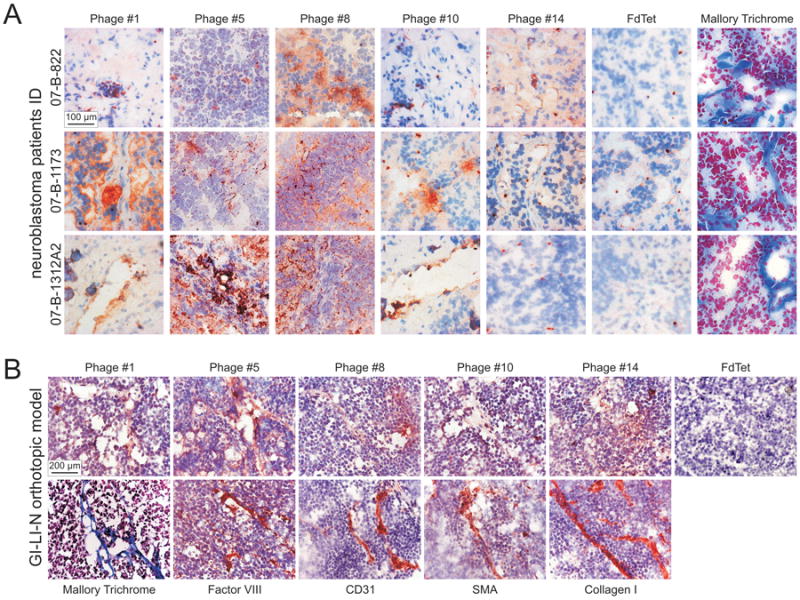
A. The 5 NB-targeting phage clones, and the negative control insertless phage (Fd-Tet) were evaluated for their specificity by phage overlay binding assays on 5-μm frozen sections from patients with stage IV NB (07-B-822, 07-B-1173 and 07-B-1312A2). Tissue-bound phage clones were detected by staining with a specific anti-bacteriophage antibody, followed by incubation with DcEnVision+ System HRP. Tissues were counterstained with hematoxylin. Overall tissue histology was evaluated by Mallory Trichrome staining. Of the 15 phage clones investigated, the 5 validated as NB-targeting (i.e., #1, #5, #8, #10, #14), and the Fd-Tet are shown. B. The 5 NB-targeting clones and the Fd-Tet were also validated by overlay binding assays on tumor derived from orthotopic implants of GI-LI-N cells in athymic mice, with the procedure described above. To identify the different cell types composing tumor mass, adjacent sections were stained with antibodies specific for Factor VIII, CD31, SMA and Collagen I. Sections were counterstained by hematoxylin. Overall tissue histology was evaluated by Mallory Trichrome staining.
To evaluate the capability of the selected phage clones to target tumors in vivo, mice, bearing in the left adrenal gland orthotopic implants of GI-LI-N cells, were injected i.v. with either NB-targeting or control insertless (Fd-Tet) phage, before being perfused and sacrificed. Phage accumulation was revealed by decoration of explanted tissues with an anti-fd bacteriophage antibody, and perivascular cells were evidenced by staining for SMA. As shown in Figure 3A, Fd-Tet did not accumulate in tumor tissues, even after 24 h of circulation; on the contrary, all the targeting phages showed specific homing to NB tumors by 10 minutes after administration. Interestingly, Phage #1, #5 and #10 progressively accumulated into the NB tumors over time, while Phage #8 and #14 were detectable at lower level in tumors explanted after 24 hours (Figure 3A). These data could support the assumption that, although all the selected phage-displayed peptides recognize NB-specific markers, they might have different targeting properties, e.g. target availability or affinity. Importantly, negligible phages accumulation was observed in healthy tissues, such as ipsilateral and controlater kidneys, liver and spleen (Figure 3B).
Figure 3. Neuroblastoma-specific phage clones target an orthotopically implanted mouse model of human NB and spare targeting healthy tissues in in vivo experiments.
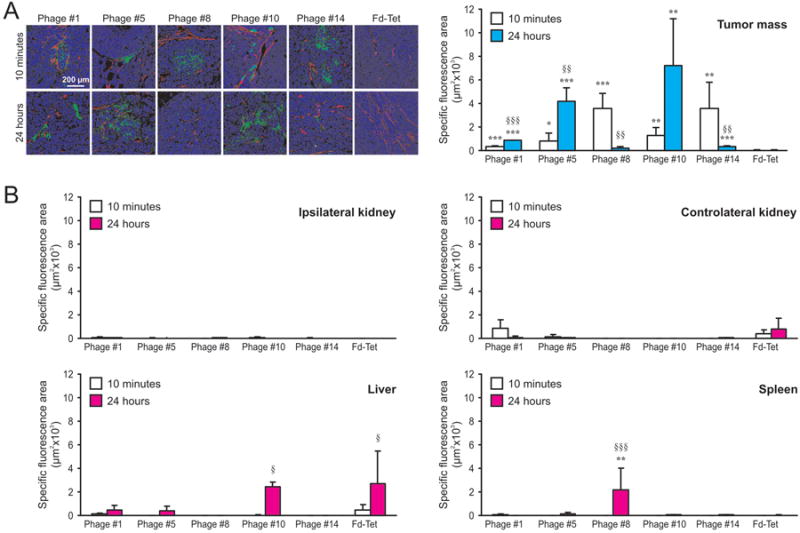
The capability of the NB-targeting phage clones to home to tumors was evaluated after their i.v. administration into mice bearing human GI-LI-N NB-derived orthotopic xenografts (3 animals/group). Insertless Fd-Tet phage was injected as a negative control. After 10 min and 24 h, mice were perfused with complete medium and euthanized. Tumor tissues were explanted, Bouin-fixed, paraffin-embedded and co-stained with anti-fd bacteriophage (green) and anti-SMA-Cy3 (red) antibodies (A). B. Experiments were performed as described in A. Healthy ipsilateral and controlateral kidneys, liver and spleen were explanted, Bouin-fixed, paraffin-embedded and stained with anti-fd bacteriophage antibody. Staining were quantified on 5 confocal images/experimental point by Image J software and presented by histograms. Symbols used to report the statistical analysis: * = peptide-displaying versus insertless phage; § = 24 h versus 10 min phage circulation. *, p<0.05; **, §§, p<0.01; ***, §§§, p<0.001.
