

Abstract
The aggregation of amyloid-β (Aβ) peptides plays a crucial role in the etiology of Alzheimer’s disease (AD). Recently, it has been reported that an A2T mutation in Aβ can protect against AD. Interestingly, a nonpolar A2V mutation also has been found to offer protection against AD in the heterozygous state, although it causes early-onset AD in homozygous carriers. Since the conformational landscape of the Aβ monomer is known to directly contribute to the early-stage aggregation mechanism, it is important to characterize the effects of the A2T and A2V mutations on Aβ1–42 monomer structure. Here, we have performed extensive atomistic replica-exchange molecular dynamics simulations of the solvated wild-type (WT), A2V, and A2T Aβ1–42 monomers. Our simulations reveal that although all three variants remain as collapsed coils in solution, there exist significant structural differences among them at shorter timescales. A2V exhibits an enhanced double-hairpin population in comparison to the WT, similar to those reported in toxic WT Aβ1–42 oligomers. Such double-hairpin formation is caused by hydrophobic clustering between the N-terminus and the central and C-terminal hydrophobic patches. In contrast, the A2T mutation causes the N-terminus to engage in unusual electrostatic interactions with distant residues, such as K16 and E22, resulting in a unique population comprising only the C-terminal hairpin. These findings imply that a single A2X (where X = V or T) mutation in the primarily disordered N-terminus of the Aβ1–42 monomer can dramatically alter the β-hairpin population and switch the equilibrium toward alternative structures. The atomistically detailed, comparative view of the structural landscapes of A2V and A2T variant monomers obtained in this study can enhance our understanding of the mechanistic differences in their early-stage aggregation.
Introduction
Alzheimer's disease (AD) is the most common form of dementia, affecting nearly 38 million people worldwide, a figure that is predicted to double over the next 20 years. The pathological hallmarks of AD are the aberrant deposition of extracellular senile plaques comprised of amyloid-β (Aβ) peptides and intracellular neurofibrillary tangles (1). Proteolytic cleavage of the amyloid precursor protein generates Aβ peptides of different lengths (39–43 residues), among which Aβ1–40 and Aβ1–42 are the major isoforms, with Aβ1–42 being more aggregation-prone and toxic than Aβ1–40 (2). The molecular process underlying AD, as outlined in the amyloid β hypothesis, involves an imbalance between production and clearance of Aβ. This imbalance results in accumulation of amyloid plaques comprised of Aβ aggregates. The abnormal aggregation of Aβ peptides into β-sheet-rich fibrils involves a heterogeneous ensemble of oligomeric intermediates, all of which are found to be neurotoxic (3). Aβ toxicity originates from a number of factors, including formation of ion channels (4), oxidative stress (5), and interaction with receptors (6). NMR studies (7–10) suggest that Aβ1–42 fibrils model as parallel-stacked hairpin-like structures. The hydrophilic N-terminus (residues 1–16) appears unstructured in those fibril structures, whereas the two hydrophobic patches (residues 17–20 and 30–40) form U-shaped conformations comprised of two intermolecular, parallel, in-register β-sheets separated by a hydrophilic turn region (residues 22–29).
Most proposed pathways for the initial stages of Aβ amyloid fibril formation suggest some conformational change (misfolding) of the intrinsically disordered Aβ monomer, resulting in an activated monomer that then recruits other Aβ molecules to form low-n oligomers (11). Based on the protease-resistant nature of residues A21–A30, this region was proposed as the nucleus site for Aβ monomer misfolding (activation) (12). A link between monomer misfolding and aggregation propensity and toxicity has been proposed based on experiments (13–16) and simulations (17–26) of the naturally occurring alloforms. Those alloforms contain substitutions that occur mainly within the turn region (residues 22–30). Because of the disordered structure of the N-terminus in the fibril structure, the role of this region in aggregation, toxicity, and pathogenesis was considered for a long time to be negligible (27) and therefore remained largely unexplored. However, several recent reports indicate the possible importance of the N-terminus in Aβ structure, association, and toxicity (28–30). A novel structural motif of triple β-sheet within Aβ1–42 amyloid oligomers has been reported with minimal exposure of hydrophobic residues, in which association was found between the N-terminus and residues 17–22 (31). Antibodies that specifically bind to the N-terminus are known to effectively interact with both soluble and insoluble forms of Aβ (32). Amyloid inhibitor tetrapeptides are also known to bind at the Aβ N-terminus (33). Two N-terminal mutations, English (H6R) and Tottori (D7N), are reported to alter the monomer misfolding and produce oligomers that are toxic compared with the wild-type (WT) (16). A few recent studies have also suggested the importance of the extreme N-terminus. Experiments reported delayed assembly kinetics and compactness of the D1Y-substituted Aβ40/42 peptides (34) and higher toxicity of an A2F variant (35), a consequence of a more hydrophobic N-terminus. It is known that the double mutation D1E/A2V affects Aβ1–40 fibrillation (36). Pyroglutamate-modified Aβ (Aβ(pE3-42)) peptides have also emerged as potential key factors in AD pathology due to their abundance in the AD brain, high aggregation propensity, stability, and cellular toxicity (37). A familial A2V mutation has been reported to cause dementia in homozygous carriers, whereas heterozygous carriers were found to be protected (38). The co-incubation of WT and A2V Aβ1–42 peptides was shown to inhibit toxicity in the same study (38). The A2V Aβ1–42 also has been shown to have a very different aggregation pathway involving annular oligomers with higher hydrophobic exposure and stronger toxicity compared to the WT; the A2V peptides more rapidly formed a structured oligomer, as demonstrated by dynamic light-scattering experiments (39).
Recently, a rare A2T variant was found to protect against AD and cognitive decline in the elderly without AD (40). This is thought to represent the first example of an Aβ variant that confers protection against AD. The prevailing hypothesis for its protective action is that the mutation (A673T in the amyloid precursor protein or A2T in Aβ), close to the β-secretase cleavage site, lowers Aβ production, thereby balancing production with clearance. However, the reported lowering in production was a moderate 20% in heterozygous carriers (40). The downstream effects of this mutation could also be vital to its protective behavior (41).
