

Abstract
The FKBP5 gene product forms part of a complex with the glucocorticoid receptor and can modulate cortisol-binding affinity. Variations in the gene have been associated with increased recurrence of depression and with rapid response to antidepressant treatment. We sought to determine whether common FKBP5 variants confer risk for bipolar disorder. We genotyped seven tag single-nucleotide polymorphisms (SNPs) in FKBP5, plus two SNPs previously associated with illness, in 317 families with 554 bipolar offspring, derived primarily from two studies. Single marker and haplotypic analyses were carried out with FBAT and EATDT employing the standard bipolar phenotype. Association analyses were also conducted using 11 disease-related variables as covariates. Under an additive genetic model, rs4713902 showed significant overtransmission of the major allele (P = 0.0001), which was consistent across the two sample sets (P=0.004 and 0.006). rs7757037 showed evidence of association that was strongest under the dominant model (P = 0.001). This result was consistent across the two datasets (P=0.017 and 0.019). The dominant model yielded modest evidence for association (P< 0.05) for three additional markers. Covariate-based analyses suggested that genetic variation within FKBP5 may influence attempted suicide and number of depressive episodes in bipolar subjects. Our results are consistent with the well-established relationship between the hypothalamic–pituitary–adrenal (HPA) axis, which mediates the stress response through regulation of cortisol, and mood disorders. Ongoing whole-genome association studies in bipolar disorder and major depression should further clarify the role of FKBP5 and other HPA genes in these illnesses.
Keywords: HPA axis, mood disorder, linkage disequilibrium
Introduction
Bipolar disorder, a psychiatric illness characterized by episodes of low mood (depression) and high mood (mania or hypomania), has a lifetime prevalence of 2–3%.1 Family, twin and adoption studies have established a substantial genetic contribution, with heritability estimates averaging about 70%.2 The high heritability has provided a strong rationale for molecular genetic studies. Childhood adversity and life events in adulthood might also contribute to the development of bipolar disorder, possibly via effects on hypothalamic–pituitary–adrenal (HPA) axis function.
The HPA axis is the part of the neuroendocrine system that regulates responses to stress, with cortisol, a glucocorticoid hormone, being the principal stress hormone within the axis. The first studies showing a relationship between elevated cortisol and depression, a phenotype closely related to bipolar disorder, were carried out in the 1950s.3,4 Many subsequent studies have shown HPA axis derangements in depression, including altered cortisol responses to suppression with dexamethasone, a synthetic cortisol analogue5,6 and to the combined dexamethasone/corticotropin-releasing hormone (Dex-CRH) challenge test.7 More recently, studies of bipolar disorder have similarly shown HPA abnormalities on the Dex-CRH challenge test.8,9
While there is ample evidence of state-like HPA changes in the setting of mood episodes, there are also studies suggesting heritable trait-like abnormalities. Studies of unaffected offspring of both depressed10 and bipolar11 parents have shown they have elevated baseline cortisol levels. Similarly, the Munich Vulnerability Study showed that unaffected first-degree relatives from families with a high genetic load for mood disorder often display abnormal responses to the Dex-CRH challenge test.12,13
Several studies have found evidence for association between variations in HPA axis genes and depression. These include two implicating the glucocorticoid receptor gene, which encodes the receptor for cortisol.14,15 Binder et al.16 reported significant associations of antidepressant response with three single-nucleotide polymorphisms (SNPs) in FKBP5, whose product forms part of a complex with the glucocorticoid receptor. The association with SNP rs1360780 was replicated in a second independent sample. The TT genotype of rs1360780 was found to correlate with increased number of depressive episodes and increased FKBP5 protein levels. Subjects with the associated TT genotype had less HPA-axis hyperactivity during their depressive episodes.
FKBP5 has been implicated as a modulator of glucocorticoid receptor function through association with heat shock protein 90, a molecular chaperone with a central role in steroid hormone signaling. Glucocorticoid resistance in neotropical primates has been attributed to overexpression of FKBP51, an ortholog of FKBP5, suggesting that this cochaperone can modulate cortisol-binding affinity.17 Additional evidence supporting the potential functional relevance of FKBP5 comes from a recent study which identified 121 genes showing significant differences in expression in the brains of mice treated with lithium as compared to controls.18 One of these genes was the mouse homolog of FKBP5, which showed a reproducible 3.2-fold increase in mice treated with lithium.
We sought to test whether common FKBP5 variants confer risk for bipolar disorder in a large family-based sample, derived primarily from two studies. Toward this end, we studied seven tag SNPs in the gene, plus two additional SNPs from the Binder et al. study, for association with illness in 317 families with 554 bipolar offspring.