Our findings suggest that, due to their substantial presence at the diseased sites at 24 h post-administration, peptides #5 and #10 have the potential to be the best performers for in vivo applications.
Production and characterization of neuroblastoma-targeted drug delivery nanocarriers
As a first step toward the engineering of peptide-based modular nanotherapeutics, we synthesized biotin-conjugated peptides corresponding to the phage-displayed, NB-targeting sequences. The maintenance of their binding properties was tested by in vitro association studies on human NB cell lines (GI-LI-N, HTLA-230 and IMR-32) and on human endothelium (HUVECs). Cells were incubated with 20, -100, and -200 μg/mL of biotin-labeled peptides for 1 h at 4°C, and their binding was evaluated by flow cytometry analysis. Compared to the scrambled (SCR) peptide, all the peptides selectively recognized NB tumor cells and, at lower extent, HUVECs (Figure 4A). Notably, as observed with the in vivo phage homing (compare to Figure 3), peptides corresponding to Phages #5 and #10 showed the highest cellular association.
Figure 4. Specific cell binding and cytotoxicity of neuroblastoma-targeted conjugates.
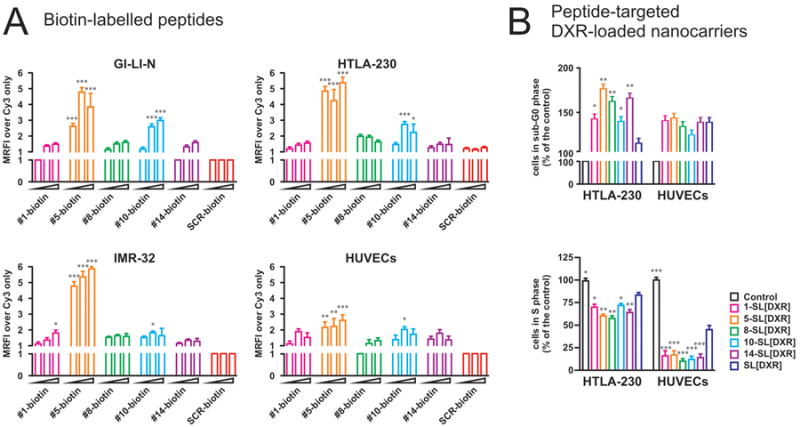
A. Biotin-conjugated peptides were investigated for their capability to bind to NB cell lines (GI-LI-N, HTLA-230 and IMR-32) and to primary endothelial cells (HUVECs). Binding was evaluated by incubation with Cy3-Streptavidin, and was quantified by flow cytometry. A scrambled peptide (SCR) was used as a negative control, and binding is expressed as mean relative fluorescence intensity (MRFI) normalized over cell staining with Cy3-labelled streptavidin (Cy3) only. Experiments were repeated 5 times. Columns depict the fold increase fluorescence intensity of each sample over control, considering 1.00 the level of control; errors bars indicate 95% C.I. *, p<0.05; **, p<0.01; ***, p<0.001 versus SCR-biotin. B. The cytostatic effects of peptide-functionalized liposomes were evaluated in HTLA-230 and HUVECs, by both PI staining (B, upper panel, to quantify cells in sub-G0) and BrdU uptake (B, lower panel, to quantify cells in S phase). Values are expressed as percent of the controls. *, p<0.05; **, p<0.01; ***, p<0.001 versus SL[DXR].
Having demonstrated the NB-targeting properties of the selected peptides, we analyzed whether they could effectively target nanocarriers encapsulating a chemotherapeutic drug, by first producing untargeted and targeted doxorubicin (DXR)-loaded liposomes (SL). Untargeted SL were not functionalized, or functionalized with a scrambled (SCR) peptide and NB-targeted SL were functionalized with peptides #1, #5, #8, #10, and #14. From this point on, these formulations will be reported as SL[DXR], SCR-SL[DXR], 1-SL[DXR], 5-SL[DXR], 8-SL[DXR], 10-SL[DXR], and 14-SL[DXR], respectively. Liposomes were typically 135 ± 10 nm in diameter, with a polydispersity of 0.06±0.01, and a z-potential value of -28±2 mV in water and of -2.4±0.7 mV in PBS. The DXR entrapment efficiency was ∼95%, with an average coupling efficiency of 10 μg peptide/μmol phospholipids (PL).