The effects of the A2V and A2T mutations on Aβ aggregation and toxicity have just started to emerge, and reports appear somewhat controversial. Two recent studies that employed thioflavin T (ThT) fluorescence monitoring suggest very different effects of the A2T mutation on Aβ1–42 aggregation (42,43). Benilova et al. showed no change in Aβ1–42 aggregation upon either A2T or A2V mutation (43). On the other hand, the results from Maloney et al. suggested a lower aggregation propensity of A2T Aβ1–42 when compared to both WT and A2V with similar aggregation profiles (42). In the context of Aβ1–40, A2T has been shown to aggregate faster (43,44) or similarly (42) compared to WT. On the other hand, A2V Aβ1–40 exhibited faster aggregation than the WT (38,42,43). The effects of those mutations on aggregation also have been analyzed in combination with the WT peptide. Although 1:1 mixture of WT/A2V Aβ1–42 showed intermediate aggregation compared to the pure solutions in a study by Messa et al. (39), the same mixture was reported by Benilova and colleagues (43) to demonstrate fast aggregation similar to that observed for WT Aβ1–42. The latter group reported that a 1:1 WT/A2T mixture exhibited no change in aggregation kinetics (43), but Maloney et al. found an intermediate effect (42).
Maloney et al. (42) also compared the toxicity of WT, A2V, and A2T peptides to cultured neurons and found that WT and A2T peptides exhibited similar toxicity, whereas A2V had somewhat lower toxicity. This is to some extent counterintuitive and contradictory to what was reported for the A2V Aβ1–42 by Di Fede et al. (38). Taken together, the effects of A2V and A2T mutations on early aggregation and toxicity are still not fully clear and seem to be very different depending on the isoform (Aβ1–40 versus Aβ1–42) and experimental conditions. It is also possible that the current experiments are not sensitive enough to capture the effects of those mutations in the more hydrophobic, aggregation-prone, and toxic Aβ1–42 isoform.
Given the protective and causative effects associated with the A2V and A2T substitutions and the established link between monomer misfolding and early aggregation, it is crucial to obtain a comparative view of the conformational landscapes of those two variants and the WT Aβ1–42 full-length monomers. Even with the sophistication of modern techniques, experimental characterization of the monomeric Aβ peptides remains challenging due to the intrinsically disordered and rapidly interchanging nature of the system. On the other hand, molecular dynamics (MD) simulations at different resolutions have been widely used to complement experiments (45) in providing detailed information on the structure of various Aβ species ranging from monomers (23,46–50) to oligomers (51,52) to protofibrils (53) to fibrils (54). Interactions of different Aβ species with toxicity and aggregation inhibitors (55–57) and with lipid bilayers (58,59) also have been studied using MD. In this study, we have performed extensive all-atom replica-exchange molecular dynamics (REMD) simulations of WT, A2V, and A2T Aβ1–42 in explicit water to characterize the monomeric ensemble. To our knowledge, this is the first study comparing the structural landscapes of the A2T and A2V Aβ1–42 monomers. Our simulations reveal an enhanced double-hairpin population in A2V due to hydrophobic clustering between the N-terminus and distant hydrophobic patches. In contrast, the A2T mutation engages the N-terminus in an unusual set of long-range electrostatic interactions with residues such as K16 and E22, resulting in a population of unique conformations that comprise only the C-terminal hairpin. These findings imply that the A2X (X = V or T) mutations situated within the mainly disordered N-terminus can dramatically alter the WT monomeric conformational landscape by globally rewiring the intramolecular interaction network within Aβ1–42.
Materials and Methods
Details of the MD simulations regarding system setup, simulation parameters, simulation length, and so forth can be found in the Supporting Material. The total number of replicas used was 64, and each replica was 175 ns long, resulting in an aggregate simulation time of 11.2 μs per system. A number of convergence checks were performed (see the Supporting Material).
Results
In this work, we compare in silico the monomeric conformational ensemble of the A2V and A2T Aβ1–42 variants with that of WT by performing extensive atomistic REMD simulations in explicit water. All three variants exhibit similar secondary structure propensity, with coils and turns being prevalent (see Fig. S3 a in the Supporting Material). The secondary-structure propensity values for WT are ∼30% coil, ∼45% turn, and ∼18% β-strand; A2V and A2T show similar values. This estimated composition for the WT system in this study agrees well with an earlier report (20), implying that the sampling obtained using the protocol described here represents the WT monomeric structure reasonably and consistently well. The ensemble-averaged secondary structure per residue for each variant is shown in Fig. 1, a–c. The residue-based secondary-structure propensity analysis (Fig. 1, a–c) suggests a primarily disordered N-terminal region (residues 1–15) for all three variants, with a strong turn character around residues 8–9 and 13–15 and a small (5–10%) β-strand propensity in the first 12 residues. Some helical structure near residues 12–16 is also observed. Residues 17–20 (the central hydrophobic cluster (CHC)) and 31–40 show high β-strand propensity (≥25%) in addition to a predominant turn around residues 23–30 in the WT (Fig. 1 a), consistent with the reported strand-loop-strand (SLS) structure of Aβ10–35 (60). Interestingly, A2V demonstrates higher β-strand propensity within residues 16–20, 30–35, and 39–41 compared with the WT. A significant increase in the turn propensity compared to the WT also can be noticed in residues 23–30 and 36–37 of A2V (Fig. 1 b). On the other hand, the β-strand propensity is found to be lower in A2T around residues 18–19, 31–32, and 36–39 (Fig. 1 c). In addition, the turns around residues 23–30 and 36–37 appear more prominent in A2T. These data indicate that a mutation in the extreme N-terminus (at position 2) can alter the secondary structural preference of the distant hydrophobic regions (the CHC and the C-terminus).
Figure 1.
