

SUMMARY
Understanding of the regulatory mechanisms of gene expression in the control of blood pressure and fluid volume is a key issue in cardiovascular medicine. Guanylyl cyclase/natriuretic peptide receptor-A (GC-A/NPRA) signalling antagonizes the physiological and pathophysiological effects mediated by the renin–angiotensin–aldosterone system (RAAS) in the regulation of cardiovascular homeostasis.
The targeted-disruption of the Npr1 gene (coding for GC-A/PRA) leads to activation of the cardiac RAAS involved in the hypertrophic remodelling process, which influences cardiac size, expression of pro-inflammatory cytokine genes and the behaviour of various hypertrophy marker genes. The Npr1 gene-knockout (Npr1−/−) mice exhibit 35–40 mmHg higher systolic blood pressure and a significantly greater heart weight to bodyweight ratio than wild-type (Npr1+/+) mice.
The expression of both angiotensin-converting enzyme (ACE) and angiotensin II AT1a receptors are significantly increased in hearts from Npr1−/− mice compared with hearts from Npr1+/+ mice. In parallel, the expression of interleukin-6 and tumour necrosis factor-α is also markedly increased in hearts from Npr1−/− mice.
These findings indicate that disruption of NPRA/cGMP signalling leads to augmented expression of the cardiac RAAS in conjunction with pro-inflammatory cytokines in Npr1-null mutant mice, which promotes the development of cardiac hypertrophy and remodelling.
Keywords: angiotensin-converting enzyme, AT1 receptor, atrial natriuretic peptide, cardiac hypertrophy, fibrosis, gene expression, gene knockout, guanylyl cyclase/natriuretic peptide receptor-A, pro-inflammatory cytokines
INTRODUCTION
The cardiac hormones atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) are expressed in high concentrations in the heart, are released into the circulation and can elicit natriuretic, diuretic, vasorelaxant and antiproliferative responses, all directed to the reduction of blood pressure and blood volume.1–6 Conversely, C-type natriuretic peptide (CNP) is found primarily in vascular endothelial cells and the central nervous system.5 Three natriuretic peptide receptor systems have been described.7 Both ANP and BNP bind to guanylyl cyclase-A/natriuretic peptide receptor-A (GC-A/NPRA), which is considered the principal natriuretic peptide hormone receptor that synthesises the intracellular second messenger cGMP.7–11 Previous studies have suggested that ANP/NPRA signalling locally antagonizes cardiac growth responses to hypertrophic stimuli.12 In particular, mice carrying targeted disruption of the Npr1 gene (encoding for NPRA) exhibit hypertension and congestive heart failure, with sudden death occurring in mice after 6 months of age.13–16 Overexpression of NPRA reduces blood pressure and attenuates hypertrophic agonist-induced cardiomyocyte growth.5,17,18 Interestingly, ANP gene delivery has been shown to attenuate cardiac hypertrophy in spontaneously hypertensive rat models.19–21 Nevertheless, the cellular mechanisms by which the ANP/NPRA system blocks cell proliferation and hypertrophic growth is not well understood.
The renin–angiotensin–aldosterone system (RAAS) plays an important role in the endocrine/paracrine regulation of arterial pressure and blood volume. Angiotensin (Ang) II, the main active component of the RAAS, is a potent vasoconstrictor; it increases sympathetic tone and elicits trophic effects in the cardiovascular remodelling process associated with hypertension.22–27 Thus, in addition to its systemic effects, AngII is implicated in the development of cardiac hypertrophy and cardiac fibrosis in humans and in experimental animal models.25,27–30 Most of the effects of AngII in the cardiovascular system are mediated through AT1 receptors.30–32 Treatment with angiotensin-converting enzyme (ACE) inhibitors or AT1 receptor antagonists effectively lowers blood pressure and prevents or ameliorates myocardial hypertrophy.24,25,33 In addition, AngII is considered to be a pro-inflammatory mediator that has a pivotal function in the inflammatory process underlying the development of vascular complications.32,34 Furthermore, AngII has been shown to mediate hypertrophic growth in neonatal and adult myocytes by activating transforming growth factor (TGF)-β and endothelin signalling pathways.35,36
At the cellular and molecular levels, ANP/NPRA signalling has been suggested to inhibit the AngII-mediated induction of protein kinase C and mitogen-activated protein kinsases in vascular smooth muscle cells and mesangial cells.37–39 Conversely, AngII has been shown to modulate and repress Npr1 gene transcription and expression.40–42 In addition, the ANP/NPRA system has been reported to have an anti-inflammatory role, inhibiting tumour necrosis factor (TNF)-α production in interferon-γ-activated macrophages and inhibiting TNF-α-induced adhesion molecule expression in endothelial cells.43,44 The present review focuses on the effect of disruption of the GC-A/NPRA signalling pathway in the activation of cardiac RAAS components and inflammatory mediators in ventricular tissues of hypertrophied hearts of mice lacking NPRA.