We first tested the NB-targeted liposomes for their effects against tumor and endothelial cells in vitro. Cells were treated for 1 h with 50 μM of DXR encapsulated in either untargeted (SL[DXR]) or peptide targeted SL before evaluating the percentage of apoptotic/hypodiploid cells. In terms of cell cycle arrest, the percentage of NB cells, i.e., HTLA-230 or GI-LI-N (not shown), in sub-G0 phase was significantly increased, compared with SL[DXR] and SCR-SL[DXR], by incubation with all the targeted formulations (5-SL[DXR], 8-SL[DXR] and 14-SL[DXR] vs SL[DXR] and SCR-SL[DXR]: p<0.01; 1-SL[DXR] and 10-SL[DXR], vs SL[DXR] and SCR-SL[DXR]: p<0.05) (Figure 4B). In contrast, the effect of both targeted and untargeted formulations on HUVECs was similar, although they were statistically significant compared to untreated cells (p<0.05). On the contrary, the evaluation of BrdU uptake showed that all the NB-targeted formulations exerted an enhanced, statistically significant anti-proliferative effect also on HUVECs by decreasing the numbers of cells in S phase, compared to both untargeted and SCR-targeted liposomes (p<0.001) (Figure 4B).
Pharmacokinetic profiles and accelerated blood clearance phenomenon of dual labeled peptide-targeted liposomes
As previously shown, long circulation times are required for small liposomes to gain access to tumor sites [25]. In order to quantify the fate of both the liposome and the drug, pharmacokinetic studies were performed using dual labeled liposomes. Specifically, [3H]-CHOL-labeled peptide-targeted liposomes, loaded with [14C]-labeled DXR were evaluated [26] in nude mice without tumors. The results of the pharmacokinetics are expressed as a percentage of the administered dose of phospholipids (PL) and of the DXR remaining in the blood (Figure 5A). The results clearly indicate that each liposomal formulation coupled to the five different peptides has good stability and long circulation times, with about 15% of both the carrier and the drug remaining in the blood 24 hours after liposome inoculation.
Figure 5. Pharmacokinetic profiles and tumor accumulation of liposomes functionalized with selected peptides in vivo.
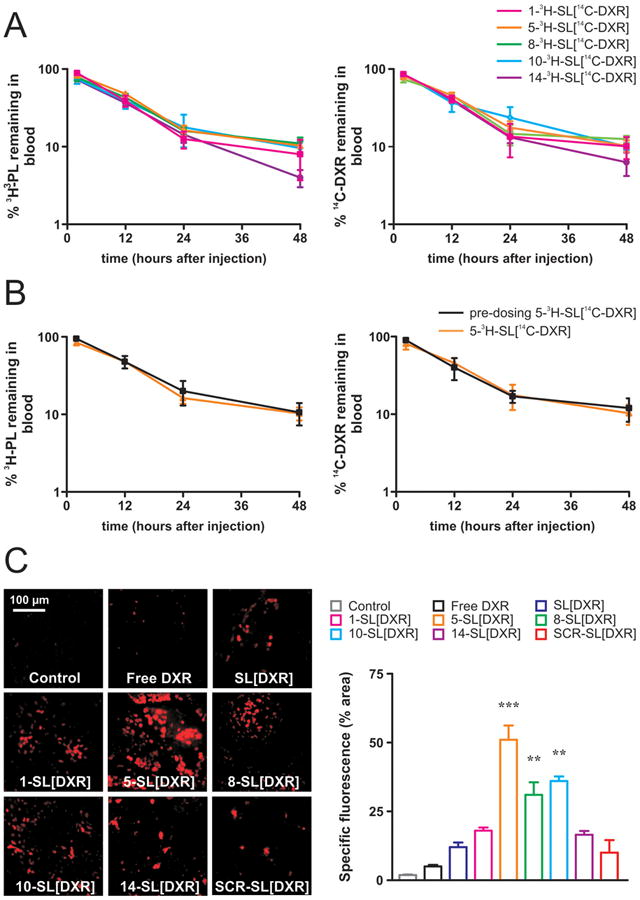
Mice were injected via the tail vein with either a single dose (A; all peptides-targeted liposomes shown) or two doses (pre-dosing, B; 05-SL[DXR] shown) of peptides-targeted, liposomes, dual labeled with the lipid tracer [3H]-CHOL (PL) and the drug tracer [14C]-DXR. At selected time points (2, 12, 24, and 48 hours) post-injection, blood was counted for 3H and 14C labels in a Packard beta-counter. Points, average of three mice; bars, ± SD. PL: phospholipids. C. Mice bearing orthotopic models of human NB were injected with a single bolus of doxorubicin (DXR) (5 mg/kg/mouse), either free (Free DXR), or encapsulated in untargeted (SL[DXR]) and peptide-targeted (1-SL[DXR], 5-SL[DXR], 8-SL[DXR], 10-SL[DXR], 14-SL[DXR]) liposomes. Scrambled peptide-targeted formulation (SCR-SL[DXR]) was used as a further control. Control mice received HEPES-buffered saline (3 animals/group). Tumors were explanted 24 h after treatment, and the presence of DXR-associated fluorescence was evaluated by fluorescence microscopy on 5-μm frozen tissue sections. Specific fluorescence is expressed by percent area of total area acquired. **, p<0.01; ***, p<0.001 versus control, SCR-SL[DXR] and SL[DXR].
Although other investigators reported that the first injection of liposomal DXR fails to produce increased clearance of the second dose [26], we also investigated the impact of the first injection of our novel peptide-targeted, DXR-loaded liposomes on inducing the accelerated blood clearance (ABC) phenomenon. As expected, no enhanced clearance of the second dose (pre-dosing) for both the carrier and the encapsulated drug was observed after the repeated injections of 5-SL [DXR] (Figure 5B) and of all the other peptide-targeted formulations (not shown), confirming that the ABC phenomenon does not occur after repeated injections of our novel targeted liposomal DXR.