Materials and methods
Sample
We selected 1188 subjects from 317 nuclear families for genotyping. They constituted all available independent trios (two parents and one affected child) and quads (two parents and two affected children) from among the subjects originally ascertained as part of the Chicago, Hopkins, NIMH Intramural Program (CHIP) bipolar disorder study,19 the Clinical Neurogenetics (CNG) Bipolar Disorder Study,20 or the National Institute of Mental Health (NIMH) Genetics Initiative Bipolar Disorder Collaborative Study.21 All were ascertained on the basis of multiple relatives affected with a major mood disorder; the detailed description of the ascertainment and assessment protocols for each of these studies can be found in the initial study reports. All subjects signed IRB-approved written informed consent forms prior to enrolling in the studies.
For the purposes of this study, we considered those with bipolar I disorder (BPI), bipolar II disorder (BPII) with recurrent major depression or schizoaffective disorder, manic or bipolar type (SA/BP) to be affected. The 1188 subjects contained 317 pedigrees and 491 offspring with BPI, 39 offspring with BPII and 24 offspring with SA/BP, resulting in 80 independent trios and 237 independent quads. The sample had 80% power to detect evidence of association for a locus of moderate effect (genotypic relative risk = 1.6–1.7) assuming an additive model, a disease prevalence of 1%, and α=0.0019 (0.05 divided by 27, representing a Bonferroni's correction for the nine markers and three genetic models tested—additive, dominant and genotypic).
The study design focused on combining the three samples to maximize power, but we also performed post hoc tests of consistency of results by dividing the data into two parts. We grouped the CHIP (71 families, 24 trios and 47 quads) and the CNG (11 families, 4 trios and 7 quads) samples because they used very similar assessment methods (probands and parents were largely interviewed using the Schedule for Affective Disorders and Schizophrenia-Lifetime Version (SADS)22 and diagnoses were made using the Research Diagnostic Criteria).23 The CHIP/CNG set was compared to the NIMH sample which used the Diagnostic Interview for Genetic Studies (DIGS)24 to make diagnoses under Diagnostic and Statistical Manual III-R or IV criteria. The NIMH sample contained 235 families, 52 trios and 183 quads. Analyses of both samples made use of the Bipolar Disorder Phenome Database,25 which has recently combined the CHIP and NIMH datasets and subjected them to a variety of quality control checks.
SNP selection
We used the HapMap database (Phase I) to identify the SNP markers necessary to capture the common genetic variation across the FKBP5 gene.26 Tag SNPs spanning the RefSeq FKBP5 transcript (115 plus 10 kb on either side) were chosen using the ldSelect program,27 which allows the user to select tag SNPs based on linkage disequilibrium (LD) parameters. We required an r2 of 0.8 and a minor allele frequency (MAF) of 0.1 in the Centre d'Etude du Polymorphisme Humain-derived HapMap (CEU) sample. Seven SNPs were chosen to adequately cover the region: rs1043805, rs7757037, rs3798346, rs9296158, rs9380525, rs7763535 and rs737054. Genotyping was conducted at Illumina using their BeadArray platform (Illumina, San Diego, CA, USA).28 One of the tag SNPs (rs7763535) failed Illumina's assay design criteria, and a second tag SNP (rs737054) failed to genotype using the GoldenGate Assay.
The two markers that could not be genotyped using the Illumina system similarly failed on the TaqMan platform. Alternatives were chosen from the HapMap database. SNP marker rs4713902 has an r2 of 1.0 with marker rs737054, and SNP marker rs6912833 has an r2 of 0.955 with marker rs7763535 in the CEU sample, indicating that these two markers were excellent replacements for the two missing tag SNPs. These two additional markers were genotyped using an ABI 7900HT and TaqMan assays (Applied Biosystems, Foster City, CA, USA).29 We also used the ABI 7900HT to genotype two additional SNPs (rs3800373 and rs1360780) previously identified as being associated with response to antidepressant treatment.16
Analytic methods
We examined the genotype data for deviations from Hardy–Weinberg equilibrium (HWE) using Haplo-view30 and screened the genotype data for Mendelian inconsistencies using family-based association test (FBAT).31 Any pedigree with a non-Mendelian inheritance at a specific SNP had its genotypes removed for that SNP.
We used the program Haploview to explore the extent of LD across the region. We evaluated pairwise measures of LD, including D′ and r2, between SNPs and assessed for evidence of haplotype-block structure. We used the ‘confidence intervals’ approach32 and ‘solid spine of LD’ approach30 to explore the putative block structure.
We tested for disease association with single SNPs using the FBAT.31 In addition, we performed genotypic transmission disequilibrium tests (TDT) using the gtrr function in STATA made available by David Clayton (http://www.gene.cimr.cam.ac.uk/clayton/software/stata). Because of their ability to capture LD more informatively, testing associations with haplotypes may be more powerful than with individual SNPs. Therefore, we also used exhaustive allelic TDT (EATDT) to test for evidence of haplotypic association.33 EATDT generates permutation-based P-values for each haplotype tested, allowing the user to correct for multiple testing while taking LD into account. SNP marker order was based on the UCSC genome database (March 2006).