SYSTOLIC BLOOD PRESSURE AND DEVELOPMENT OF CARDIAC HYPERTROPHY AND FIBROSIS IN Npr1 GENE-DISRUPTED MICE
The Npr1−/− mutant mice exhibit significantly increased systolic blood pressure (SBP) and an increased heart weight (HW) to bodyweight (BW) ratio compared with Npr1+/+ control mice.16,45 Table 1 lists the SBP, HW/BW ratio and indices of fibrosis in hearts from both Npr1+/+ and Npr1−/− mice. On average, SBP, HW/BW ratio, total collagen and fibrosis are significantly higher in hearts from Npr1−/− mutant mice compared with age-matched wild-type control mice.16 The coronary vessel wall thickness and myocyte cross-sectional area were also significantly increased in mutant mice compared with wild-type mice.13,14,45 Interestingly, the antihypertensive drugs captopril and hydralazine were effective in reducing the elevated SBP in mutant mice.16 However, captopril alone attenuated the HW/BW ratio while significantly reducing fibrosis and collagen content, suggesting the involvement of the RAAS in the hypertrophic growth of mutant mice hearts. These studies also demonstrated a correlation between decreased ventricular cGMP and increased ventricular AngII. Previously it was shown that Npr1−/− mice have cardiac hypertrophy disproportionate to their increased blood pressure, suggesting that the ANP/NPRA system exerts a local antihypertrophic effect on cardiac cells.46 Furthermore, mouse pups lacking NPRA showed a significant increase in heart size compared with age-matched wild-type control pups as early as 16–30 days after birth, supporting the notion that blood pressure alone is not likely to be the primary cause for the development of cardiac hypertrophy in Npr1−/− mutant mice.13,47,48 In a previous study, the ACE inhibitor captopril was shown to reduce blood pressure and decrease the HW/BW ratio in Npr1−/− mice.16 However, another study47 reported that enalapril was able to reduce blood pressure only and not the HW/BW ratio in Npr1−/− mice. We believe that the discrepancy between these two studies may be due to the different methodologies used for drug delivery in Npr1 mice.16,47
Table 1.
Systolic blood pressure, heart rate, heart weight to bodyweight ratio, cardiac collagen content, cGMP, angiotensin II, Thiobarbituric acid-reactive substances and fibrosis in Npr1+/+ and Npr1−/− mice treated with and without antihypertensive drugs
| Untreated | Hydralazine | Captopril | ||||
|---|---|---|---|---|---|---|
| Npr1+/+ | Npr1−/− | Npr1+/+ | Npr1−/− | Npr1+/+ | Npr−/− | |
| SBP (mmHg) | 100 ± 4 | 130 ± 8*** | 97 ± 3 | 110 ± 6**†† | 98 ± 4 | 106 ± 5**†† |
| HW/BW ratio | 4.6 ± 0.3 | 7.5 ± 0.5* | 4.7 ± 0.4 | 7.1 ± 0.7* | 4.4 ± 0.2 | 5.3 ± 0.7*† |
| cGMP (pmol/mg protein) | 26 ± 5 | 5 ± 1*** | 25 ± 3 | 6 ± 1** | 28 ± 6 | 10 ± 2† |
| AngII (pmol/mg protein) | 15 ± 2 | 32 ± 2*** | 14 ± 1 | 28 ± 3** | 12 ± 1 | 17 ± 2†† |
| TBARS (nmol MDA/mg protein) | 25 ± 3 | 38 ± 3*** | 23 ± 2 | 32 ± 2* | 22 ± 2 | 23 ± 2†† |
| W/L ratio | 0.10 ± 0.02 | 0.25 ± 0.07*** | 0.10 ± 0.03 | 0.2 ± 0.5** | 0.1 ± 0.3 | 0.13 ± 0.03†† |
| MCA (μm2) | 415 ± 22 | 915 ± 73*** | 413 ± 18 | 825 ± 34** | 409 ± 16 | 562 ± 82*†† |
| Collagen (mg/g tissue) | 1.4 ± 0.1 | 3.5 ± 0.7*** | 1.3 ± 0.3 | 2.4 ± 0.2** | 1.2 ± 0.3 | 1.6 ± 0.4**†† |
| Fibrosis (%) | 0.8 ± 0.3 | 25 ± 3*** | 0.9 ± 0.3 | 24 ± 2** | 1.0 ± 0.2 | 7.6 ± 0.9**†† |
To examine the local effect of the ANP/NPRA system in counteracting the effect of AngII in vivo, conditional cardiomyocyte (CM)-restricted disruption of Npr1 (CM-Npr1−/−) mice have also been used.49 In that study, the authors reported that the hypertensive responses of CM-Npr1−/− mice to exogenous AngII were not altered; however, AngII-induced cardiac hypertrophy and fibrosis were greatly enhanced. Their findings also indicated that ANP/NPRA signalling can moderate cardiac events independent of its blood pressure-lowering effects, even though the higher basal blood pressure and higher left ventricular weight themselves contribute towards the increased susceptibility to heart failure in Npr1−/− mice. In ANP gene-knockout (−/−) mice with volume overload due to an aortocaval fistula, higher ventricular weight and wall thickness were observed compared with control ANP wild-type (+/+) mice;50 despite this, blood pressure did not differ significantly, suggesting that ANP can exert an antihypertrophic effect independent of blood pressure.50 Similarly, a Npr1 gene-knockout aortocaval fistula mouse model produced a greater degree of congestive heart failure and impaired haemodynamics with increased left and right ventricular weights compared with sham-operated control mice.51 Those authors suggested that, in the absence of NPRA signalling, volume overload induced a greater degree of heart failure.