Peptide-targeted drug delivery nanocarriers have potentiated therapeutic efficacy
To evaluate the in vivo localization of liposomes-entrapped DXR, we exploited its intrinsic fluorescence as an indicator of the drug amounts that reached the tumors. For these studies, mice bearing orthotopic models of human NB were injected with a single bolus of DXR (5 mg/kg), either free, or encapsulated in untargeted or targeted SL labeled with either scrambled peptide or NB peptide, and tumors were explanted 24 h after treatment. Control mice received saline solution. All the targeted formulations specifically accumulated into the tumor masses (Figure 5C). Significantly increased amounts of DXR were delivered by the liposomes functionalized with peptide #5 (5.1x compared to the SCR-SL[DXR]), #8 (3.1x) and #10 (3.6x). The results of these tumor accumulation studies strongly support the use of 5-SL[DXR], 8-SL[DXR], and 10-SL[DXR], either individually or in combination, for targeted therapeutic approaches against NB.
In a first set of therapeutic experiments we transduced GI-LI-N cells with the reporter gene luciferase before inoculation into the left adrenal glands of athymic mice. Injection of the various DXR formulations started 21 days after cell implant, and tumor growth was monitored by bioluminescence (BLI) once a week five days after each treatment (day 26, 33 and 40). In this experimental setting (Figure 6A), free, as well as untargeted (SL) or scrambled peptide-targeted (SCR-SL) DXR had very weak, if any, therapeutic readout. Conversely, treatment with almost all the targeted formulations resulted in a delay of tumor progression; 5-SL[DXR] and 10-SL[DXR] proved to be the most efficient targeting systems, leading to a strong and long-lasting, statistically significant inhibition of primary tumor growth (day 40, p<0.001 vs SL[DXR]). Also 8-SL[DXR] and 14-SL[DXR] showed a statistically significant tumor growth arrest, but to a lower extent, compared with untargeted liposomes (p<0.05 vs SL[DXR]). The long-lasting anti-tumor response caused by 5-SL[DXR] and 10-SL[DXR] was confirmed by x-ray analysis of NB-bearing mice, performed one month after the end of the treatment (day 65). Figure 6A clearly shows abdominal tumor growth in mice injected with SL[DXR], whereas tumors were barely detectable in both 5-SL[DXR] and 10-SL[DXR] treated mice.
Figure 6. Liposomes functionalized with selected peptides show potentiated therapeutic efficacy.
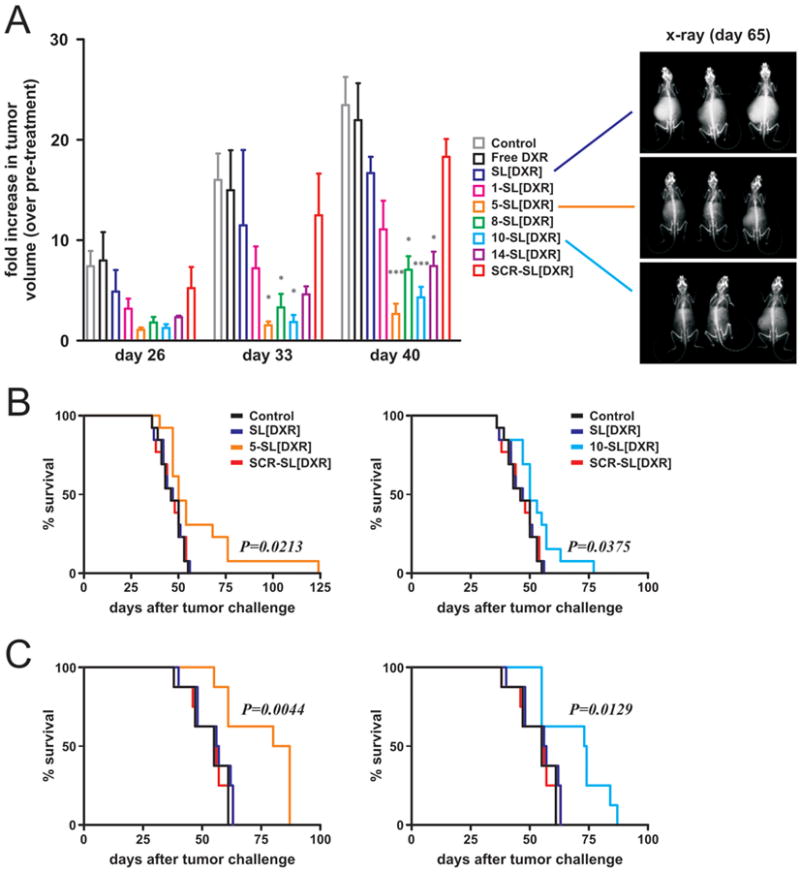
A. Time-dependent anti-tumor effects of DXR-loaded, peptide-targeted nanocarriers evaluated by the use of a luciferase-transfected human NB cell line (GI-LI-N) orthotopically inoculated in animals. Treatment started twenty-one days (T21) after tumor cell injection. Animals (5 animals/group) were treated i.v., once a week for 3 weeks, with 5 mg/kg of DXR free, or encapsulated in either non-targeted or peptide-targeted nanocarriers. SCR peptide-targeted nanocarriers were used as a control. Control mice received HEPES-buffered saline. Tumor growth was monitored by BLI at day 26, 33, and 40 after tumor challenge. Values are reported as fold increase in tumor volume compared to pre-treatment (day 20). Exemplary pictures of x-ray acquisitions one month after the end of treatment (day 65), corresponding to three mice treated with SL[DXR], 5-SL[DXR] and 10-SL[DXR] are shown. B, C. The therapeutic effect of the targeted formulations was evaluated in terms of overall survival in two animal models: HTLA-230 cells inoculated i.v. into the tail vein (pseudo-metastatic model, 13 mice/group, B), GI-LI-N cells implanted into the adrenal gland (orthotopic model, 8 mice/group, C). Statistical analysis: A), p versus SL[DXR]; B, C), p versus control, SL[DXR] and SCR-SL[DXR].