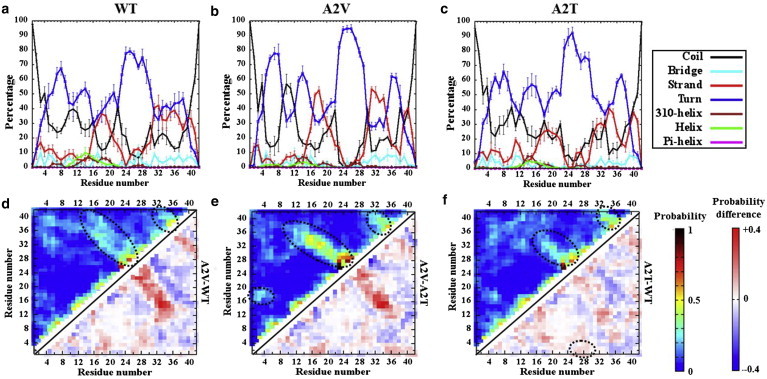
Secondary and tertiary structure. (Top) Secondary structure per residue averaged over the production ensemble: (a) WT, (b) A2V, and (c) A2T. Colors used are shown on the right. The standard deviations were estimated by splitting the data in four equal sets. (Bottom) Tertiary contact maps: (d) WT, (e) A2V, and (f) A2T. (Upper triangle) Ensemble-averaged probability of interresidue Cα-Cα contact formations (nonsequential contacts, i.e. |i-j| ≥3, are only shown) are colored according to the color map shown on the right. (Lower triangle) Arithmetic difference between the contact probabilities for each pair of Aβ variants colored according to the color map shown on the right. Black dotted circles highlight the two sets of antidiagonal contacts as well as the crucial contacts involving residues 1–5, which help discriminating the variants. To see this figure in color, go online.
The Cα-contact map (Fig. 1 d, upper triangle) reveals the presence of strong antidiagonal contacts between the CHC and C-terminus, suggesting a β-hairpin formation in the WT, consistent with earlier simulations (60). β-hairpin formation of residues 17–36 during early Aβ aggregation has been further supported by an affibody-Aβ complex structure (61) and by the presence of a turn-like structure at residues 25–29 in Aβ oligomers (62). Additional antidiagonal contacts appear prevalent in the C-terminal region between residues 33–36 and 39–41, indicative of a second β-hairpin motif. These results imply a double-hairpin topology, in agreement with the simulations performed by Rosenman et al. (20). Long-range interactions between two termini (residues 1–8 and 32–42) are observed in addition to some contacts between the CHC and the extreme N-terminus (residues 1–5). Highly probable local contacts consistently appear in the turn regions.
Fig. 1 e (upper triangle) reveals a more robust nature of those β-hairpin interactions in A2V compared to the WT, suggesting a favored population of double-hairpin structures in A2V. The long-range interactions between the N-terminus (residues 1–4) and the CHC are stronger in A2V (Fig. 2 b, upper triangle) compared with the WT. The difference contact map (Fig. 1 d, lower triangle) further reveals a register shift toward the N-terminus in A2V compared with the WT for both sets of antidiagonal interactions.
Figure 2.
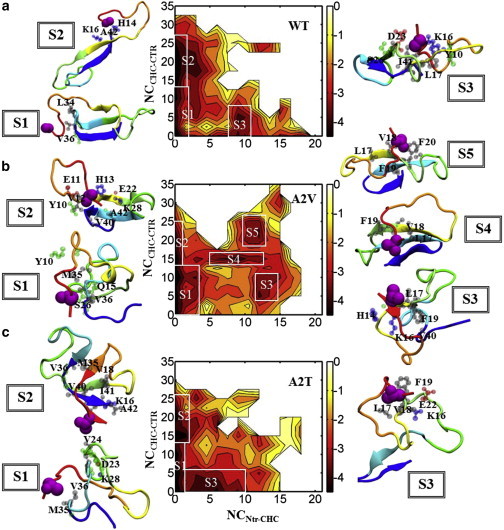
Potential of mean force (PMF) plots: (a) WT, (b) A2V, and (c) A2T. Two-dimensional PMF plots as a function of number of contacts between the Ntr (residues 1–5) and the CHC (residues 16–21), NCNtr-CHC, and number of contacts between the CHC and the C-terminal region (residues 31–42), NCCHC-CTR. Each contour level represents 0.5 kcal/mol. White squares denote the discrete regions on the PMF plots individually representing ≥5% of the production ensemble. The representative conformation of the largest cluster for each of those regions is shown using cartoon representation colored with a red-to-blue rainbow spectrum from the N- to C-terminus. Residue 2 is shown in purple spheres, whereas the residues forming long-range contacts with the N-tr are shown using CPK representation. A 5 Å cutoff between heavy atoms is used for this purpose. The residues are colored according to their types (hydrophobic, gray; polar, green; basic, blue; acidic, red). To see this figure in color, go online.
In the A2T variant (Fig. 1 f, upper triangle), the antiparallel β-hairpin interactions between the CHC and residues 30–35 are weaker than in the WT, whereas the C-terminal β-hairpin appears more robust. Also, a cluster of contacts between residues 5–7 and the CHC is observed in A2T. Interactions between residues 1–4 and 34–42 appear weaker in A2T than in the WT (Fig. 1 f, lower triangle). However, residues 1–4 of A2T make contact more frequently with the 23–30 turn (Fig. 1 f, lower triangle, dashed circle); in general, those contacts are weak. At least 25% of those contacts occur with a ≥10% probability for the A2T variant, compared to 7% probability in the WT and A2V. Taken together, these results suggest that the A2V/T substitution primarily alters 1) the β-hairpin interactions between the CHC and the C-terminus, termed NCCHC-CTR and 2) the interactions between the N-terminus (Ntr) (residues 1–5) and the CHC, termed NCNtr-CHC.
In the following, we compare the potential mean force (PMF) plots (Fig. 2) to discriminate between the conformational ensembles of different variants. It should be noted that as all three peptides are intrinsically disordered, it is nontrivial to choose an optimal set of reaction coordinates for defining their free-energy landscapes. Since from the above analysis, the two sets of contacts, namely NCNtr-CHC and NCCHC-CTR, appear to capture notable differences in the tertiary contact probabilities among variants, we estimated the PMFs as a function of them (see the Supporting Material). The WT PMF plot reveals distinct features of the most populated regions (regions S1–S3) (Fig. 2 a, white boxes) of the landscape. The S1 region corresponds to the structures with strongly limited NCNtr-CHC and NCCHC-CTR contacts. Structures with robust CHC-CTR interaction accompanied by a few (≤5) Ntr-CHC contacts populate the S2 region. In contrast, S3 conformations exhibit stronger Ntr-CHC interaction but lack substantial CHC-CTR contacts, and they are much less populated (compared to S1 and S2). We also performed a cluster analysis with a 3 Å Cα-RMSD cutoff distance on those discrete regions. The definition criteria, population size, and cluster analysis results for those regions are listed in Table S1, demonstrating that those structures all together represent >75% of the total ensemble for each system. The total number of clusters and the size of the top five clusters show that the S1 structures are more disordered than S2 structures, whereas S3 can be well represented by a single cluster. The representative structure for the largest cluster of each region is shown in Fig. 2 a. Additional representative structures, as well as the tertiary contact maps and the residue-based secondary-structure propensities, can be found in Fig. S4. Those data confirm that the WT S1 conformations are mostly disordered and show very little secondary and tertiary structure (Fig. S4 a). The only structured region in the S1 conformations is the C-terminus, with a 20% β-hairpin population. In contrast, both sets of hairpin contacts are evident in S2 structures, the C-terminal hairpin being slightly weaker than the CHC-CTR one (Fig. S4 b). Interestingly, the minor S3 population consists of interaction of the 23–30 turn with both termini, which is absent in S1 and S2 structures (Fig. S4 c). Representative conformations illustrated in Figs. 2 a and S4 support these results.