GENE EXPRESSION AND ROLES OF ACE AND AT1 RECEPTORS IN BLOOD PRESSURE REGULATION AND CARDIAC REMODELING IN Npr1 GENE-DISRUPTED MICE
The ventricular expression of ACE and AT1a were found to be up-regulated by almost three- to fourfold in hearts from Npr1-null mutant mice compared with hearts from wild-type mice.16 Surprisingly, both AngII content and AT1a mRNA expression were found to be increased in hearts from mutant mice, further suggesting that RAAS components were significantly activated in the absence of Npr1−/− signalling. Accordingly, the increased AngII levels in hearts from mutant mice were found to be negatively correlated with ventricular cGMP levels, further supporting the finding that the ANP/NPRA signalling system antagonizes RAAS signalling in a tissue-specific manner.45,52 Although systemic and renal AngII levels are increased in newborn mouse pups lacking NPRA, both circulating and intrarenal AngII and renin levels were reduced in adult Npr1-null mutant mice compared with age-matched Npr1+/+ mice.52 It has been suggested that the possible cause for the reduction in circulating and renal AngII and renin levels may be the increased arterial pressure, which would lead to baroreceptor activation and reflex neural inhibition of renin release in the absence of a functional Npr1 gene in adult mice.4,45,52 It is implied that the cardiac RAAS is activated in hearts from adult mutant mice, even though the circulating levels of AngII are decreased, further supporting previous studies indicating that the cardiac RAAS can be activated independently of and dissociated from the circulating RAAS.53,54 The evidence also suggests that tissue levels of RAAS components are greatly increased in contrast with circulating levels of RAAS components in AngII-infused hypertensive rat models with elevated arterial pressure.55
It has been demonstrated that all the components of the RAAS can be synthesised in local tissues and that locally produced AngII can serve as a paracrine and/or autocrine growth stimulatory factor.52,55–57 It has also been shown that inhibition of plasma renin activity with salt overload does not affect ventricular remodelling after myocardial infarction in rats; however, the local cardiac RAAS is activated, which plays a predominant role in local adaptation of the heart after myocardial infarction.58 Our studies have established that the cardiac RAAS is greatly activated and plays a critical role in the ventricular remodelling process in mice lacking NPRA.45
In another mouse model lacking NPRA, blockade of the AT1 receptor reduced the cardiac hypertrophy and fibrosis to a great extent, suggesting a role for AT1 receptor function in cardiac remodelling in Npr1−/− mice.59 Interestingly, coactivation of ACE and AT1a expression has been reported in ventricular tissue of NG-nitro-L-arginine methyl ester (L-NAME)-induced hypertensive rat models.33 Treatment with ACE or AT1 receptor antagonists reduced the inflammatory phenotype in the arterial vessel wall of L-NAME-induced hypertensive rat models.33,60 Previous studies have demonstrated that treatment with captopril, but not hydralazine, significantly attenuates indices of cardiac hypertrophy and coronary vessel wall thickening in Npr1−/− mutant mice (Table 1).
The increased expression of both ACE and AT1a receptor mRNA in hearts of Npr1−/− mice was significantly reduced by captopril, but not hydralazine, treatment compared with wild-type mice (Fig. 1a–d). Increased expression of AT1a (Fig. 2a,e) and nuclear factor (NF)-κB (Fig. 2b,f) immunoreactivity was also evident in the vascular tissue of hearts from Npr1−/− mutant mice compared with hearts from wild-type mice. Masson’s trichrome staining showed increased perivascular fibrosis in the vascular tissue of hearts from mutant mice compared with hearts from wild-type mice (Fig. 2c,g). Toluidine blue staining of infiltrating mast/monocyte cells showed no significant increase in the number of these cells in the arteries of Npr1−/− compared with Npr1+/+ mice (Fig. 2d,h). Figure 2i shows the average mast/monocyte positive cell infiltration per section in mutant and wild-type mice. It was concluded that Npr1−/− mutant mice hearts showed no significant increase in the infiltration of mast/monocyte cells compared with wild-type mice hearts.16 However, blockade of AngII seems to contribute to improvement in the cardiac remodelling and inflammatory process in Npr1−/− mice. Because both captopril and hydralazine equally attenuate SBP, but only captopril significantly decreases the HW/BW ratio, it is implied that AngII elicits cardiac remodelling in a blood pressure-independent manner.16 Similarly, the mineralocorticoid receptor blocker eplerenone has been shown to attenuate cardiac remodelling in Npr1−/− mice in a blood pressure-independent manner.61 In another model of Npr1-null mutant mice in which both Npr1 and AT1 receptor genes are ablated, the HW/BW ratio and levels of fibrosis were similar to those of wild-type mice and it was suggested that cardiac remodelling in Npr1−/− mice occurs in an AT1 receptor-dependent manner.59
Fig. 1.

Expression profiles of (a,b) angiotensin-converting enzyme (ACE) and (c,d) angiotensin AT1a receptor genes in hearts from Npr1+/+ and Npr1−/− mice. (a,c) Representative reverse transcription–polymerase chain reaction (RT-PCR) mRNA expression analysis of ACE and AT1a receptor expression in Npr1+/+ and Npr1−/− mice with and without antihypertensive drug treatment. UT, untreated; HYZ, hydralazine; CAP, captopril. (b,d) Densitometry analysis of mRNA transcripts normalized against the expression of GAPDH. The RT-PCR was performed using gene-specific primers. Values are expressed as the mean ± SEM (n = 8/group). ***P < 0.001. Reproduced with permission from Vellaichamy et al.16
Fig. 2.