Covariate analysis
We also tested for evidence of association using 11 clinical covariates. Nine covariates were available on all affected individuals from the CHIP and NIMH studies (except for 10 subjects who were not interviewed): sex, age of onset (dichotomous and continuous), history of attempted suicide, history of psychosis, history of hospitalization, number of depressive episodes (continuous), number of manic episodes (continuous) and weeks of most severe depression (continuous). Two more clinical covariates derived from the DIGS interview were rapid cycling and rapid switching. The former was defined as four or more syndromal mood episodes in a year, while the latter was defined as a subjective sense of one's mood switching back and forth quickly from feeling high to feeling depressed or normal. A total of 165 CHIP subjects interviewed with the SADS did not have data available on these two covariates.
Association tests with single SNPs were carried out using the genotypic TDT as implemented in STATA 9.2, modified to include covariates by M Daniele Fallin and K Lasseter. We formally tested for differences across subgroups by fitting to the entire sample a conditional logistic regression model that included a term for the covariate by genotype interaction. Likelihood ratio tests were then used to test whether the model including the interaction term provided a better fit to the data than a model without the interaction term, suggesting heterogeneity in the observed association.
Results
Figure 1 illustrates the organization of the FKBP5 gene, the location of the nine SNP markers we studied and the extent of LD in the region in our sample. The nine SNP markers are contained in two haplotype blocks as defined by the ‘confidence intervals’ approach (Figure 1) and one haplotype block as defined by the ‘solid spine’ method. These nine markers tag 78% of the common SNPs (MAF 0.1) at an r2 of 0.8 found in FKBP5 in CEU samples from Phase II of the HapMap project.
Figure 1.
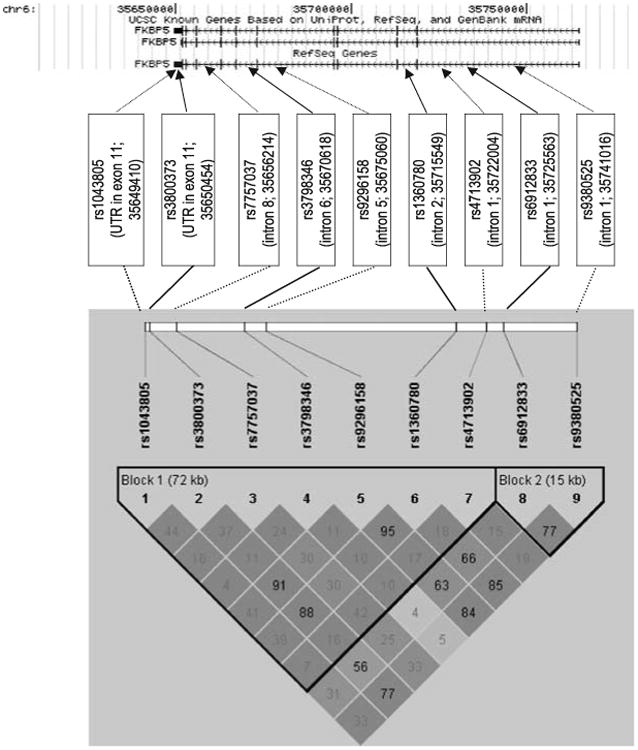
The FKBP5 chromosomal location, gene structure, single-nucleotide polymorphism (SNP) locations and linkage disequilibrium (LD) structure are depicted in the figure. The chromosomal location and gene structure were taken from the UCSC genome browser (March 2006 Build; NCBI Build 36.1). The RefSeq transcript has 11 exons and spans 115kb from 35 649 346–35 764 692 on chromosome 6. The locations of the study's nine FKBP5 SNPs are indicated by arrows. The LD structure for the nine FKBP5 SNPs was determined using our own genotype data in the family sample. The LD diagram demonstrates that the nine markers belong to two haplotype blocks as determined by the ‘confidence intervals’ method.32 High pairwise LD (D′) between markers is illustrated with dark shading. The r2 scores for the marker pairs are listed in the corresponding blocks.
The genotypes for the nine SNPs were analyzed with FBAT using both an additive and a dominant genetic model. Under the additive model (Table 1), marker rs4713902 showed a significant overtransmission of the major allele (465 transmissions observed; 424 transmissions expected) resulting in a significant P-value (P=0.0001) which remained significant (P = 0.003) after Bonferroni's correction for 27 tests (see Materials and methods). Marker rs7757037 also showed evidence of an overtransmission of the major allele (447 transmissions observed; 414 transmissions expected) resulting in a significant P-value (P = 0.005) that was no longer significant after the conservative Bonferroni's correction (P = 0.14).