The S1 and S2 regions in A2V (Figs. 2 b and S5, a and b) and A2T (Figs. 2 c and S6, a and b) monomers show similar features overall when compared to the WT. Interestingly, A2V samples additional structures containing significant Ntr-CHC and CHC-CTR interactions (Fig. 2 b, S4 and S5 regions); such conformations are rarely observed for the WT and A2T. The representative structures from those regions imply that a nonpolar amino acid at position 2 (i.e., Val) forms a hydrophobic cluster involving hydrophobic side chains from CHC, resulting in further stabilization of CHC-CTR contacts (Fig. 2 b). Accordingly, the secondary-structure analyses and tertiary contact maps for S4 and S5 structures (Fig. 3, middle and right) reveal double-hairpin-like conformations. Those conformations are regularly populated in A2V (S4 + S5 represent 15% of the total ensemble (Table S1)), as compared with the WT. Lowering of disorder can be seen in those double-hairpin conformations, as revealed from the total number of clusters and the size of the top five clusters (Table S1). Fig. 3 further shows that both sets of hairpin interactions experience a subtle register shift toward the N-terminus in a subpopulation of those double-hairpin structures (the S4 region). The Ntr-CHC contacts are also found in the S3 structures of A2V; however, in those structures, the C-terminus remains in a collapsed coil structure (Fig. 3, left).
Figure 3.
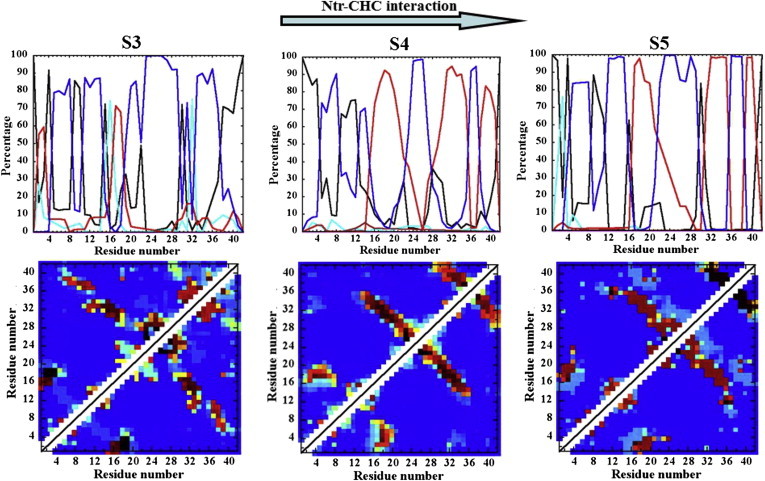
Analysis of the A2V conformations with enhanced Ntr-CHC interaction. The residue-based secondary structure and tertiary Cα-Cα contact maps for A2V conformations corresponding to the S3 (left), S4 (middle), and S5 (right) regions. S4 and S5 structures demonstrate double-hairpin topology triggered by Ntr-CHC contacts. Color schemes used are the same as in Figs. 1 and 2. The top arrow indicates the relative probability of Ntr-CHC interaction. To see this figure in color, go online.
In sharp contrast, A2T more frequently visits conformations with significant Ntr-CHC interaction but much weaker CHC-CTR contacts (Fig. 2 c, S3 region). Interestingly, this region of the landscape is rarely populated for both WT and A2V, but the population for A2T is a significant 17% of the total ensemble (Table S1). Further analysis reveals a weaker CHC-CTR interaction resulting in the loss of the first hairpin present in those unique S3 structures (Figs. 2 c and 4, a and b). Consequently, those structures contain only the C-terminal β-hairpin (Fig. 4, a and b). The side-chain-side-chain contact map illustrates strong interaction between the N-terminus (residues 1–6) and residues 16–22 (Fig. S7); however, residue T2 appears to be weakly involved in those interactions and remains solvent-exposed. In particular, strong D1-K16 and R5-E22 side-chain-side-chain contacts are observed. Accordingly, the representative structure illustrates an ionic network comprised of residues D1, E3, R5, K16, and E22 (Fig. 4 c). The salt-bridge population analysis (Table S2) indicates that such ionic interaction becomes more probable in the presence of a solvent-exposed threonine at position 2. For example, the D1-K16 salt bridge is populated with a probability of 34% in S3 structures. At the same time, the formation of the E22-K28 salt bridge becomes less probable (5% (Table S2)) in S3 structures compared to what is observed in S1 and S2 (35% and 12% probabilities, respectively). Taken together, the altered ionic network between the N-terminus and distant charged residues, such as K16 and E22, appears to hinder the formation of the first hairpin interaction in the S3 structures of A2T.
Figure 4.
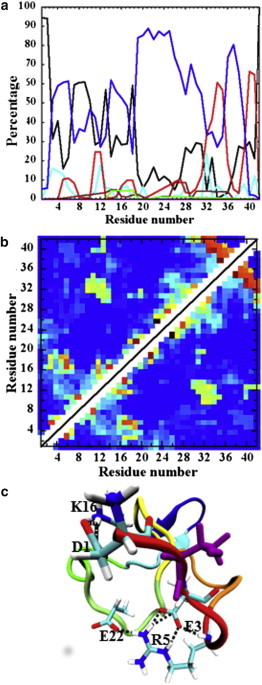
Unique A2T conformations with altered ionic network. (a) Residue-based secondary structure, (b) tertiary Cα-Cα contact map, and (c) ionic network within the representative conformation of the largest S3 cluster. The residues, i.e., D1, E3, R5, K16, and E22, forming long-range ionic interaction, are shown using licorice representation. Hydrogen bonds are shown in black dotted lines. Residue Thr2 is shown in purple. Color schemes used are the same as in Figs. 1 and 2. To see this figure in color, go online.