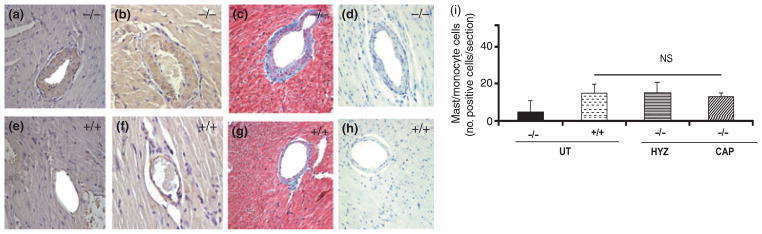
Immunohistochemical analysis of angiotensin AT1a receptor and nuclear factor (NF)-κB protein expression in coronary vessels of hearts from Npr1+/+ and Npr1−/− mice. (a,e) Increased AT1a receptor protein expression, (b,f) NF-κB protein expression, (c,g) perivascular fibrosis and (d,h) the number of inflammatory mast/monocyte infiltrating cells in coronary vessels of hearts from Npr1−/− (a–d) and Npr1+/+ (e–h) mice. (i) Mast/monocyte positive cells infiltration/section. Values are expressed as the mean ± SEM (n = 8). UT, untreated; HYZ, hydralazine; CAP, captopril. Reproduced with permission from Vellaichamy et al.16
CARDIAC REMODELLING AND VENTRICULAR CYTOKINE GENE EXPRESSION IN Npr1 GENE-DISRUPTED MICE
Previous findings have shown that disruption of ANP/NPRA signalling results in augmented expression of both circulating and ventricular cytokines (interleukin (IL)-2, IL-6 and TNF-α), suggesting that activation of the inflammatory process in vascular and ventricular tissues of hearts from Npr1−/− mutant mice is correlated with enhanced cardiac fibrosis and hypertrophic growth, independent of blood pressure.13,16 It has been proposed that pro-inflammatory cytokines such as IL-6 and TNF-α are important mediators in the development of cardiac hypertrophy and heart failure.62–66 Cytokines have been shown to be expressed within the heart in response to either mechanical overload or ischaemic injury.64,66–68 In hearts from Npr1−/− mice, significant upregulation was observed in the ventricular expression of cytokine genes compared with hearts from age-matched wild-type mice.16 Parallel increases of almost fourfold in plasma levels of IL-6 and TNF-α occurred in Npr1−/− mice. The mechanisms underlying the stimulation of ventricular and plasma concentrations of inflammatory markers are not well understood. Participation of early gene activation has been proposed, with progressive mechanical stress associated with hypertension and cardiovascular events.13 In previous studies, the reduced expression of inflammatory markers was more pronounced in captopril-treated Npr1−/− mice than in hydralazine-treated mice,16 suggesting that haemodynamic overload may not to be the sole mechanism accounting for improvement in the inflammatory process in these hearts.
We have reported that a significant increase occurrs in NF-κB levels and inhibitory κB kinase-β (IKK-β) activity in hearts from Npr1−/− mice compared with Npr1+/+ mice, suggesting that the NF-κB signalling pathway is activated in the mutant mouse heart.13,16 The increased NF-κB binding activity was positively correlated with increased expression of cytokine genes in hearts from Npr1-null mice. Recent studies by several groups have implicated the activation of NF-κB as a causal event in cardiac hypertrophy.69–71 We have also shown that ventricular levels of thiobarbituric acid-reactive substances (TBARS) are increased in hearts from Npr1-null mutant mice compared with hearts from wild-type mice, indicating an increase in overall oxidative stress.16 The increased levels of TBARS, an end-product of lipid peroxidation, have been considered as a potential marker for oxidative stress in the pathophysiology of many forms of hypertension.72,73 Furthermore, treatment with free radical-scavenging agents such as tempol (4-hydroxy tetramethylpiperidine-1-oxyl) significantly ameliorates oxidative stress, blood pressure and hypertrophic growth in different hypertensive models.74 It has been reported that AngII functions a strong activator of reactive oxygen species and NF-κB, mediated by its binding to the AT1a receptor in vivo and in vitro.30,75,76
Conversely, it has been shown that the ANP/NPRA system attenuates the production of inflammatory mediators such as TNF-α and IL-6 by regulating the NF-κB pathway.44,77 Furthermore, a recent study demonstrated that augmentation of the second messenger cGMP via chronic inhibition of cGMP-specific phosphodiesterase 5A attenuates load-induced hypertrophy, fibrosis and myocardial dysfunction independent of changes in load, suggesting that cGMP locally antagonizes hypertrophic mediators and signalling pathways.78 Immunohistochemical analyses have demonstrated increased expression of AT1 receptors and NF-κB in the vascular wall of hearts from Npr1−/− mutant mice, suggesting that vascular wall remodelling and medial thickening play a crucial role in the development of hypertrophy in Npr1−/− mutant mice.16 Figure 3 shows the signalling pathway proposed to be involved in the cardiac remodelling process in the absence of NPRA/cGMP signalling. Previous findings have implicated disruption of ANP/NPRA/cGMP signalling leading to increased activation of the RAAS and pro-inflammatory cytokines, in turn leading to medial thickening and perivascular fibrosis. These changes promote cardiac remodelling, hypertrophy and heart failure.13,16 Together, those studies have clearly demonstrated that NF-κB signalling is critically involved in the activation of inflammatory cytokine gene expression in the vascular and ventricular tissues of hearts from Npr1−/− mutant mice.
Fig. 3.