Table 1. Family-based association results.
| Marker | Allele frequencies (%) | Genetic modela | S/E(S)b major allele | P-valuec major allele | S/E(S) minor allele | P-value minor allele |
|---|---|---|---|---|---|---|
| rs1043805 | A (85) | Additive | 331/336 | P=0.564 | 157/152 | P=0.564 |
| T (15) | Dominant | 40/43 | P=0.365 | 137/135 | P=0.791 | |
| rs3800373 | A (72) | Additive | 396/415.5 | P=0.060 | 264/244.5 | P=0.060 |
| C (28) | Dominant | 77/90.25 | P=0.010 | 182/175.75 | P=0.452 | |
| rs7757037 | G (52) | Additive | 447/414 | P=0.005 | 371/404 | P=0.005 |
| A (48) | Dominant | 175/167.25 | P=0.310 | 147/172.25 | P=0.001 | |
| rs3798346 | A (78) | Additive | 413/427.5 | P=0.137 | 225/210.5 | P=0.137 |
| G (22) | Dominant | 51/56.5 | P=0.177 | 174/165 | P= 0.285 | |
| rs9296158 | G (70) | Additive | 430/449 | P=0.083 | 294/275 | P=0.083 |
| A (30) | Dominant | 94/107 | P=0.020 | 200/194 | P=0.487 | |
| rs1360780 | C (70) | Additive | 402/417.5 | P=0.141 | 274/258.5 | P=0.141 |
| T (30) | Dominant | 87/97.25 | P=0.057 | 182/176.75 | P=0.526 | |
| rs4713902 | T (70) | Additive | 465/424 | P=0.0001 | 225/266 | P=0.0001 |
| C (30) | Dominant | 109/96.25 | P=0.019 | 147/175.25 | P=0.0007 | |
| rs6912833 | T (73) | Additive | 360/369.5 | P=0.335 | 234/224.5 | P=0.335 |
| A (27) | Dominant | 78/84 | P=0.228 | 160/156.5 | P= 0.654 | |
| rs9380525 | C (68) | Additive | 419/436.5 | P=0.110 | 305/287.5 | P=0.110 |
| G (32) | Dominant | 100/111.75 | P=0.044 | 192/186.25 | P=0.496 |
P-values for the recessive model are the inverse for those of the dominant model. For example, the recessive P-value for marker rs7757037 would be P = 0.001 for the major allele and P=0.310 for the minor allele.
S, test statistic for observed number of transmitted alleles as calculated by FBAT; E(S), expected value of S under the null hypothesis as calculated by FBAT.
P-values, asymptotic P-values as calculated by FBAT.
The dominant genetic model yielded additional evidence for association (Table 1). Five markers had nominal evidence for association (P<0.05), including rs4713902 (P=0.0007), rs7757037 (P=0.001), rs3800373 (P= 0.010), rs9296158 (P=0.020) and rs9380525 (P=0.044). The findings for markers rs4713902 and rs7757037 remained significant after Bonferroni's correction (P = 0.02 and 0.03, respectively).
Genotypic TDT results were calculated for each of the nine SNPs (Table 2). As with the allelic association analysis, the strongest finding was at rs4713902 (P=0.001). Markers rs7757037, rs3800373, rs9296158 and rs9380525 had evidence for genotypic association using the standard bipolar phenotype.
Table 2. Genotypic association results.
| Marker | Allele frequencies (%) | Genotypic association resultsa odds ratios (95% CI) | |||
|---|---|---|---|---|---|
|
|
|||||
| Homozygous major allele (reference) | Heterozygous | Homozygous minor allele | P-valueb | ||
| rs1043805 | A (85) | 1.0 | 0.78 (0.49, 1.23) | 0.76 (0.47, 1.24) | 0.53 |
| T (15) | |||||
| rs3800373 | A (72) | 1.0 | 1.05 (0.82, 1.34) | 1.66 (1.11, 2.46) | 0.03 |
| C (28) | |||||
| rs7757037 | G (52) | 1.0 | 0.68 (0.53, 0.87) | 0.63 (0.45, 0.89) | 0.007 |
| A (48) | |||||
| rs3798346 | A (78) | 1.0 | 0.75 (0.44, 1.29) | 0.67 (0.38, 1.19) | 0.35 |
| G (22) | |||||
| rs9296158 | G (70) | 1.0 | 1.04 (0.82, 1.32) | 1.53 (1.05, 2.22) | 0.05 |
| A (30) | |||||
| rs1360780 | C (70) | 1.0 | 1.04 (0.81, 1.33) | 1.81 (0.97, 2.13) | 0.13 |
| T (30) | |||||
| rs4713902 | T (70) | 1.0 | 0.69 (0.55, 0.88) | 0.47 (0.30, 0.74) | 0.001 |
| C (30) | |||||
| rs6912833 | T (73) | 1.0 | 0.80 (0.58, 1.10) | 0.77 (0.52, 1.15) | 0.36 |
| A (27) | |||||
| rs9380525 | C (68) | 1.0 | 0.73 (0.54, 0.97) | 0.69 (0.48, 1.01) | 0.09 |
| G (32) | |||||
Abbreviation: CI, confidence interval.
Odds ratios are calculated with respect to the reference group (the homozygous major allele for each marker). Odds ratios > 1.0 indicate that the genotype under examination (homozygous minor allele or heterozygous) is associated with higher risk. Odds ratios <1 indicate that the homozygous major allele is associated with higher risk.