Discussion
Given the reported protective and causative effects associated with A2V and A2T substitutions vis à vis AD, it is of importance to understand the differences in the self-assembly process of those variant peptides, which is intimately associated with the disease pathogenesis. The first step toward this goal is to characterize the conformational landscapes of those variant monomers and compare them with the WT. To our knowledge, this is the first study to compare the structural landscapes of the A2T and A2V Aβ1–42 monomers. Our findings from extensive REMD simulations report significant differences in terms of the transient structure population in those variants, which is modulated by interactions of the N-terminus with hydrophobic residues (the CHC) in the case of A2V or with charged residues (K16 and E22) in the case of A2T.
The more persistent β-hairpin character in the hydrophobic regions that lead to a double-hairpin topology in the A2V monomer, as revealed in our simulations, could have important implications in determining the aggregation and toxicity of A2V. This monomer has been reported to follow a peculiar aggregation behavior that leads to the formation of annular structures with a higher hydrophobicity profile and hence higher toxicity in vitro (39) as well as in vivo (63). A2V also showed greater β-conformation propensity during aggregation (64). The formation of β-structures is believed to be important in amyloid aggregation and toxicity. For example, β-hairpin monomeric conformations in amyloid peptides are found to represent metastable states that interconvert slowly and thus can act as the seed for nucleation (65). Experiments have also highlighted the importance of β-hairpin stabilization in the Aβ monomer in inducing a toxic soluble oligomer population (61,66,67). NMR and atomic force microscopy (AFM) experiments (68) have suggested the presence of double-hairpin conformations within toxic Aβ1–42 oligomers. Double-cysteine mutants (Aβ40cc and Aβ42cc) with overly stable β-hairpin monomeric conformation have been shown to lower fibril formation and increase populations of toxic β-sheet oligomers and/or protofibrils (67). A structural comparison of monomeric Aβ1–42 and fibrillar Aβ1–42 suggests that the monomeric hairpin conformation needs to retain sufficient flexibility to undergo a concerted conformational change toward the extended parallel β-sheet structure found in fibrils (61), as also observed in MD simulations of Aβ10–35 dimers and trimers (66). The fact that double-hairpin structures were only observed in Aβ1–42 and not in Aβ1–40 has been linked to the higher aggregation propensity and toxicity of the longer isoform (20). Taken together, our simulations, combined with results of previous studies, imply that the transiently formed double-hairpin structures of the A2V monomer might contribute to the stabilization of small, toxic oligomers composed of β-structures, thus inhibiting fibrillation. Thus, a single-point nonpolar substitution at position 2 of Aβ1–42 might act as a switch to redirect the peptides to aggregate via a neurotoxic pathway by stabilizing the β-hairpin conformation (Fig. 5). Our results are consistent with simulations of the shorter A2V Aβ1–28 monomer, which indicated that a valine at position 2 can favor Ntr-CHC interaction (69). Nevertheless, the simulations of the full-length A2V peptide presented here demonstrate how the hydrophobic Ntr-CHC interaction can lead to enhanced CHC-CTR hairpin population, leading to the double-hairpin topology (Fig. 5).
Figure 5.
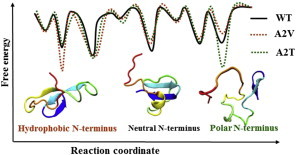
Schematic illustration of the Aβ1–42 N-terminus influence on the monomeric landscape. The curves show free energy of the variant monomers in one dimension. Based on the analysis, the reaction coordinate might represent a combination of the CHC-CTR interaction and Ntr-peptide interaction. The N-terminal substitution, depending on its hydrophobic or polar nature, can switch the relative population among different β-hairpin conformational states present in the WT landscape. It should be noted that this is an overly simplified representation of the Aβ1–42 landscape, and many other conformational states are present due to the intrinsically disordered nature of the system. To see this figure in color, go online.
Our simulations further suggest a competition between specific hydrophobic and electrostatic interactions modulated by the N-terminus within the Aβ1–42 monomer, which in turn regulates the delicate balance among the local minima present in its frustrated conformational landscape (Fig. 5). As discussed above, the N-terminus in the A2V variant stabilizes the double-hairpin conformation by clustering with distant hydrophobic residues (Fig. 5). In contrast, the A2T variant, with its more polar N-terminus, leads to unique structure populations that are rarely seen for the WT and A2V. Although Thr-2 itself minimally interacts with the rest of the protein in those conformations, its presence allows the N-terminus (e.g., residues D1, E3, and R5) to form an unusual set of ionic interactions with distant residues, such as K16 and E22. This effect impairs the formation of the E22-K28 salt-bridge that is known to be crucial for CHC-CTR hairpin formation (18). In consequence, those structures comprise only the C-terminal short β-hairpin (Fig. 5).
Mutations at residues E22 (E22K, E22G, and E22Q) and D23 (D23N) that affect Aβ aggregation and cause early-onset AD (15) are reported to alter the WT 21–30 turn structure in simulations (70). Two N-terminal mutations (H6R and D7N) also have been found to affect the extent of β-structure and salt-bridge network within the monomer (71,72). Nevertheless, those mutations directly change the charge distribution within the peptide, which is not the case for A2T. At first glance, a threonine at position 2 would not be expected to have a dramatic effect on the Aβ1–42 conformational landscape by altering the electrostatic interaction network. It appears that Thr-2 changes the local conformational preference of the N-terminus, consequently promoting alternative electrostatic interactions that compete with those required for turn nucleation and CHC-CTR hairpin formation. This leads to population of rare and unique conformations (S3 structures). Since β-hairpin abundance has been linked with aggregation mechanisms and toxicity associated with AD (17,73), the unique A2T conformations devoid of the CHC-CTR β-hairpin that also have an altered electrostatic interaction network might contribute toward its slower aggregation (42). It is noteworthy that a threonine in position 2 appears to introduce more disorder within Aβ1–42, whereas a valine at the same position induces some order, which overall is consistent with the intrinsic (dis)order-promoting nature of those amino acids (74). However, our simulations reveal that this effect arises from the global interaction changes triggered by a single amino acid substitution. Interestingly, conformation-specific antibody staining experiments imply the involvement of the N-terminus in tertiary interactions in the oligomers of both A2V and A2T variants (43). Our simulations demonstrate that such an interaction is also present at the monomer level and is strikingly different between the two variants, which might play a critical role in their early aggregation by populating different monomer structures. Since the Aβ N-terminus also has been suggested to be crucial in binding with membrane (75) and with some toxic Aβ receptors (76), the altered interaction pattern observed in the A2V and A2T monomers may also affect their toxicity (77).