Diagram of the proposed signalling pathway involved in the cardiac remodelling process in the absence of natriuretic peptide receptor-A (NPRA)/cGMP signalling. Disruption of atrial natriuretic peptide (ANP)/NPRA signalling leads to increased activation of the renin–angiotensin system (RAS). Activation of the RAS has been shown to activate increased generation of reactive oxygen species (ROS). This leads to activation and dissociation of nuclear factor (NF)-kB and activator protein (AP)-1 transcription factor subunits from the complex in the cytoplasm and translocation into the nucleus, which activates pro-inflammatory cytokine genes, leading to medial thickening and perivascular fibrosis, in turn promoting cardiac remodelling, hypertrophy and heart failure. KHD, kinase homology domain; GC, guanylate cyclase; AngII, angiotensin II. Reproduced with permission from Vellaichamy et al.16
CONCLUSION AND PERSPECTIVES
In conclusion, the evidence suggests that disruption of the ANP/NPRA signalling pathway results in increased blood pressure and cardiac hypertrophy associated with augmented expression of cytokines, such as IL-6 and TNF-α. Furthermore, both antihypertensive drugs captopril and hydralazine are effective in reducing the elevated SBP, but captopril alone attenuated fibrosis and collagen content, implicating direct involvement of RAAS components in hypertrophic growth of Npr1-null mice, independent of increased blood pressure. Interestingly, captopril treatment also reduced the augmented expression of cytokines in Npr1−/− mice, suggesting that AngII is involved in the activation of cardiac inflammatory mediators that may be associated with pathways leading to cardiac hypertrophy and congestive heart failure. Indeed, future studies are needed to explore molecular targets of natriuretic peptides and their receptor systems in the treatment, diagnosis and prevention of high blood pressure and cardiovascular events.
Acknowledgments
The authors extend special thanks to Dr Bharat B Aggarwal (Department of Experimental Therapeutics and Cytokine Research Laboratory, MD Anderson Cancer Center, Houston, TX, USA) and Dr Susan L Hamilton (Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX, USA) for providing their facilities at our disposal during the period of our displacement due to Hurricane Katrina. The authors’ research described herein was supported by grants from the National Institutes of Health (HL-57531 and HL-62147).
List of abbreviations
- ACE
Angiotensin-converting enzyme
- AngII
Angiotensin II
- ANP
Atrial natriuretic peptide
- BNP
B-Type natriuretic peptide
- CNP
C-Type natriuretic peptide
- GC-A/NPRA
Guanylyl cyclase-A/natriuretic peptide receptor-A
- IKK-β
Inhibitory κB kinase-β
- IL
Interleukin
- L-NAME
NG-Nitro-L-arginine methyl ester
- NF-κB
Nuclear factor-κB
- RAAS
Renin–angiotensin–aldosterone system
- SBP
Systolic blood pressure
- TBARS
Thiobarbituric acid-reactive substances
- TNF-α
Tumour necrosis factor-α
References
- 1.de Bold AJ. Atrial natriuretic factor: A hormone produced by the heart. Science. 1985;230:767–70. doi: 10.1126/science.2932797. [DOI] [PubMed] [Google Scholar]
- 2.Garbers DL. Guanylyl cyclase receptors and their endocrine, paracrine, and autocrine ligands. Cell. 1992;71:1–4. doi: 10.1016/0092-8674(92)90258-e. [DOI] [PubMed] [Google Scholar]
- 3.Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med. 1998;339:321–8. doi: 10.1056/NEJM199807303390507. [DOI] [PubMed] [Google Scholar]
- 4.Shi SJ, Vellaichamy E, Chin SY, Smithies O, Navar LG, Pandey KN. Natriuretic peptide receptor A mediates renal sodium excretory responses to blood volume expansion. Am J Physiol Renal Physiol. 2003;285:F694–702. doi: 10.1152/ajprenal.00097.2003. [DOI] [PubMed] [Google Scholar]
- 5.Pandey KN. Emerging roles of antriuretic peptides and their receptors in pathophysiology of hypertension and cardiovascular regulation. J Am Soc Hypertens. 2008;2:210–26. doi: 10.1016/j.jash.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubattu S, Bigatti G, Evangelista A, et al. Association of atrial natriuretic peptide and type A natriuretic peptide receptor gene polymorphisms with left ventricular mass in human essential hypertension. J Am Coll Cardiol. 2006;48:499–505. doi: 10.1016/j.jacc.2005.12.081. [DOI] [PubMed] [Google Scholar]
- 7.Pandey KN. Biology of natriuretic peptides and their receptors. Peptides. 2005;26:901–32. doi: 10.1016/j.peptides.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 8.Drewett JG, Garbers DL. The family of guanylyl cyclase receptors and their ligands. Endocr Rev. 1994;15:135–62. doi: 10.1210/edrv-15-2-135. [DOI] [PubMed] [Google Scholar]
- 9.Pandey KN, Pavlou SN, Inagami T. Identification and characterization of three distinct atrial natriuretic factor receptors. Evidence for tissue-specific heterogeneity of receptor subtypes in vascular smooth muscle, kidney tubular epithelium, and Leydig tumor cells by ligand binding, photo-affinity labeling, and tryptic proteolysis. J Biol Chem. 1988;263:13406–13. [PubMed] [Google Scholar]
- 10.Pandey KN, Singh S. Molecular cloning and expression of murine guanylate cyclase/atrial natriuretic factor receptor cDNA. J Biol Chem. 1990;265:12342–8. [PubMed] [Google Scholar]
