

Abstract
Protein-protein docking programs can give valuable insights into the structure of protein complexes in the absence of an experimental complex structure. Web interfaces can facilitate the use of docking programs by structural biologists. Here, we present an easy web interface for protein-protein docking with the ATTRACT program. While aimed at nonexpert users, the web interface still covers a considerable range of docking applications. The web interface supports systematic rigid-body protein docking with the ATTRACT coarse-grained force field, as well as various kinds of protein flexibility. The execution of a docking protocol takes up to a few hours on a standard desktop computer.
Main Text
Protein-protein interactions are abundant in the cell and are involved in many important biological processes. Structural data at atomistic resolution are required to understand the assembly of proteins into complexes carrying out specific cellular functions. This resolution can be obtained by experimental methods such as x-ray crystallography, nuclear magnetic resonance spectroscopy, and more recently, cryo-electron microscopy (cryo-EM). However, these methods often have limitations regarding protein size, flexibility, and/or strength of the interaction. These limitations can be alleviated by combining experimental knowledge with computational methods such as protein-protein docking. The CAPRI challenge (1) has shown that protein-protein docking programs are often able to accurately predict the structure of an unknown protein complex from the known structures of its constituents. Moreover, many docking programs can now easily be accessed via web interfaces and servers (2–9). With the large user base acquired by many of these web services, docking has been established as a valuable tool for the structural biology community.
The ATTRACT docking program (10) has been used successfully to predict complex structures in various rounds of CAPRI (11–13). Characteristic features of ATTRACT are its coarse-grained force field (10,14), the use of protein flexibility throughout the docking search (12,15), and the simultaneous docking of any number of protein bodies (16). ATTRACT has been applied to the docking of proteins to DNA (17), RNA (18), and small ligands (19), as well as the assembly of large molecular machines using cryo-EM data (16). All-atom flexible refinement of ATTRACT-generated models can be performed by the iATTRACT protocol (20). Thus, ATTRACT can tackle a large variety of docking problems, due to an extensive set of features and options.
ATTRACT is implemented as a suite of command line tools and options that can be combined at will. Therefore, ATTRACT is typically invoked via a custom, hand-written shell script. Although this approach is very flexible, it limits the accessibility of ATTRACT to expert users only. However, with the SPYDER framework (used in similar web servers (6,21)), it is possible to generate docking protocols automatically, based on a set of parameters that can be edited in a web browser.
Here, we present an easy web interface for setting up protein docking in the program ATTRACT. While aimed at nonexpert users, the protocols that can be generated still cover a considerable range of docking applications. The web interface supports systematic rigid-body protein docking, as well as various kinds of protein flexibility. The execution of a docking protocol takes just a few minutes to a few hours on a standard desktop machine. The web interface and a detailed manual are available at www.attract.ph.tum.de.
ATTRACT Easy web interface
The ATTRACT easy web interface provides a convenient way to set up an ab initio two-body protein-protein docking protocol. On the one hand, it is sufficient to provide just a Protein Data Bank (PDB) file for both protein partners. On the other hand, a number of options are available (but not required) to customize the protocol. For example, the web interface offers several possibilities to include protein flexibility in the docking search. If an induced fit model of binding is hypothesized, the “Harmonic Modes” option can be enabled, selecting collective modes that will be calculated from an elastic network model (19). The protein will then be deformed along these modes during the docking. Alternatively, an ensemble of multiple rigid conformations can be provided, allowing the most likely conformation to be selected during the docking. Regardless, the initial docking search may be followed by a flexible refinement using the iATTRACT protocol (20). Finally, for benchmarking purposes, the docking results can be assessed against a user-supplied reference structure with the same statistics as used in CAPRI. A detailed overview of all parameters used in the ATTRACT easy web interface can be found in the online manual (under the Help menu). A flowchart illustrating the usage of the ATTRACT Easy web interface is hown in Fig. 1.
Figure 1.
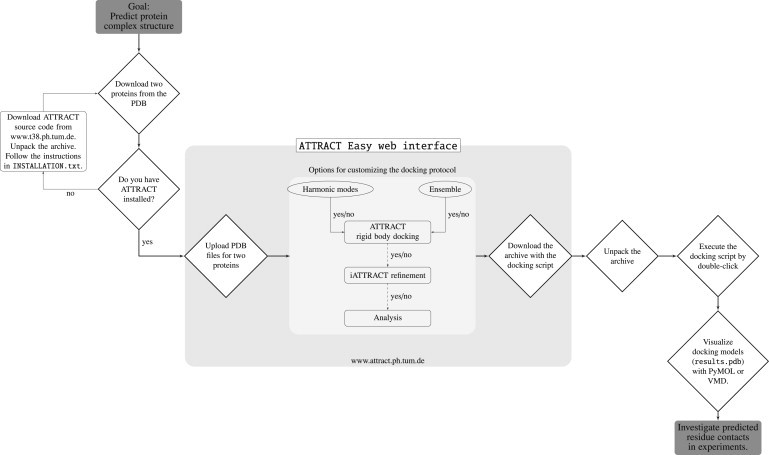
A flow chart illustrating the usage of the ATTRACT easy web interface.