Robust P-values are estimates of the overall trend for the data as calculated by the gtrr function using robust variance estimates.
We also performed post hoc tests of consistency of results by dividing the data into two parts. We grouped the CHIP and the CNG samples together and compared them to the NIMH sample. The association signals for rs4713902 and rs7757037 were supported by both sample sets. Marker rs4713902 had P-values of P=0.006 for the CHIP/CNG samples and P=0.004 for the NIMH sample using the additive model, and marker rs7757037 had P-values of P=0.017 for the CHIP/CNG samples and P=0.019 for the NIMH sample under the dominant model.
We used EATDT to test for haplotypic association. EATDT, which looks at all combinations of adjacent SNPs, tested 130 haplotypes and SNPs in the analysis. Eleven haplotypes with permutation P-values <0.05 were identified. The two strongest haplotypes, each with a permutation P=0.0002, covered the following SNPs: (a) rs9296158–rs1360780– rs4713902; and (b) rs1360780–rs4713902.
To compare our results to previous studies, we also conducted covariate-based association analyses. Toward this end, the genotypic analyses were repeated using 11 covariates. Four markers had evidence for association that was significantly influenced by the attempted suicide covariate (rs1043805, P=0.012; rs3800373, P= 0.022; rs9296158, P=0.021 and rs1360780, P=0.028), and three markers had evidence for association that significantly corresponded with the number of past depressive episodes (rs1043805, P=0.005; rs9296158, P=0.021 and rs1360780, P = 0.04).
Discussion
Five SNPs from the FKBP5 gene (rs4713902, rs7757037, rs9296158, rs3800373 and rs9380525) showed evidence for association with bipolar disorder in a family sample with 317 bipolar pedigrees and 554 affected offspring. The strongest signal was at rs4713902 (P=0.0001) which remained significant after correction for the nine markers and three models tested and was consistent across the two studies that comprised the sample. In addition, covariate-based analyses in the family sample identified four SNPs within FKBP5 (rs1043805, rs3800373, rs9296158 and rs1360780) whose association with bipolar disorder differed depending upon the attempted suicide covariate and/or the number of depressive episodes in bipolar subjects.
Our results are consistent with the findings of Binder et al.16 In that study, markers rs3800373 and rs1360780 showed evidence for association in depressed subjects stratified according to antidepressant response. Marker rs1360780 (TT genotype) was also associated with increased number of depressive episodes, increased intracellular FKBP5 protein expression and a decreased response in the Dex-CRH test. In our study, marker rs1360780 had a trend toward an association (P=0.057) with the standard bipolar disorder phenotype and showed evidence of association with number of depressive episodes. Our findings also implicate rs3800373; the CC genotype is the risk genotype in our study and the antidepressant responsive genotype in the Binder et al. study. Our strongest SNP, rs4713902, is 6.5 kb from rs1360780, though it is only in modest LD (r2 = 0.18). Marker rs4713902 was chosen to replace rs737054 with which it is in perfect LD. Of potential functional significance, rs737054 lies within a highly conserved region34 of intron 5 that is predicted to have high regulatory potential.35
Gawlik et al.36 attempted to replicate the Binder et al. findings by genotyping three associated SNPs from that study in a sample containing 248 subjects with depression or bipolar disorder and 188 controls. No evidence for allelic or genotypic association was found in the case–control analysis. The authors also tested for evidence of association with five disease variables, including age at onset, age at first hospitalization, duration of inpatient treatments, duration of disease and number of inpatient treatments. One genotype and one haplotype were nominally associated with longer duration of disease (P = 0.01 and 0.045, respectively). Fallin et al.37 genotyped five FKBP5 SNPs in 323 BPI trios of Ashkenazi descent and found no evidence of association. These two negative studies may have been the result of genetic heterogeneity, insufficient marker density and/or smaller sample sizes.
While our family sample benefits from a design that is immune to issues of population stratification, which can complicate the analysis and interpretation of case–control studies, family-based designs may be more vulnerable to issues arising from genotyping error. In particular, undetected genotyping errors can lead to false positive family-based association signals at markers with unequal allele frequencies, leading to apparent overtransmission of the common alleles and undertransmission of the minor alleles.38 While this pattern was seen in the additive model for both of the two strongest association signals in our study, these SNPs showed no evidence of inheritance inconsistencies, blind duplicate discordance or deviation from HWE, making genotyping error an unlikely explanation for our results.
Our results should be considered in light of several limitations. First, our FKBP5 tag SNPs were selected using data from an earlier version of the HapMap project (Phase I), resulting in incomplete coverage for the common genetic variation that has subsequently been identified. Second, we have not assayed rare variation in FKBP5 that might influence susceptibility to illness. Third, we were not able to test for correlation of positive genotypes with variation in measures of HPA axis function, as these have not been obtained in this sample. Fourth, although we had only 1.6% missing data for our strongest marker (rs4713902), if there were substantial bias in the pattern of missing genotypes, this could reduce the strength of our results.