If the interaction between the extreme N-terminus and the rest of the peptide is indeed critical for aggregation and toxicity, then it may be possible, for example, to use small organic molecules (78) and peptides (79,80) that would bind to Aβ monomers and/or oligomers and inhibit aggregation and/or toxicity by intervening in or altering this interaction. The role of the extreme N-terminus in modulating the Aβ1–42 monomeric hairpin structure revealed here may guide the design of inhibitory peptides, including variants of the WT (1DAEFRH6) sequence (64).
Conclusions
Taken together, our findings suggest that the effect of the second amino acid on the Aβ1–42 monomer structure is highly complex and sequence-dependent. An enhanced double-hairpin population similar to those reported in toxic WT Aβ1–42 oligomers is found in the A2V monomer. Hydrophobic clustering between the N-terminus and the central and C-terminus hydrophobic patches promotes such double-hairpin formation in A2V. In contrast, the A2T mutation triggers unusual ionic interactions of the N-terminus with K16 and E22, thereby impeding CHC-CTR hairpin formation. Consequently, a unique population comprising only the C-terminal hairpin is observed. Although further investigation is needed to obtain a complete molecular picture of the relationship between monomer misfolding, aggregation, and toxicity, and protection against or causation of AD by these N-terminal variants, the simulations described here clearly show that single A2V and A2T substitutions can alter the structural landscape of the Aβ1–42 monomer by shifting the equilibrium to different conformational states.
Acknowledgments
We thank Mirco Sorci for useful discussions and Camilo A. Jimenez Cruz for comments on the manuscript.
Support from IBM BlueGene Science Program is acknowledged.
Supporting Material
References
- 1.Selkoe D.J. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Klein A.M., Kowall N.W., Ferrante R.J. Neurotoxicity and oxidative damage of β amyloid 1–42 versus β amyloid 1–40 in the mouse cerebral cortex. Ann. N. Y. Acad. Sci. 1999;893:314–320. doi: 10.1111/j.1749-6632.1999.tb07845.x. [DOI] [PubMed] [Google Scholar]
- 3.Shankar G.M., Bloodgood B.L., Sabatini B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonda D.J., Wang X., Smith M.A. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59:290–294. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Sakono M., Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 7.Petkova A.T., Yau W.-M., Tycko R. Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato T., Kienlen-Campard P., Smith S.O. Inhibitors of amyloid toxicity based on β-sheet packing of Aβ40 and Aβ42. Biochemistry. 2006;45:5503–5516. doi: 10.1021/bi052485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lührs T., Ritter C., Riek R. 3D structure of Alzheimer’s amyloid-β1–42 fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petkova A.T., Ishii Y., Tycko R. A structural model for Alzheimer’s β -amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor B.M., Sarver R.W., Epps D.E. Spontaneous aggregation and cytotoxicity of the β-amyloid Aβ1–40: a kinetic model. J. Protein Chem. 2003;22:31–40. doi: 10.1023/a:1023063626770. [DOI] [PubMed] [Google Scholar]
- 12.Lazo N.D., Grant M.A., Teplow D.B. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14:1581–1596. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hori Y., Hashimoto T., Iwatsubo T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007;282:4916–4923. doi: 10.1074/jbc.M608220200. [DOI] [PubMed] [Google Scholar]
- 14.Bitan G., Kirkitadze M.D., Teplow D.B. Amyloid β -protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murakami K., Irie K., Shirasawa T. Neurotoxicity and physicochemical properties of Aβ mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer’s disease. J. Biol. Chem. 2003;278:46179–46187. doi: 10.1074/jbc.M301874200. [DOI] [PubMed] [Google Scholar]
- 16.Ono K., Condron M.M., Teplow D.B. Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 2010;285:23186–23197. doi: 10.1074/jbc.M109.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang M., Teplow D.B. Amyloid β-protein monomer folding: free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 2008;384:450–464. doi: 10.1016/j.jmb.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baumketner A., Bernstein S.L., Shea J.E. Amyloid β-protein monomer structure: a computational and experimental study. Protein Sci. 2006;15:420–428. doi: 10.1110/ps.051762406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urbanc B., Cruz L., Stanley H.E. In silico study of amyloid β-protein folding and oligomerization. Proc. Natl. Acad. Sci. USA. 2004;101:17345–17350. doi: 10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenman D.J., Connors C.R., García A.E. Aβ monomers transiently sample oligomer and fibril-like configurations: ensemble characterization using a combined MD/NMR approach. J. Mol. Biol. 2013;425:3338–3359. doi: 10.1016/j.jmb.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam A.R., Teplow D.B., Urbanc B. Effects of the Arctic (E22—>G) mutation on amyloid β-protein folding: discrete molecular dynamics study. J. Am. Chem. Soc. 2008;130:17413–17422. doi: 10.1021/ja804984h. [DOI] [PubMed] [Google Scholar]
- 22.Krone M.G., Baumketner A., Shea J.E. Effects of familial Alzheimer’s disease mutations on the folding nucleation of the amyloid β-protein. J. Mol. Biol. 2008;381:221–228. doi: 10.1016/j.jmb.2008.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ball K.A., Phillips A.H., Head-Gordon T. Homogeneous and heterogeneous tertiary structure ensembles of amyloid-β peptides. Biochemistry. 2011;50:7612–7628. doi: 10.1021/bi200732x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cote S., Derreumaux P., Mousseau N. Distinct morphologies for amyloid β protein monomer: Aβ1–40, Aβ1–42, and Aβ1–40 (D23N) J. Chem. Theory Comput. 2011;7:2584–2592. doi: 10.1021/ct1006967. [DOI] [PubMed] [Google Scholar]
- 25.Mitternacht S., Staneva I., Irbäck A. Comparing the folding free-energy landscapes of Aβ42 variants with different aggregation properties. Proteins. 2010;78:2600–2608. doi: 10.1002/prot.22775. [DOI] [PubMed] [Google Scholar]