In a second set of experiments, the best candidates among the novel peptide-targeted formulations were tested for their potential to increase the life span of NB-bearing mice. Two aggressive animal models of human NB were used, with cells inoculated either i.v. in the tail vein (HTLA-230 cells, Figure 6B), to mimic the spreading of tumor cells and the minimal residual disease, or orthotopically in the adrenal gland (GI-LI-N cells, Figure 6C), to reflect the growth of advanced NB in children (large adrenal gland tumors and small metastatic lesions). In both models, 5-SL[DXR] and 10-SL[DXR] formulations led to a significant increase in the survival of NB-bearing mice, when compared to control animals or to animals treated with DXR, either free (not shown) or encapsulated in untargeted (SL[DXR]) or scrambled targeted (SCR-SL[DXR]) formulations (Figure 6B-C).
Biodistribution profiles of peptide 5 and 10-targeted liposomes
Biodistribution (BD) experiments of the most promising (in terms of anti-tumor efficacy) peptide 5- and 10-targeted DXR-loaded dual labeled liposomes were performed in orthotopically injected NB-bearing mice. BD profiles were evaluated in the tumor and in the liver, spleen, kidney and heart, with the latter being an important indicator of DXR-related cardiotoxicity (Richardson et al. Blood Rev 1997). Figure 1 supplementary shows a time-dependent tumor uptake of each liposomal formulation, while negligible accumulation was revealed in the heart at all time points, confirming that both 5- and 10-SL[DXR] minimized nonspecific heart uptake. As expected, a time-dependent liver and spleen accumulation of the injected 3H-labeled carrier was seen for each formulation tested. Accordingly with the liposomal stability results showed in the pharmacokinetics profiles (Figure 5A-B), also similar extent of the liposomal loaded-14C-DXR (not shown) were found in each of the analyzed tissues.
Discussion
Here we report the identification of five novel peptide ligands selected by combined in vitro/ex-vivo phage display screenings in preclinical models of human neuroblastoma (NB). We show that corresponding peptide-targeted drug delivery nanocarriers counteract NB progression in biologically relevant animal models, with a substantial improvement in their therapeutic efficacy, compared to both free drug and untargeted systems.
Chemotherapy remains the main treatment for most cancer types; however, there is still no clinically available antineoplastic drug capable of a highly selective action on the tumor mass. Despite some positive hints of anti-cancer selectivity for targeted antibodies and small-molecule inhibitors, the pitfalls of these approaches (low responsiveness, primary and secondary resistance, high costs) are much more limiting than initially expected. Nanotechnology is providing powerful tools for cancer treatment and prevention, but the successful achievement of a reduced toxicity in chemotherapy-driven side effects is not yet satisfactorily correlated with a sustained increase in overall patient survival [27]. For all these reasons, widespread efforts are presently focused on the development of drug-targeting strategies that would enhance the efficacy of therapeutic agents (by increasing local concentration) while reducing their toxic side effects (by decreasing systemic exposure).
In tumors, both epithelial and stromal (e.g., endothelial) cells express surface markers that are accessible from the circulation, but are undetectable, inactive or inaccessible in normal tissues [28, 29]. These markers can be exploited for a selective delivery of anticancer agents to the tumor sites, therefore combining selectivity of the drug nanocarriers with longevity in the bloodstream. Considering the variety of cell types and of signaling pathways involved in the crosstalk between the tumor and its microenvironment, it is reasonable to expect that a multi-target approach would lead to a substantially increased therapeutic efficacy. Screening phage-display peptide libraries on intact cells or tissues allows the identification of specific peptide ligands to be exploited for systemic targeting throughout the circulation [30].
We have recently demonstrated that two phage display-derived peptides, discovered previously in other tumor models, and recognizing separate epitopes of the tumor vasculature, increased the therapeutic effects of chemotherapy against preclinical models of human NB when coupled at the external surface of liposomal formulations [18]. We here report the identification of new peptide sequences obtained by direct screening of phage libraries on NB cells and fresh tumors from NB-bearing mice. To our knowledge, such an approach, aimed at selecting specific NB-targeting moieties, has not been applied before.
We validated 5 novel peptide sequences, namely #1-YEGLISR, #5-HSYWLRS, #8-WSWPREL, #10-ALAAHKL (selected as ligands for NB microenvironment) and #14-KSFFLSH (selected as a ligand for the whole NB tumor mass) (Figure 1), demonstrating their specific recognition of NB tumors in different preclinical settings. The overlay binding assays performed on biopsies from NB patients (Figure 2A) showed that, although Phage #1, #5, #8, and #10 preferentially target NB microenvironment, and while Phage #14 binds NB epithelium, none of these phages univocally associates to specific tumor components. In the case of overlay binding assays on mouse tissues (Figure 2B), the availability of a larger number of samples allowed a further characterization of the NB masses by staining for Factor VIII (endothelial cells, stroma), CD31 (endothelial/hematopoietic cells), SMA (perivascular cells, cancer-associated fibroblasts), and collagen I (stroma, cancer-associated fibroblasts), for comparison with the tissue binding of selected phage. The results of this second panel of overlay assays substantially confirmed the staining patterns observed on human tissues, suggesting that the molecular target(s) of the selected NB-binding peptides might be expressed, although in different amounts, by different cell types. These results are consistent with the fact that relatively low-stringency phage display outputs reflect the non-discrete nature of biomarker distribution in normal/tumor tissues. In other words, with the screening approach described here it is possible to identify ligands not only for receptors that are absent in control and present in target tissues, but also (and more generally) that are just enriched in target tissues.