The ATTRACT web interface is not a web server: it returns a docking protocol (shell script) for execution on a local machine. ATTRACT is easily installed under LINUX or a similar operating system; in addition, we provide an ATTRACT VirtualBox that can be launched from within any operating system. Once downloaded, the docking script is launched by a simple double-click. Thus, the user retains full control over the docking process. As the user gains more experience with ATTRACT, the generated docking script may become a starting point for modification, adding, e.g., symmetry, clustering, experimental restraints, or multibody docking to the protocol. Thus, the easy web interface provides a user-friendly, general-purpose entry point for protein docking with ATTRACT. In the future, we plan to develop advanced web interfaces that expose more of the functionality of ATTRACT, as well as specialized web interfaces, e.g., for protein-nucleic acid docking and for assembly of proteins into cryo-EM maps (ATTRACT-EM).
Application to viral chemokine binding protein M3 interacting with chemokine MCP-1
Chemokines are a group of small signaling proteins that induce chemotaxis in nearby cells. In the case of an inflammation, chemokines are secreted by the cell and establish concentration gradients by attaching to the extracellular matrix thereby recruiting leukocytes and triggering the immune response. The murine gammaherpes virus-68 (γHV68) evades detection by the immune system with the help of the chemokine binding protein M3. The M3 protein is critical to the induction of lethal meningitis by γHV68 (22). γHV68 is closely related to human herpes virus 8 and Epstein-Barr virus (23).
The ATTRACT easy web interface was used to set up a docking run and predict the complex structure of viral chemokine binding protein M3 from HV68 with murine chemokine MCP-1. For both individual protein partners, experimental structures were available (PDB 1MKF and PDB 1DOL) (24,25). The protein complex has also been previously characterized (PDB 1ML0) (24) and will be used as a reference to assess the performance of the docking and the quality of the prediction. To account for protein flexibility, five soft harmonic modes (15,19) were specified for each protein partner.
As the top-ranked model, a structure of two-star CAPRI quality was obtained (interface root-mean-squared deviation (iRMSD) 1.8 Å) and the top 100 ATTRACT models contained several other two-star solutions. One of these had an iRMSD of 1.4 Å and retrieved >70% of the native contacts (Fig. 2). Because the iRMSD difference (1) between bound and unbound protein structures of this complex is 1.14 Å, this value is also the theoretical limit for rigid body docking of the unbound protein structures. Both the binding site and the correct binding mode are successfully predicted by ATTRACT. Previously, mutational experiments revealed an important role for the residue Y13 in the MCP-1 N-loop (24). This contact was also identified by the best ATTRACT models (Fig. 2) including the top-ranked complex. Crucial interface residues can thus be predicted and docking-derived information about native contacts could be explored in selected mutational experiments.
Figure 2.
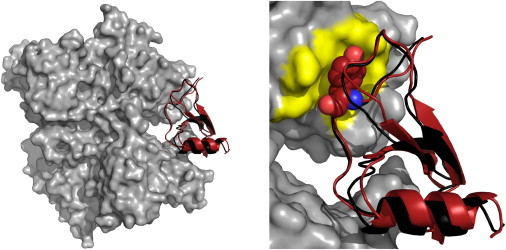
The ATTRACT results for docking viral chemokine binding protein M3 from herpes virus with chemokine MCP-1. (Gray) Receptor protein; (red) ligand protein. The ATTRACT model is superimposed on the interface region of the bound complex structure (PDB 1ML0). (Black) Ligand protein of the crystal structure. A more detailed view of the interface region depicts a tyrosine residue whose importance to binding was determined in mutational experiments. (Yellow) Binding pocket. The images were produced using the program PYMOL (26). To see this figure in color, go online.
Availability and requirements
The ATTRACT web interface is available at www.attract.ph.tum.de and open to all users. The web interface requires a local installation of ATTRACT: the ATTRACT source code and installation instructions are available at www.attract.ph.tum.de/services/ATTRACT/attract.tgz. Alternatively, a (zipped) ATTRACT VirtualBox file is available at www.attract.ph.tum.de/services/ATTRACT/ATTRACT.vdi.gz. We recommend at least 4 GB memory to run ATTRACT. Two example cases are available as web pages with filled-in parameters. The chemokine case described above is available at www.attract.ph.tum.de/cgi/services/ATTRACT/demo-chemokine.py. On a standard desktop computer, docking takes ∼2–3 h. A simple rigid-body docking of xylanase is available at www.attract.ph.tum.de/cgi/services/ATTRACT/demo-xylanase.py, taking ∼10–30 min on a desktop computer.
Acknowledgments
The authors acknowledge Marc van Dijk for the visual design of the ATTRACT website, and the members of the Zacharias group for testing and feedback.
This work was supported by grant No. ZA153/19-2 from the Deutsche Forschungsgemeinschaft and the Excellence Cluster at the Center of Integrated Protein Science Munich, funded by the Deutsche Forschungsgemeinschaft. Computer resources were provided by grant No. pr86pu from the Leibnitz Rechenzentrum, Germany.