- 26.Urbanc B., Cruz L., Dokholyan N.V. Molecular dynamics simulation of amyloid β dimer formation. Biophys. J. 2004;87:2310–2321. doi: 10.1529/biophysj.104.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda T., Klimov D.K. Probing the effect of amino-terminal truncation for Aβ1–40 peptides. J. Phys. Chem. B. 2009;113:6692–6702. doi: 10.1021/jp9016773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheidt H.A., Morgado I., Huster D. Dynamics of amyloid β fibrils revealed by solid-state NMR. J. Biol. Chem. 2011;287:2012–2021. doi: 10.1074/jbc.M111.308619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lv Z., Roychaudhuri R., Lyubchenko Y.L. Mechanism of amyloid β-protein dimerization determined using single-molecule AFM force spectroscopy. Sci. Rep. 2013;3:2880. doi: 10.1038/srep02880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarkar B., Mithu V.S., Maiti S. Significant structural differences between transient amyloid-β oligomers and less-toxic fibrils in regions known to harbor familial Alzheimer’s mutations. Angew. Chem. Int. Ed. Engl. 2014;53:6888–6892. doi: 10.1002/anie.201402636. [DOI] [PubMed] [Google Scholar]
- 31.Ma B., Nussinov R. Polymorphic triple β-sheet structures contribute to amide hydrogen/deuterium (H/D) exchange protection in the Alzheimer amyloid β42 peptide. J. Biol. Chem. 2011;286:34244–34253. doi: 10.1074/jbc.M111.241141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zago W., Buttini M., Kinney G.G. Neutralization of soluble, synaptotoxic amyloid β species by antibodies is epitope specific. J. Neurosci. 2012;32:2696–2702. doi: 10.1523/JNEUROSCI.1676-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Du Z., Bitan G. C-terminal tetrapeptides inhibit Aβ42-induced neurotoxicity primarily through specific interaction at the N-terminus of Aβ42. J. Med. Chem. 2011;54:8451–8460. doi: 10.1021/jm200982p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maji S.K., Ogorzalek Loo R.R., Teplow D.B. Amino acid position-specific contributions to amyloid β-protein oligomerization. J. Biol. Chem. 2009;284:23580–23591. doi: 10.1074/jbc.M109.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luheshi L.M., Tartaglia G.G., Crowther D.C. Systematic in vivo analysis of the intrinsic determinants of amyloid β pathogenicity. PLoS Biol. 2007;5:e290. doi: 10.1371/journal.pbio.0050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qahwash I., Weiland K.L., Yan R. Identification of a mutant amyloid peptide that predominantly forms neurotoxic protofibrillar aggregates. J. Biol. Chem. 2003;278:23187–23195. doi: 10.1074/jbc.M213298200. [DOI] [PubMed] [Google Scholar]
- 37.Jawhar S., Wirths O., Bayer T.A. Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. J. Biol. Chem. 2011;286:38825–38832. doi: 10.1074/jbc.R111.288308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Fede G., Catania M., Tagliavini F. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messa M., Colombo L., Salmona M. The peculiar role of the A2V mutation in amyloid-β (Aβ) 1–42 molecular assembly. J. Biol. Chem. 2014;289:24143–24152. doi: 10.1074/jbc.M114.576256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jonsson T., Atwal J.K., Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 41.De Strooper B., Voet T. Alzheimer’s disease: A protective mutation. Nature. 2012;488:38–39. doi: 10.1038/488038a. [DOI] [PubMed] [Google Scholar]
- 42.Maloney J.A., Bainbridge T., Atwal J.K. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 2014;289:30990–31000. doi: 10.1074/jbc.M114.589069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benilova I., Gallardo R., De Strooper B. The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 2014;289:30977–30989. doi: 10.1074/jbc.M114.599027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meinhardt J., Tartaglia G.G., Fändrich M. Similarities in the thermodynamics and kinetics of aggregation of disease-related Aβ (1–40) peptides. Protein Sci. 2007;16:1214–1222. doi: 10.1110/ps.062734207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma B., Nussinov R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol. 2006;10:445–452. doi: 10.1016/j.cbpa.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 46.Kirshenbaum K., Daggett V. pH-dependent conformations of the amyloid β (1–28) peptide fragment explored using molecular dynamics. Biochemistry. 1995;34:7629–7639. doi: 10.1021/bi00023a009. [DOI] [PubMed] [Google Scholar]
- 47.Baumketner A., Shea J.-E. The structure of the Alzheimer amyloid β10–35 peptide probed through replica-exchange molecular dynamics simulations in explicit solvent. J. Mol. Biol. 2007;366:275–285. doi: 10.1016/j.jmb.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 48.Rojas A.V., Liwo A., Scheraga H.A. A study of the α-helical intermediate preceding the aggregation of the amino-terminal fragment of the β amyloid peptide (Aβ1–28) J. Phys. Chem. B. 2011;115:12978–12983. doi: 10.1021/jp2050993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flöck D., Colacino S., Di Nola A. Misfolding of the amyloid β-protein: a molecular dynamics study. Proteins. 2006;62:183–192. doi: 10.1002/prot.20683. [DOI] [PubMed] [Google Scholar]
- 50.Lin Y.-S., Bowman G.R., Pande V.S. Investigating how peptide length and a pathogenic mutation modify the structural ensemble of amyloid β monomer. Biophys. J. 2012;102:315–324. doi: 10.1016/j.bpj.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gnanakaran S., Nussinov R., García A.E. Atomic-level description of amyloid β-dimer formation. J. Am. Chem. Soc. 2006;128:2158–2159. doi: 10.1021/ja0548337. [DOI] [PubMed] [Google Scholar]
- 52.Zhu X., Bora R.P., Prabhakar R. Dimerization of the full-length Alzheimer amyloid β-peptide (Aβ42) in explicit aqueous solution: a molecular dynamics study. J. Phys. Chem. B. 2012;116:4405–4416. doi: 10.1021/jp210019h. [DOI] [PubMed] [Google Scholar]