Globally, our results show that #5-HSYWLRS is the most promising ligand for future developments, in terms of both in vitro binding and in vivo targeting of different models of human NB. Interestingly, this peptide shares similarity with the FF/YXLRS motif, isolated by Karjalainen K. et al [31], and characterized as a specific ligand of neuropilin-1 (NRP-1), despite the lack of the canonical NRP-1-recognition sequences R/KXXR/K and RRXR also identified by phage display [32, 33]. Karjalainen K. and colleagues showed that this sequence is responsible for the enhanced internalization of an apoptosis-inducing moiety into leukemia and lymphoma cells, leading to increased tumor cell killing. Although a deeper investigation is needed to understand whether the flanking regions in #5-HSYWLRS influence its binding capability, we may speculate that this peptide might recognize the NRP-1 receptor expressed in NB tumors [34], thus mediating a specific targeting drug delivery, when coupled at the external surface of doxorubicin (DXR)-loaded liposomes.
We used two aggressive models of human NB to investigate the efficacy of our targeted formulation, mimicking either the spreading of tumor cells and minimal residual disease (i.v. cell inoculation), or the growth of advanced NB in children that appear as large adrenal gland tumors and small metastatic lesions (orthotopic implant into the adrenal gland) [16, 19]. Remarkably, #5-HSYWLRS and #10-ALAAHKL confer to the cognate-targeted formulations a significant therapeutic efficacy in the two preclinical NB models that were tested (Figure 6A-C). This result indicates that either peptide is a valuable targeting moiety for the delivery of encapsulated DXR to circulating tumor cells and established tumor masses. #14-KSFFLSH- and, accordingly with its tumor accumulation ability (Figure 5C), #8-WSWPREL-targeted liposomes were capable of partially arresting orthotopically implanted NB tumors (Figure 6A), without, however, any prolonged survival time showed. These data support the assumption that, although all the selected phage-displayed peptides recognize NB-specific markers, they might have different targeting properties, e.g. target availability or affinity. Conversely, #1-YEGLISR peptide sequence, did not confer to cognate targeted nanocarriers a statistically significant and long lasting therapeutic efficacy, at least at the doses and schedule treatment used. However, considering the overall capability of homing to tumors in our NB murine model all these peptides, alone or in combination, might be exploited for the development of nanotechnology-based molecular imaging systems of NB or used in combined therapeutic setting. Indeed, the multi-targeting approach (theragnostics) to improve, for instance, magnetic resonance imaging of angiogenesis with a liposomal contrast agent, is a promising clinical strategy [35].
In this paper we demonstrated that the enhanced permeability and retention (EPR) effect was very weak on each neuroblastoma animal model used. Our results seem to be in accordance with the recent concept on EPR [36]. EPR heterogeneity effect in different tumors as well as limited experimental data from patients on the effectiveness of this mechanism, seems to hamper the progress in developing drugs using this approach. Our data are also in agreement with our past and recent published findings, in which we showed that nanocarriers were always able to yield increased anti-neuroblastoma effects via an active targeting, when compared to the passive, EPR-driven, anti-tumor efficacy [16, 37].
Despite the absence of long-term survivors in NB-bearing mice treated with single formulations, the identification of various NB peptide ligands opens the way for developing, in the next future, simultaneous or sequential multi-targeting systems for the treatment of NB, as proposed by Ferrari's team for other cancers [38-40]. The use of peptides against different receptors may promote, in principle, synergistic targeting effects, and therefore, might improve the therapeutic response to anticancer nanodrugs. This view is supported by recent findings showing that hitting tumor endothelial and perivascular cells with separate peptide-targeted nanoparticles [18], or tumor cells and tumor microenvironment with the same nanoparticles [41], results in an improvement in the therapeutic activity of anticancer drug-loaded liposomes. Further investigation is necessary to identify the receptors recognized by the peptides discovered in the present work. Undoubtedly, these notions would be of extreme importance, particularly by informing the choice of the optimal treatment schedule in a multi-target therapy setting, thereby avoiding redundancy of targets and increasing the anti-tumor effects.
Finally, the most promising, in terms of anti-tumor efficacy, peptide 5- and 10-targeted DXR-loaded liposomes showed good stability and long circulation times, and showed time-dependent tumor accumulation, while sparing heart uptake. These data confirm the importance of performing phage display screening on preclinical models of human NB, an approach that enabled the finding of more selective tumor-targeting moieties that can be expected to substantially improve current therapeutic regimens, and have the potential of being translated into the clinical practice.
Supplementary Material
{kind=link}
Acknowledgments
Work supported by Associazione Italiana per la Ricerca sul Cancro (My First AIRC Grant, (MFAG) to Pastorino F. and to Marchiò S.), and IG to Ponzoni M.); Fondazione Umberto Veronesi (to Pastorino F.); Italian Ministry of Health, Finanziamento Ricerca Corrente 2010, Ministero Salute (contributo per la ricerca intramurale), Istituto Giannina Gaslini; Banca d'Alba, Piedmont Region (Finalized Health Research Under 40); Piedmont Foundation for Cancer Research (FPRC) Intramural Grant 5×1000 2008 (to Marchiò S.). Loi M. is a recipient of a Fondazione Italiana per la Ricerca sul Cancro (FIRC) fellowship; Di Paolo D. is a recipient of a Fondazione Umberto Veronesi fellowship.