- 11.Pandey KN, Nguyen HT, Sharma GD, Shi SJ, Kriegel AM. Ligand-regulated internalization, trafficking, and down-regulation of guanylyl cyclase/atrial natriuretic peptide receptor-A in human embryonic kidney 293 cells. J Biol Chem. 2002;277:4618–27. doi: 10.1074/jbc.M106436200. [DOI] [PubMed] [Google Scholar]
- 12.Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest. 1998;101:812–18. doi: 10.1172/JCI119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J Biol Chem. 2005;280:19230–42. doi: 10.1074/jbc.M411373200. [DOI] [PubMed] [Google Scholar]
- 14.Oliver PM, Fox JE, Kim R, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci USA. 1997;94:14730–5. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellmers LJ, Scott NJ, Piuhola J, et al. Npr1-regulated gene pathways contributing to cardiac hypertrophy and fibrosis. J Mol Endocrinol. 2007;38:245–57. doi: 10.1677/jme.1.02138. [DOI] [PubMed] [Google Scholar]
- 16.Vellaichamy E, Zhao D, Somanna N, Pandey KN. Genetic disruption of guanylyl cyclase/natriuretic peptide receptor-A upregulates ACE and AT1 receptor gene expression and signaling: Role in cardiac hypertrophy. Physiol Genomics. 2007;31:193–202. doi: 10.1152/physiolgenomics.00079.2007. [DOI] [PubMed] [Google Scholar]
- 17.Oliver PM, John SW, Purdy KE, et al. Natriuretic peptide receptor 1 expression influences blood pressures of mice in a dose-dependent manner. Proc Natl Acad Sci USA. 1998;95:2547–51. doi: 10.1073/pnas.95.5.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zahabi A, Picard S, Fortin N, Reudelhuber TL, Deschepper CF. Expression of constitutively active guanylate cyclase in cardiomyocytes inhibits the hypertrophic effects of isoproterenol and aortic constriction on mouse hearts. J Biol Chem. 2003;278:47694–9. doi: 10.1074/jbc.M309661200. [DOI] [PubMed] [Google Scholar]
- 19.Klinger JR, Petit RD, Warburton RR, Wrenn DS, Arnal F, Hill NS. Neutral endopeptidase inhibition attenuates development of hypoxic pulmonary hypertension in rats. J Appl Physiol. 1993;75:1615–23. doi: 10.1152/jappl.1993.75.4.1615. [DOI] [PubMed] [Google Scholar]
- 20.Louzier V, Eddahibi S, Raffestin B, et al. Adenovirus-mediated atrial natriuretic protein expression in the lung protects rats from hypoxia-induced pulmonary hypertension. Hum Gene Ther. 2001;12:503–13. doi: 10.1089/104303401300042401. [DOI] [PubMed] [Google Scholar]
- 21.Lin KF, Chao J, Chao L. Atrial natriuretic peptide gene delivery attenuates hypertension, cardiac hypertrophy, and renal injury in salt-sensitive rats. Hum Gene Ther. 1998;9:1429–38. doi: 10.1089/hum.1998.9.10-1429. [DOI] [PubMed] [Google Scholar]
- 22.Baker KM, Booz GW, Dostal DE. Cardiac actions of angiotensin II. Role of an intracardiac renin–angiotensin system. Annu Rev Physiol. 1992;54:227–41. doi: 10.1146/annurev.ph.54.030192.001303. [DOI] [PubMed] [Google Scholar]
- 23.Morgan HE, Baker KM. Cardiac hypertrophy. Mechanical, neural, and endocrine dependence. Circulation. 1991;83:13–25. doi: 10.1161/01.cir.83.1.13. [DOI] [PubMed] [Google Scholar]
- 24.Pfeffer JM, Fischer TA, Pfeffer MA. Angiotensin-converting enzyme inhibition and ventricular remodeling after myocardial infarction. Annu Rev Physiol. 1995;57:805–26. doi: 10.1146/annurev.ph.57.030195.004105. [DOI] [PubMed] [Google Scholar]
- 25.Pfeffer JM, Pfeffer MA, Mirsky I, Braunwald E. Regression of left ventricular hypertrophy and prevention of left ventricular dysfunction by captopril in the spontaneously hypertensive rat. Proc Natl Acad Sci USA. 1982;79:3310–14. doi: 10.1073/pnas.79.10.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev. 2000;52:11–34. [PubMed] [Google Scholar]
- 27.Burnier M, Zanchi A. Blockade of the renin–angiotensin–aldosterone system: A key therapeutic strategy to reduce renal and cardiovascular events in patients with diabetes. J Hypertens. 2006;24:11–25. doi: 10.1097/01.hjh.0000191244.91314.9d. [DOI] [PubMed] [Google Scholar]
- 28.Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–71. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 29.Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angioten-sin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–84. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- 30.Weber KT. Extracellular matrix remodeling in heart failure: A role for de novo angiotensin II generation. Circulation. 1997;96:4065–82. doi: 10.1161/01.cir.96.11.4065. [DOI] [PubMed] [Google Scholar]
- 31.Timmermans PB, Wong PC, Chiu AT, et al. Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev. 1993;45:205–51. [PubMed] [Google Scholar]
- 32.Usui M, Egashira K, Tomita H, et al. Important role of local angiotensin II activity mediated via type 1 receptor in the pathogenesis of cardiovascular inflammatory changes induced by chronic blockade of nitric oxide synthesis in rats. Circulation. 2000;101:305–10. doi: 10.1161/01.cir.101.3.305. [DOI] [PubMed] [Google Scholar]
- 33.Takemoto M, Egashira K, Tomita H, et al. Chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade: Effects on cardiovascular remodeling in rats induced by the long-term blockade of nitric oxide synthesis. Hypertension. 1997;30:1621–7. doi: 10.1161/01.hyp.30.6.1621. [DOI] [PubMed] [Google Scholar]