Additional data regarding the role of FKBP5 in mood disorders should be forthcoming soon as several whole-genome association studies are currently being conducted or planned using the bipolar disorder and recurrent major depression phenotypes. The first published genome-wide association study of bipolar disorder,39 which used a DNA pooling strategy, detected nominally significant evidence of association with rs7757037 in one sample and with rs3798346 in both samples studied. A second genome-wide association study, from the Wellcome Trust Case–Control Consortium, identified one SNP within FKBP5 (rs16878806) with nominal evidence for association with bipolar disorder.40 Marker rs16878806 lies in close proximity to rs7757037 and is in strong LD with it.
If our results and those of Binder et al. are correct, and FKBP5 does play a role in susceptibility to bipolar disorder as well as in recurrence of depression and rapidity of response to antidepressant medication, this confluence of associations would be consistent with prior clinical observations. Recurrence of episodes has been reported to be greater in bipolar disorder than in unipolar depression.41 A rapid response to antidepressants could be an indication of a bipolar diathesis given that some patients experience antidepressant-induced shifts from depression into hypomania or mania.42
In conclusion, common variants of FKBP5 were associated with bipolar disorder and with a feature of illness, number of depressive episodes, implicated in a prior study of depression. This result is consistent with the well-established relationship between the HPA axis, which mediates the stress response, and mood disorders. Whole-genome association studies that are ongoing in bipolar disorder and major depression should further clarify the role of FKBP5 and other HPA genes in these illnesses.
Acknowledgments
This work was supported by grants from the National Institute of Mental Health (R01 MH-042243 and R01 MH-061613), the Charles A Dana Foundation Consortium on the Genetic Basis of Manic Depressive Illness, the National Alliance for Research on Schizophrenia and Depression, the Alex Brown Foundation and the Stanley Medical Research Institute. Dr Willour and Dr Potash were also supported by Margaret Ann Price Investigatorships. Some DNA samples were prepared and distributed by Rutgers University under a contract from the NIMH. We are grateful to Yuqing Huo, Kuangyi Miao and Brandie Craighead for their contributions. We are also grateful to the many interviewers and diagnosticians who contributed to this project, and to the families who devoted their time and effort to the study.
The Bipolar Disorder Phenome Group consists of Francis McMahon, Jo Steele, Justin Pearl, Layla Kassem, Victor Lopez from the Genetic Basis of Mood and Anxiety Disorders Unit, Mood and Anxiety Program, National Institute of Mental Health, National Institutes of Health, Bethesda, MD; James Potash, Dean MacKinnon, Erin Miller, Jennifer Toolan from the Department of Psychiatry, Johns Hopkins School of Medicine, Baltimore, MD; Peter Zandi from the Department of Mental Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD; Thomas Schulze from the Division of Genetic Epidemiology in Psychiatry, Central Institute of Mental Health, Ruprecht-Karls-University of Heidelberg, Mannheim, Germany; Evaristus Nwulia from the Department of Psychiatry, Howard University Hospital, Washington, DC; Sylvia Simpson from the Department of Psychiatry, University of Colorado at Denver, Denver, CO.
Some of the data and biomaterials were collected in four projects that participated in the NIMH Bipolar Disorder Genetics Initiative from 1991–1998. The Principal Investigators and Co-Investigators were Indiana University, Indianapolis, IN, U01 MH46282, John Nurnberger, MD, PhD, Marvin Miller, MD and Elizabeth Bowman, MD; Washington University, St Louis, MO, U01 MH46280, Theodore Reich, MD, Allison Goate, PhD and John Rice, PhD; Johns Hopkins University, Baltimore, MD U01 MH46274, J Raymond DePaulo, Jr, MD, Sylvia Simpson, MD, MPH and Colin Stine, PhD; NIMH Intramural Research Program, Clinical Neurogenetics Branch, Bethesda, MD, Elliot Gershon, MD, Diane Kazuba, BA and Elizabeth Maxwell, MSW. Other data and biomaterials were collected in 10 NIMH Bipolar Disorder Genetics Initiative projects from 1999–2003. The Principal Investigators and Co-Investigators were Indiana University, Indianapolis, IN, R01 MH59545, John Nurnberger, MD, PhD, Marvin J Miller, MD, Elizabeth S Bowman, MD, N Leela Rau, MD, P Ryan Moe, MD, Nalini Samavedy, MD, Rif El-Mallakh, MD (at University of Louisville), Husseini Manji, MD (at Wayne State University), Debra A Glitz, MD (at Wayne State University), Eric T Meyer, MS, Carrie Smiley, RN, Tatiana Foroud, PhD, Leah Flury, MS, Danielle M Dick, PhD, Howard Edenberg, PhD; Washington University, St Louis, MO, R01 MH059534, John Rice, PhD, Theodore Reich, MD, Allison Goate, PhD, Laura Bierut, MD; Johns Hopkins University, Baltimore, MD, R01 MH59533, Melvin McInnis MD, J. Raymond DePaulo, Jr, MD, Dean F MacKinnon, MD, Francis M Mondimore, MD, James B Potash, MD, Peter P Zandi, PhD, Dimitrios Avramopoulos, PhD and Jennifer Payne, MD; University of Pennsylvania, PA, R01 MH59553, Wade Berrettini MD, PhD; University of California at Irvine, CA, R01 MH60068, William Byerley MD and Mark Vawter MD; University of Iowa, IA, R01 MH059548, William Coryell MD and Raymond Crowe MD; University of Chicago, IL, R01 MH59535, Elliot Gershon, MD, Judith Badner PhD, Francis McMahon MD, Chunyu Liu PhD, Alan Sanders MD, Maria Caserta, Steven Dinwiddie MD, Tu Nguyen, Donna Harakal; University of California at San Diego, CA, R01 MH59567, John Kelsoe, MD, Rebecca McKinney, BA; Rush University, IL, R01 MH059556, William Scheftner MD, Howard M Kravitz, DO, MPH, Diana Marta, BS, Annette Vaughn-Brown, MSN, RN and Laurie Bederow, MA; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis J McMahon, MD, Layla Kassem, PsyD, Sevilla Detera-Wadleigh, PhD, Lisa Austin, PhD, Dennis L Murphy, MD.