Thank to Murgia D. and Candiano G. for technical assistance, and Biobanca Istituto Giannina Gaslini for the human samples.
Bibliography
- 1.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zage PE, Louis CU, Cohn SL. New aspects of neuroblastoma treatment: ASPHO 2011 symposium review. Pediatr Blood Cancer. 2011;58:1099–1105. doi: 10.1002/pbc.24116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clin Cancer Res. 2012;18:2740–2753. doi: 10.1158/1078-0432.CCR-11-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma-preclinical and clinical advancements. Nat Rev Clin Oncol. 2012;9:391–399. doi: 10.1038/nrclinonc.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carol H, Boehm I, Reynolds CP, Kang MH, Maris JM, Morton CL, Gorlick R, Kolb EA, Keir ST, Wu J, Wozniak AE, Yang Y, Manfredi M, Ecsedy J, Wang J, Neale G, Houghton PJ, Smith MA, Lock RB. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol. 2011;68:1291–1304. doi: 10.1007/s00280-011-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furman WL, McGregor LM, McCarville MB, Onciu M, Davidoff AM, Kovach S, Hawkins D, McPherson V, Houghton PJ, Billups CA, Wu J, Stewart CF, Santana VM. A single-arm pilot phase II study of gefitinib and irinotecan in children with newly diagnosed high-risk neuroblastoma. Invest New Drugs. 2011 doi: 10.1007/s10637-011-9724-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner LM, Danks MK. New therapeutic targets for the treatment of high-risk neuroblastoma. J Cell Biochem. 2009;107:46–57. doi: 10.1002/jcb.22094. [DOI] [PubMed] [Google Scholar]
- 8.Morton CL, Maris JM, Keir ST, Gorlick R, Kolb EA, Billups CA, Wu J, Smith MA, Houghton PJ. Combination testing of cediranib (AZD2171) against childhood cancer models by the pediatric preclinical testing program. Pediatr Blood Cancer. 2011;58:566–571. doi: 10.1002/pbc.23159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verissimo CS, Molenaar JJ, Fitzsimons CP, Vreugdenhil E. Neuroblastoma therapy: what is in the pipeline? Endocr Relat Cancer. 2011;18:R213–231. doi: 10.1530/ERC-11-0251. [DOI] [PubMed] [Google Scholar]
- 10.Koshkaryev A, Sawant R, Deshpande M, Torchilin V. Immunoconjugates and long circulating systems: Origins, current state of the art and future directions. Adv Drug Deliv Rev. 2012 doi: 10.1016/j.addr.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen TM, Cullis PR. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv Drug Deliv Rev. 2012 doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 12.Marchio S, Arap W, Pasqualini R. Targeting the extracellular signature of metastatic colorectal cancers. Expert Opin Ther Targets. 2009;13:363–379. doi: 10.1517/14728220902762910. [DOI] [PubMed] [Google Scholar]
- 13.Corti A, Pastorino F, Curnis F, Arap W, Ponzoni M, Pasqualini R. Targeted drug delivery and penetration into solid tumors. Med Res Rev. 2011 doi: 10.1002/med.20238. [DOI] [PubMed] [Google Scholar]
- 14.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, Ruoslahti E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–727. [PMC free article] [PubMed] [Google Scholar]
- 15.Marchio S, Lahdenranta J, Schlingemann RO, Valdembri D, Wesseling P, Arap MA, Hajitou A, Ozawa MG, Trepel M, Giordano RJ, Nanus DM, Dijkman HB, Oosterwijk E, Sidman RL, Cooper MD, Bussolino F, Pasqualini R, Arap W. Aminopeptidase A is a functional target in angiogenic blood vessels. Cancer Cell. 2004;5:151–162. doi: 10.1016/s1535-6108(04)00025-x. [DOI] [PubMed] [Google Scholar]
- 16.Pastorino F, Brignole C, Marimpietri D, Cilli M, Gambini C, Ribatti D, Longhi R, Allen TM, Corti A, Ponzoni M. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003;63:7400–7409. [PubMed] [Google Scholar]
- 17.Pastorino F, Di Paolo D, Piccardi F, Nico B, Ribatti D, Daga A, Baio G, Neumaier CE, Brignole C, Loi M, Marimpietri D, Pagnan G, Cilli M, Lepekhin EA, Garde SV, Longhi R, Corti A, Allen TM, Wu JJ, Ponzoni M. Enhanced antitumor efficacy of clinical-grade vasculature-targeted liposomal doxorubicin. Clin Cancer Res. 2008;14:7320–7329. doi: 10.1158/1078-0432.CCR-08-0804. [DOI] [PubMed] [Google Scholar]
- 18.Loi M, Marchio S, Becherini P, Di Paolo D, Soster M, Curnis F, Brignole C, Pagnan G, Perri P, Caffa I, Longhi R, Nico B, Bussolino F, Gambini C, Ribatti D, Cilli M, Arap W, Pasqualini R, Allen TM, Corti A, Ponzoni M, Pastorino F. Combined targeting of perivascular and endothelial tumor cells enhances anti-tumor efficacy of liposomal chemotherapy in neuroblastoma. J Control Release. 2010;145:66–73. doi: 10.1016/j.jconrel.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 19.Pastorino F, Brignole C, Marimpietri D, Sapra P, Moase EH, Allen TM, Ponzoni M. Doxorubicin-loaded Fab' fragments of anti-disialoganglioside immunoliposomes selectively inhibit the growth and dissemination of human neuroblastoma in nude mice. Cancer Res. 2003;63:86–92. [PubMed] [Google Scholar]