- 34.Sun Y, Zhang J, Lu L, Bedigian MP, Robinson AD, Weber KT. Tissue angiotensin II in the regulation of inflammatory and fibrogenic components of repair in the rat heart. J Lab Clin Med. 2004;143:41–51. doi: 10.1016/j.lab.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Sano M, Fukuda K, Kodama H, et al. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem. 2000;275:29717–23. doi: 10.1074/jbc.M003128200. [DOI] [PubMed] [Google Scholar]
- 36.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–63. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 37.Pandey KN, Nguyen HT, Li M, Boyle JW. Natriuretic peptide receptor-A negatively regulates mitogen-activated protein kinase and proliferation of mesangial cells: Role of cGMP-dependent protein kinase. Biochem Biophys Res Commun. 2000;271:374–9. doi: 10.1006/bbrc.2000.2627. [DOI] [PubMed] [Google Scholar]
- 38.Kumar R, Cartledge WA, Lincoln TM, Pandey KN. Expression of guanylyl cyclase-A/atrial natriuretic peptide receptor blocks the activation of protein kinase C in vascular smooth muscle cells. Role of cGMP and cGMP-dependent protein kinase. Hypertension. 1997;29:414–21. doi: 10.1161/01.hyp.29.1.414. [DOI] [PubMed] [Google Scholar]
- 39.de Arriba G, Barrio V, Olivera A, Rodriguez-Puyol D, Lopez-Novoa JM. Atrial natriuretic peptide inhibits angiotensin II-induced contraction of isolated glomeruli and cultured glomerular mesangial cells of rats: The role of calcium. J Lab Clin Med. 1988;111:466–74. [PubMed] [Google Scholar]
- 40.Garg R, Pandey KN. Regulation of guanylyl cyclase/natriuretic peptide receptor-A gene expression. Peptides. 2005;26:1009–23. doi: 10.1016/j.peptides.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 41.Arise KK, Pandey KN. Inhibition and down-regulation of gene transcription and guanylyl cyclase activity of NPRA by angiotensin II involving protein kinase C. Biochem Biophys Res Commun. 2006;349:131–5. doi: 10.1016/j.bbrc.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Garg R, Pandey KN. Angiotensin II-mediated negative regulation of Npr1 promoter activity and gene transcription. Hypertension. 2003;41:730–6. doi: 10.1161/01.HYP.0000051890.68573.94. [DOI] [PubMed] [Google Scholar]
- 43.Vollmar AM. The role of atrial natriuretic peptide in the immune system. Peptides. 2005;26:1086–94. doi: 10.1016/j.peptides.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 44.Tsukagoshi H, Shimizu Y, Kawata T, et al. Atrial natriuretic peptide inhibits tumor necrosis factor-alpha production by interferon-gamma-activated macrophages via suppression of p38 mitogen-activated protein kinase and nuclear factor-kappa B activation. Regul Pept. 2001;99:21–9. doi: 10.1016/s0167-0115(01)00218-x. [DOI] [PubMed] [Google Scholar]
- 45.Zhao D, Vellaichamy E, Somanna NK, Pandey KN. Guanylyl cyclase/natriuretic peptide receptor-A gene disruption causes increased adrenal angiotensin II and aldosterone levels. Am J Physiol Renal Physiol. 2007;293:F121–7. doi: 10.1152/ajprenal.00478.2006. [DOI] [PubMed] [Google Scholar]
- 46.Kishimoto I, Rossi K, Garbers DL. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc Natl Acad Sci USA. 2001;98:2703–6. doi: 10.1073/pnas.051625598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knowles JW, Esposito G, Mao L, et al. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A-deficient mice. J Clin Invest. 2001;107:975–84. doi: 10.1172/JCI11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ellmers LJ, Knowles JW, Kim HS, Smithies O, Maeda N, Cameron VA. Ventricular expression of natriuretic peptides in Npr1(−/−) mice with cardiac hypertrophy and fibrosis. Am J Physiol Heart Circ Physiol. 2002;283:H707–14. doi: 10.1152/ajpheart.00677.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kilic A, Bubikat A, Gassner B, Baba HA, Kuhn M. Local actions of atrial natriuretic peptide counteract angiotensin II stimulated cardiac remodeling. Endocrinology. 2007;148:4162–9. doi: 10.1210/en.2007-0182. [DOI] [PubMed] [Google Scholar]
- 50.Mori T, Chen YF, Feng JA, Hayashi T, Oparil S, Perry GJ. Volume overload results in exaggerated cardiac hypertrophy in the atrial natriuretic peptide knockout mouse. Cardiovasc Res. 2004;61:771–9. doi: 10.1016/j.cardiores.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 51.Nishikimi T, Hagaman JR, Takahashi N, et al. Increased susceptibility to heart failure in response to volume overload in mice lacking natriuretic peptide receptor-A gene. Cardiovasc Res. 2005;66:94–103. doi: 10.1016/j.cardiores.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 52.Shi SJ, Nguyen HT, Sharma GD, Navar LG, Pandey KN. Genetic disruption of atrial natriuretic peptide receptor-A alters renin and angioten-sin II levels. Am J Physiol Renal Physiol. 2001;281:F665–73. doi: 10.1152/ajprenal.2001.281.4.F665. [DOI] [PubMed] [Google Scholar]
- 53.Unger T, Li J. The role of the renin–angiotensin–aldosterone system in heart failure. J Renin Angiotensin Aldosterone Syst. 2004;5 (Suppl 1):S7–10. doi: 10.3317/jraas.2004.024. [DOI] [PubMed] [Google Scholar]
- 54.Morgan TO, Aubert JF, Wang Q. Sodium, angiotensin II, blood pressure, and cardiac hypertrophy. Kidney Int Suppl. 1998;67:S213–15. doi: 10.1046/j.1523-1755.1998.06751.x. [DOI] [PubMed] [Google Scholar]