References
- 1.Kessler RC, Akiskal HS, Angst J, Guyer M, Hirschfeld RM, Merikangas KR, et al. Validity of the assessment of bipolar spectrum disorders in the WHO CIDI 3.0. J Affect Disord. 2006;96:259–269. doi: 10.1016/j.jad.2006.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet. 2003;123C:48–58. doi: 10.1002/ajmg.c.20013. [DOI] [PubMed] [Google Scholar]
- 3.Board F, Persky H, Hamburg DA. Psychological stress and endocrine functions; blood levels of adrenocortical and thyroid hormones in acutely disturbed patients. Psychosom Med. 1956;18:324–333. doi: 10.1097/00006842-195607000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Board F, Wadeson R, Persky H. Depressive affect and endocrine functions; blood levels of adrenal cortex and thyroid hormones in patients suffering from depressive reactions. AMA Arch Neurol Psychiatry. 1957;78:612–620. [PubMed] [Google Scholar]
- 5.Carroll BJ, Martin FI, Davies B. Resistance to suppression by dexamethasone of plasma 11-O.H.C.S. levels in severe depressive illness. Br Med J. 1968;3:285–287. doi: 10.1136/bmj.3.5613.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson JC, Davis JM. DST studies in psychotic depression: a metaanalysis. Am J Psychiatry. 1997;154:1497–1503. doi: 10.1176/ajp.154.11.1497. [DOI] [PubMed] [Google Scholar]
- 7.Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res. 1994;28:341–356. doi: 10.1016/0022-3956(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 8.Rybakowski JK, Twardowska K. The dexamethasone/corticotropin-releasing hormone test in depression in bipolar and unipolar affective illness. J Psychiatr Res. 1999;33:363–370. doi: 10.1016/s0022-3956(99)00014-x. [DOI] [PubMed] [Google Scholar]
- 9.Watson S, Gallagher P, Ritchie JC, Ferrier IN, Young AH. Hypothalamic-pituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry. 2004;184:496–502. doi: 10.1192/bjp.184.6.496. [DOI] [PubMed] [Google Scholar]
- 10.Mannie ZN, Harmer CJ, Cowen PJ. Increased waking salivary cortisol levels in young people at familial risk of depression. Am J Psychiatry. 2007;164:617–621. doi: 10.1176/ajp.2007.164.4.617. [DOI] [PubMed] [Google Scholar]
- 11.Ellenbogen MA, Hodgins S, Walker CD, Couture S, Adam S. Daytime cortisol and stress reactivity in the offspring of parents with bipolar disorder. Psychoneuroendocrinology. 2006;31:1164–1180. doi: 10.1016/j.psyneuen.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Holsboer F, Lauer CJ, Schreiber W, Krieg JC. Altered hypothalamic-pituitary-adrenocortical regulation in healthy subjects at high familial risk for affective disorders. Neuroendocrinology. 1995;62:340–347. doi: 10.1159/000127023. [DOI] [PubMed] [Google Scholar]
- 13.Modell S, Lauer CJ, Schreiber W, Huber J, Krieg JC, Holsboer F. Hormonal response pattern in the combined DEX-CRH test is stable over time in subjects at high familial risk for affective disorders. Neuropsychopharmacology. 1998;18:253–262. doi: 10.1016/S0893-133X(97)00144-9. [DOI] [PubMed] [Google Scholar]
- 14.van Rossum EF, Binder EB, Majer M, Koper JW, Ising M, Modell S, et al. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol Psychiatry. 2006;59:681–688. doi: 10.1016/j.biopsych.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 15.van West D, Van Den EF, Del Favero J, Souery D, Norrback KF, Van Duijn C, et al. Glucocorticoid receptor gene-based SNP analysis in patients with recurrent major depression. Neuropsychopharmacology. 2006;31:620–627. doi: 10.1038/sj.npp.1300898. [DOI] [PubMed] [Google Scholar]
- 16.Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Putz B, et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36:1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds PD, Ruan Y, Smith DF, Scammell JG. Glucocorticoid resistance in the squirrel monkey is associated with overexpression of the immunophilin FKBP51. J Clin Endocrinol Metab. 1999;84:663–669. doi: 10.1210/jcem.84.2.5429. [DOI] [PubMed] [Google Scholar]
- 18.McQuillin A, Rizig M, Gurling H. A microarray gene expression study of the effects lithium carbonate on mouse brain mRNA in order to understand the neurobiology of mood stabilisation and treatment of bipolar affective disorder. Pharmacogenetic Genomics. 2007;17:605–617. doi: 10.1097/FPC.0b013e328011b5b2. [DOI] [PubMed] [Google Scholar]