- 20.Marimpietri D, Brignole C, Nico B, Pastorino F, Pezzolo A, Piccardi F, Cilli M, Di Paolo D, Pagnan G, Longo L, Perri P, Ribatti D, Ponzoni M. Combined therapeutic effects of vinblastine and rapamycin on human neuroblastoma growth, apoptosis, and angiogenesis. Clin Cancer Res. 2007;13:3977–3988. doi: 10.1158/1078-0432.CCR-06-2757. [DOI] [PubMed] [Google Scholar]
- 21.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 22.Smith GP, Scott JK. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993;217:228–257. doi: 10.1016/0076-6879(93)17065-d. [DOI] [PubMed] [Google Scholar]
- 23.Pastorino F, Brignole C, Di Paolo D, Nico B, Pezzolo A, Marimpietri D, Pagnan G, Piccardi F, Cilli M, Longhi R, Ribatti D, Corti A, Allen TM, Ponzoni M. Targeting liposomal chemotherapy via both tumor cell-specific and tumor vasculature-specific ligands potentiates therapeutic efficacy. Cancer Res. 2006;66:10073–10082. doi: 10.1158/0008-5472.CAN-06-2117. [DOI] [PubMed] [Google Scholar]
- 24.Brignole C, Marimpietri D, Pastorino F, Nico B, Di Paolo D, Cioni M, Piccardi F, Cilli M, Pezzolo A, Corrias MV, Pistoia V, Ribatti D, Pagnan G, Ponzoni M. Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J Natl Cancer Inst. 2006;98:1142–1157. doi: 10.1093/jnci/djj309. [DOI] [PubMed] [Google Scholar]
- 25.Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54:987–992. [PubMed] [Google Scholar]
- 26.Ishida T, Atobe K, Wang X, Kiwada H. Accelerated blood clearance of PEGylated liposomes upon repeated injections: effect of doxorubicin-encapsulation and high-dose first injection. J Control Release. 2006;115:251–258. doi: 10.1016/j.jconrel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–1370. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 30.Trepel M, Pasqualini R, Arap W. Chapter 4. Screening phage-display Peptide libraries for vascular targeted peptides. Methods Enzymol. 2008;445:83–106. doi: 10.1016/S0076-6879(08)03004-8. [DOI] [PubMed] [Google Scholar]
- 31.Karjalainen K, Jaalouk DE, Bueso-Ramos CE, Zurita AJ, Kuniyasu A, Eckhardt BL, Marini FC, Lichtiger B, O'Brien S, Kantarjian HM, Cortes JE, Koivunen E, Arap W, Pasqualini R. Targeting neuropilin-1 in human leukemia and lymphoma. Blood. 2011;117:920–927. doi: 10.1182/blood-2010-05-282921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong TM, Chen YL, Wu YY, Yuan A, Chao YC, Chung YC, Wu MH, Yang SC, Pan SH, Shih JY, Chan WK, Yang PC. Targeting neuropilin 1 as an antitumor strategy in lung cancer. Clin Cancer Res. 2007;13:4759–4768. doi: 10.1158/1078-0432.CCR-07-0001. [DOI] [PubMed] [Google Scholar]
- 34.Fakhari M, Pullirsch D, Abraham D, Paya K, Hofbauer R, Holzfeind P, Hofmann M, Aharinejad S. Selective upregulation of vascular endothelial growth factor receptors neuropilin-1 and -2 in human neuroblastoma. Cancer. 2002;94:258–263. doi: 10.1002/cncr.10177. [DOI] [PubMed] [Google Scholar]
- 35.Kluza E, van der Schaft DW, Hautvast PA, Mulder WJ, Mayo KH, Griffioen AW, Strijkers GJ, Nicolay K. Synergistic targeting of alphavbeta3 integrin and galectin-1 with heteromultivalent paramagnetic liposomes for combined MR imaging and treatment of angiogenesis. Nano Lett. 2010;10:52–58. doi: 10.1021/nl902659g. [DOI] [PubMed] [Google Scholar]
- 36.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013;73:2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pastorino F, Di Paolo D, Loi M, Becherini P, Caffa I, Zorzoli A, Marimpietri D, Carosio R, Perri P, Montaldo PG, Brignole C, Pagnan G, Ribatti D, Allen TM, Ponzoni M. Recent advances in targeted anti-vasculature therapy: the neuroblastoma model. Curr Drug Targets. 2009;10:1021–1027. doi: 10.2174/138945009789577954. [DOI] [PubMed] [Google Scholar]
- 38.Blanco E, Sangai T, Hsiao A, Ferrati S, Bai L, Liu X, Meric-Bernstam F, Ferrari M. Multistage delivery of chemotherapeutic nanoparticles for breast cancer treatment. Cancer Lett. 2012 doi: 10.1016/j.canlet.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 39.Blanco E, Hsiao A, Ruiz-Esparza GU, Landry MG, Meric-Bernstam F, Ferrari M. Molecular-targeted nanotherapies in cancer: enabling treatment specificity. Mol Oncol. 2011;5:492–503. doi: 10.1016/j.molonc.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Multistage nanovectors: from concept to novel imaging contrast agents and therapeutics. Acc Chem Res. 2011;44:979–989. doi: 10.1021/ar200077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moura V, Lacerda M, Figueiredo P, Corvo ML, Cruz ME, Soares R, de Lima MC, Simoes S, Moreira JN. Targeted and intracellular triggered delivery of therapeutics to cancer cells and the tumor microenvironment: impact on the treatment of breast cancer. Breast Cancer Res Treat. 2012;133:61–73. doi: 10.1007/s10549-011-1688-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.