- 55.Dostal DE. The cardiac renin–angiotensin system: Novel signaling mechanisms related to cardiac growth and function. Regul Pept. 2000;91:1–11. doi: 10.1016/s0167-0115(99)00123-8. [DOI] [PubMed] [Google Scholar]
- 56.Pandey KN, Misono KS, Inagami T. Evidence for intracellular formation of angiotensins: Coexistence of renin and angiotensin-converting enzyme in Leydig cells of rat testis. Biochem Biophys Res Commun. 1984;122:1337–43. doi: 10.1016/0006-291x(84)91238-5. [DOI] [PubMed] [Google Scholar]
- 57.Pandey KN, Inagami T. Regulation of renin angiotensins by gonadotropic hormones in cultured murine Leydig tumor cells. Release of angiotensin but not renin. J Biol Chem. 1986;261:3934–8. [PubMed] [Google Scholar]
- 58.de Resende MM, Mill JG. Effect of high salt intake on local renin–angiotensin system and ventricular dysfunction following myocardial infarction in rats. Clin Exp Pharmacol Physiol. 2007;34:274–9. doi: 10.1111/j.1440-1681.2007.04556.x. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Kishimoto I, Saito Y, et al. Guanylyl cyclase-A inhibits angiotensin II type 1A receptor-mediated cardiac remodeling, an endogenous protective mechanism in the heart. Circulation. 2002;106:1722–8. doi: 10.1161/01.cir.0000029923.57048.61. [DOI] [PubMed] [Google Scholar]
- 60.Luvara G, Pueyo ME, Philippe M, et al. Chronic blockade of NO synthase activity induces a proinflammatory phenotype in the arterial wall: Prevention by angiotensin II antagonism. Arterioscler Thromb Vasc Biol. 1998;18:1408–16. doi: 10.1161/01.atv.18.9.1408. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Q, Saito Y, Naya N, et al. The specific mineralocorticoid receptor blocker eplerenone attenuates left ventricular remodeling in mice lacking the gene encoding guanylyl cyclase-A. Hypertens Res. 2008;31:1251–6. doi: 10.1291/hypres.31.1251. [DOI] [PubMed] [Google Scholar]
- 62.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995;92:4862–6. doi: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 64.Oral H, Sivasubramanian N, Dyke DB, et al. Myocardial proinflammatory cytokine expression and left ventricular remodeling in patients with chronic mitral regurgitation. Circulation. 2003;107:831–7. doi: 10.1161/01.cir.0000049745.38594.6d. [DOI] [PubMed] [Google Scholar]
- 65.Testa M, Yeh M, Lee P, et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28:964–71. doi: 10.1016/s0735-1097(96)00268-9. [DOI] [PubMed] [Google Scholar]
- 66.Vanderheyden M, Paulus WJ, Voss M, et al. Myocardial cytokine gene expression is higher in aortic stenosis than in idiopathic dilated cardiomyopathy. Heart. 2005;91:926–31. doi: 10.1136/hrt.2004.035733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ono K, Matsumori A, Shioi T, Furukawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts: Possible implication in left ventricular remodeling. Circulation. 1998;98:149–56. doi: 10.1161/01.cir.98.2.149. [DOI] [PubMed] [Google Scholar]
- 68.Serneri GG, Modesti PA, Boddi M, et al. Cardiac growth factors in human hypertrophy. Relations with myocardial contractility and wall stress. Circ Res. 1999;85:57–67. doi: 10.1161/01.res.85.1.57. [DOI] [PubMed] [Google Scholar]
- 69.Frantz S, Fraccarollo D, Wagner H, et al. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res. 2003;57:749–56. doi: 10.1016/s0008-6363(02)00723-x. [DOI] [PubMed] [Google Scholar]
- 70.Li Y, Ha T, Gao X, et al. NF-kappaB activation is required for the development of cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol. 2004;287:H1712–20. doi: 10.1152/ajpheart.00124.2004. [DOI] [PubMed] [Google Scholar]
- 71.Purcell NH, Molkentin JD. Is nuclear factor kappaB an attractive therapeutic target for treating cardiac hypertrophy? Circulation. 2003;108:638–40. doi: 10.1161/01.CIR.0000085362.40608.DD. [DOI] [PubMed] [Google Scholar]
- 72.Hussain T, Lokhandwala MF. Renal dopamine receptor function in hypertension. Hypertension. 1998;32:187–97. doi: 10.1161/01.hyp.32.2.187. [DOI] [PubMed] [Google Scholar]
- 73.Ozkaya YG, Agar A, Yargicoglu P, et al. The effect of exercise on brain antioxidant status of diabetic rats. Diabetes Metab. 2002;28:377–84. [PubMed] [Google Scholar]
- 74.Muhlbauer B, Kuster E, Luippold G. Dopamine D(3) receptors in the rat kidney: Role in physiology and pathophysiology. Acta Physiol Scand. 2000;168:219–23. doi: 10.1046/j.1365-201x.2000.00665.x. [DOI] [PubMed] [Google Scholar]
- 75.Esteban V, Lorenzo O, Ruperez M, et al. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol. 2004;15:1514–29. doi: 10.1097/01.asn.0000130564.75008.f5. [DOI] [PubMed] [Google Scholar]
- 76.Zhang L, Ma Y, Zhang J, Cheng J, Du J. A new cellular signaling mechanism for angiotensin II activation of NF-kappaB: An IkappaB-independent, RSK-mediated phosphorylation of p65. Arterioscler Thromb Vasc Biol. 2005;25:1148–53. doi: 10.1161/01.ATV.0000164624.00099.e7. [DOI] [PubMed] [Google Scholar]
- 77.Kiemer AK, Weber NC, Vollmar AM. Induction of IkappaB: Atrial natriuretic peptide as a regulator of the NF-kappaB pathway. Biochem Biophys Res Commun. 2002;295:1068–76. doi: 10.1016/s0006-291x(02)00807-0. [DOI] [PubMed] [Google Scholar]
- 78.Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–22. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]