- 19.McInnis MG, Lan TH, Willour VL, McMahon FJ, Simpson SG, Addington AM, et al. Genome-wide scan of bipolar disorder in 65 pedigrees: supportive evidence for linkage at 8q24, 18q22, 4q32, 2p12, and 13q12. Mol Psychiatry. 2003;8:288–298. doi: 10.1038/sj.mp.4001277. [DOI] [PubMed] [Google Scholar]
- 20.Detera-Wadleigh SD, Badner JA, Berrettini WH, Yoshikawa T, Goldin LR, Turner G, et al. A high-density genome scan detects evidence for a bipolar-disorder susceptibility locus on 13q32 and other potential loci on 1q32 and 18p11.2. Proc Natl Acad Sci USA. 1999;96:5604–5609. doi: 10.1073/pnas.96.10.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.NIMH Genetics Initiative Bipolar Group. Genomic survey of bipolar illness in the NIMH genetics initiative pedigrees: a preliminary report. Am J Med Genet. 1997;74:227–237. doi: 10.1002/(sici)1096-8628(19970531)74:3<227::aid-ajmg1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 22.Endicott J, Spitzer RL. A diagnostic interview: the schedule for affective disorders and schizophrenia. Arch Gen Psychiatry. 1978;35:837–844. doi: 10.1001/archpsyc.1978.01770310043002. [DOI] [PubMed] [Google Scholar]
- 23.Spitzer RL, Endicott J. Research Diagnostic Criteria (RDC) for a Selected Group of Functional Disorders. New York State Psychiatric Institute, Biometrics Research; 1975. [Google Scholar]
- 24.Nurnberger JIJ, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, et al. Diagnostic interview for genetic studies. Rationale, unique features, and training. NIMH Genetics Initiative. Arch Gen Psychiatry. 1994;51:849–859. doi: 10.1001/archpsyc.1994.03950110009002. [DOI] [PubMed] [Google Scholar]
- 25.Potash JB, Toolan J, Steele J, Miller EB, Pearl J, Zandi PP, et al. The bipolar disorder phenome database: a resource for genetic studies. Am J Psychiatry. 2007;164:1229–1237. doi: 10.1176/appi.ajp.2007.06122045. [DOI] [PubMed] [Google Scholar]
- 26.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oliphant A, Barker DL, Stuelpnagel JR, Chee MS. BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques. 2002;56–58(Suppl):60–61. [PubMed] [Google Scholar]
- 29.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 30.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 31.Rabinowitz D, Laird N. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000;50:211–223. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- 32.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 33.Lin S, Chakravarti A, Cutler DJ. Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat Genet. 2004;36:1181–1188. doi: 10.1038/ng1457. [DOI] [PubMed] [Google Scholar]
- 34.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC. Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 2005;15:1051–1060. doi: 10.1101/gr.3642605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gawlik M, Moller-Ehrlich K, Mende M, Jovnerovski M, Jung S, Jabs B, et al. Is FKBP5 a genetic marker of affective psychosis? A case control study and analysis of disease related traits. BMC Psychiatry. 2006;6:52. doi: 10.1186/1471-244X-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, et al. Bipolar I disorder and schizophrenia: A 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell AA, Cutler DJ, Chakravarti A. Undetected genotyping errors cause apparent overtransmission of common alleles in the transmission/disequilibrium test. Am J Hum Genet. 2003;72:598–610. doi: 10.1086/368203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2007 May 8; doi: 10.1038/sj.mp.4002012. e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wellcome Trust Case-Control Consortium. Genome-wide association study of 14000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodwin FK, Jamison KR. Manic-Depressive Illness. Second. Oxford University Press; New York: 2007. p. 126. [Google Scholar]
- 42.Altshuler LL, Post RM, Leverich GS, Mikalauskas K, Rosoff A, Ackerman L. Antidepressant-induced mania and cycle acceleration: a controversy revisited. Am J Psychiatry. 1995;152:1130–1138. doi: 10.1176/ajp.152.8.1130. [DOI] [PubMed] [Google Scholar]