References
- 1.Lensink M.F., Wodak S.J. Docking, scoring, and affinity prediction in CAPRI. Proteins. 2013;81:2082–2095. doi: 10.1002/prot.24428. [DOI] [PubMed] [Google Scholar]
- 2.Comeau S.R., Gatchell D.W., Camacho C.J. CLUSPRO: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:W96–W99. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schneidman-Duhovny D., Inbar Y., Wolfson H.J. PATCHDOCK and SYMMDOCK: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tovchigrechko A., Vakser I.A. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006;34:W310–W314. doi: 10.1093/nar/gkl206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lyskov S., Gray J.J. The ROSETTADOCK server for local protein-protein docking. Nucleic Acids Res. 2008;36:W233–W238. doi: 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Vries S.J., van Dijk M., Bonvin A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 7.Macindoe G., Mavridis L., Ritchie D.W. HEXSERVER: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010;38:W445–W449. doi: 10.1093/nar/gkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torchala M., Moal I.H., Bates P.A. SWARMDOCK: a server for flexible protein-protein docking. Bioinformatics. 2013;29:807–809. doi: 10.1093/bioinformatics/btt038. [DOI] [PubMed] [Google Scholar]
- 9.Pierce B.G., Wiehe K., Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30:1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zacharias M. Protein-protein docking with a reduced protein model accounting for side-chain flexibility. Protein Sci. 2003;12:1271–1282. doi: 10.1110/ps.0239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zacharias M. ATTRACT: protein-protein docking in CAPRI using a reduced protein model. Proteins. 2005;60:252–256. doi: 10.1002/prot.20566. [DOI] [PubMed] [Google Scholar]
- 12.May A., Zacharias M. Protein-protein docking in CAPRI using ATTRACT to account for global and local flexibility. Proteins. 2007;69:774–780. doi: 10.1002/prot.21735. [DOI] [PubMed] [Google Scholar]
- 13.de Vries S., Zacharias M. Flexible docking and refinement with a coarse-grained protein model using ATTRACT. Proteins. 2013;81:2167–2174. doi: 10.1002/prot.24400. [DOI] [PubMed] [Google Scholar]
- 14.Fiorucci S., Zacharias M. Binding site prediction and improved scoring during flexible protein-protein docking with ATTRACT. Proteins. 2010;78:3131–3139. doi: 10.1002/prot.22808. [DOI] [PubMed] [Google Scholar]
- 15.May A., Zacharias M. Energy minimization in low-frequency normal modes to efficiently allow for global flexibility during systematic protein-protein docking. Proteins. 2008;70:794–809. doi: 10.1002/prot.21579. [DOI] [PubMed] [Google Scholar]
- 16.de Vries S.J., Zacharias M. ATTRACT-EM: a new method for the computational assembly of large molecular machines using cryo-EM maps. PLoS ONE. 2012;7:e49733. doi: 10.1371/journal.pone.0049733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Setny P., Bahadur R.P., Zacharias M. Protein-DNA docking with a coarse-grained force field. BMC Bioinformatics. 2012;13:228. doi: 10.1186/1471-2105-13-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Setny P., Zacharias M. A coarse-grained force field for protein-RNA docking. Nucleic Acids Res. 2011;39:9118–9129. doi: 10.1093/nar/gkr636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.May A., Zacharias M. Accounting for global protein deformability during protein-protein and protein-ligand docking. Biochim. Biophys. Acta. 2005;1754:225–231. doi: 10.1016/j.bbapap.2005.07.045. [DOI] [PubMed] [Google Scholar]
- 20.Schindler C.E., De Vries S.J., Zacharias M. iATTRACT: simultaneous global and local interface optimization for protein-protein docking refinement. Proteins. 2014 doi: 10.1002/prot.24728. Published online November 17, 2014. [DOI] [PubMed] [Google Scholar]
- 21.de Vries S.J., Bonvin A.M. CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE. 2011;6:e17695. doi: 10.1371/journal.pone.0017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Berkel V., Levine B., Virgin H.W., 4th Critical role for a high-affinity chemokine-binding protein in γ-herpesvirus-induced lethal meningitis. J. Clin. Invest. 2002;109:905–914. doi: 10.1172/JCI14358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Virgin H.W., 4th, Latreille P., Speck S.H. Complete sequence and genomic analysis of murine γ-herpesvirus 68. J. Virol. 1997;71:5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander J.M., Nelson C.A., Fremont D.H. Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell. 2002;111:343–356. doi: 10.1016/s0092-8674(02)01007-3. [DOI] [PubMed] [Google Scholar]
- 25.Lubkowski J., Bujacz G., Wlodawer A. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat. Struct. Biol. 1997;4:64–69. doi: 10.1038/nsb0197-64. [DOI] [PubMed] [Google Scholar]
- 26.Schrödinger L.L.C. Schrodinger; New York: 2014. The PYMOL Molecular Graphics System, Ver. 1.7r0. [Google Scholar]