- 53.Fawzi N.L., Kohlstedt K.L., Head-Gordon T. Protofibril assemblies of the arctic, Dutch, and Flemish mutants of the Alzheimer’s Aβ1–40 peptide. Biophys. J. 2008;94:2007–2016. doi: 10.1529/biophysj.107.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buchete N.-V., Tycko R., Hummer G. Molecular dynamics simulations of Alzheimer’s β-amyloid protofilaments. J. Mol. Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 55.Convertino M., Pellarin R., Caflisch A. 9,10-Anthraquinone hinders β-aggregation: how does a small molecule interfere with Aβ-peptide amyloid fibrillation? Protein Sci. 2009;18:792–800. doi: 10.1002/pro.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chebaro Y., Jiang P., Derreumaux P. Structures of Aβ17–42 trimers in isolation and with five small-molecule drugs using a hierarchical computational procedure. J. Phys. Chem. B. 2012;116:8412–8422. doi: 10.1021/jp2118778. [DOI] [PubMed] [Google Scholar]
- 57.Das P., Kang S.G., Belfort G. Interaction of amyloid inhibitor proteins with amyloid β peptides: insight from molecular dynamics simulations. PLoS ONE. 2014;9:e113041. doi: 10.1371/journal.pone.0113041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jang H., Zheng J., Nussinov R. Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 2007;93:1938–1949. doi: 10.1529/biophysj.107.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jang H., Connelly L., Nussinov R. Mechanisms for the insertion of toxic, fibril-like β-amyloid oligomers into the membrane. J. Chem. Theory Comput. 2013;9:822–833. doi: 10.1021/ct300916f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han W., Wu Y.D. A strand-loop-strand structure is a possible intermediate in fibril elongation: long time simulations of amyloid-β peptide (10–35) J. Am. Chem. Soc. 2005;127:15408–15416. doi: 10.1021/ja051699h. [DOI] [PubMed] [Google Scholar]
- 61.Hoyer W., Grönwall C., Härd T. Stabilization of a β-hairpin in monomeric Alzheimer’s amyloid-β peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. USA. 2008;105:5099–5104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu L., Edalji R., Olejniczak E.T. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 63.Diomede L., Di Fede G., Salmona M. Expression of A2V-mutated Aβ in Caenorhabditis elegans results in oligomer formation and toxicity. Neurobiol. Dis. 2014;62:521–532. doi: 10.1016/j.nbd.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Di Fede G., Catania M., Tagliavini F. Good gene, bad gene: new APP variant may be both. Prog. Neurobiol. 2012;99:281–292. doi: 10.1016/j.pneurobio.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 65.Qiao Q., Bowman G.R., Huang X. Dynamics of an intrinsically disordered protein reveal metastable conformations that potentially seed aggregation. J. Am. Chem. Soc. 2013;135:16092–16101. doi: 10.1021/ja403147m. [DOI] [PubMed] [Google Scholar]
- 66.Larini L., Shea J.-E. Role of β-hairpin formation in aggregation: the self-assembly of the amyloid-β25–35 peptide. Biophys. J. 2012;103:576–586. doi: 10.1016/j.bpj.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sandberg A., Luheshi L.M., Härd T. Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc. Natl. Acad. Sci. USA. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahmed M., Davis J., Smith S.O. Structural conversion of neurotoxic amyloid-β1–42 oligomers to fibrils. Nat. Struct. Mol. Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nguyen P.H., Tarus B., Derreumaux P. Familial Alzheimer A2 V mutation reduces the intrinsic disorder and completely changes the free energy landscape of the Aβ1–28 monomer. J. Phys. Chem. B. 2014;118:501–510. doi: 10.1021/jp4115404. [DOI] [PubMed] [Google Scholar]
- 70.Murray M.M., Krone M.G., Bowers M.T. Amyloid β-protein: experiment and theory on the 21–30 fragment. J. Phys. Chem. B. 2009;113:6041–6046. doi: 10.1021/jp808384x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Viet M.H., Nguyen P.H., Li M.S. Effect of the English familial disease mutation (H6R) on the monomers and dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2014;5:646–657. doi: 10.1021/cn500007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Viet M.H., Nguyen P.H., Derreumaux P. Effect of the Tottori familial disease mutation (D7N) on the monomers and dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2013;4:1446–1457. doi: 10.1021/cn400110d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sgourakis N.G., Yan Y., Garcia A.E. The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Le Gall T., Romero P.R., Dunker A.K. Intrinsic disorder in the protein data bank. J. Biomol. Struct. Dyn. 2007;24:325–342. doi: 10.1080/07391102.2007.10507123. [DOI] [PubMed] [Google Scholar]
- 75.Urbanc B., Betnel M., Bitan G. Structural basis for Aβ1–42 toxicity inhibition by Aβ C-terminal fragments: discrete molecular dynamics study. J. Mol. Biol. 2011;410:316–328. doi: 10.1016/j.jmb.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kam T.-I., Song S., Jung Y.K. FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer’s disease. J. Clin. Invest. 2013;123:2791–2802. doi: 10.1172/JCI66827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hashimoto Y., Matsuoka M. A mutation protective against Alzheimer’s disease renders amyloid β precursor protein incapable of mediating neurotoxicity. J. Neurochem. 2014;130:291–300. doi: 10.1111/jnc.12717. [DOI] [PubMed] [Google Scholar]
- 78.Bleiholder C., Do T.D., Bowers M.T. Ion mobility spectrometry reveals the mechanism of amyloid formation of Aβ25–35 and its modulation by inhibitors at the molecular level: epigallocatechin gallate and scyllo-inositol. J. Am. Chem. Soc. 2013;135:16926–16937. doi: 10.1021/ja406197f. [DOI] [PubMed] [Google Scholar]
- 79.Gessel M.M., Wu C., Bowers M.T. Aβ39–42 modulates Aβ oligomerization but not fibril formation. Biochemistry. 2012;51:108–117. doi: 10.1021/bi201520b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Esteras-Chopo A., Morra G., Colombo G. A molecular dynamics study of the interaction of D-peptide amyloid inhibitors with their target sequence reveals a potential inhibitory pharmacophore conformation. J. Mol. Biol. 2008;383:266–280. doi: 10.1016/j.jmb.2008.07.076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.