

Abstract
Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase that regulates cell signaling, proliferation, migration, and development. A major mechanism of regulation of FAK activity is an intramolecular autoinhibitory interaction between two of its domains—the catalytic and FERM domains. Upon cell adhesion to the extracellular matrix, FAK is being translocated toward focal adhesion sites and activated. Interactions of FAK with phosphoinositide phosphatidylinsositol-4,5-bis-phosphate (PIP2) are required to activate FAK. However, the molecular mechanism of the activation remains poorly understood. Recent fluorescence resonance energy transfer experiments revealed a closure of the FERM-kinase interface upon ATP binding, which is reversed upon additional binding of PIP2. Here, we addressed the allosteric regulation of FAK by performing all-atom molecular-dynamics simulations of a FAK fragment containing the catalytic and FERM domains, and comparing the dynamics in the absence or presence of ATP and PIP2. As a major conformational change, we observe a closing and opening motion upon ATP and additional PIP2 binding, respectively, in good agreement with the fluorescence resonance energy transfer experiments. To reveal how the binding of the regulatory PIP2 to the FERM F2 lobe is transduced to the very distant F1/N-lobe interface, we employed force distribution analysis. We identified a network of mainly charged residue-residue interactions spanning from the PIP2 binding site to the distant interface between the kinase and FERM domains, comprising candidate residues for mutagenesis to validate the predicted mechanism of FAK activation.
Introduction
Cells in multicellular organisms are required to perceive their microenvironment by correctly responding to various stimuli. Focal adhesion kinase (FAK), containing numerous binding sites for signaling and adaptor proteins, has been identified as a hub at the crossroads of multiple signaling pathways coupling extracellular and cytosolic signals at focal adhesions (FAs) (1). However, as of this writing, the mechanism of how FAK conformation and function are coupled through allosteric regulation has only been partially uncovered.
FAK is a 120-kDa multidomain protein belonging to the nonreceptor tyrosine kinase family. As shown in Fig. 1, it contains an N-terminal three-lobed 4.1, ezrin, radixin, moesin homology (FERM) domain, followed by a 50-residue linker, a central kinase domain, a 220-residue disordered proline-rich region, and a C-terminal focal-adhesion targeting (FAT) domain. The FAT domain is involved in FAK targeting to focal adhesion sites (2), and the FERM domain exerts its role as an autoinhibitor of the kinase domain, thus regulating the protein’s catalytic activity (3). When FAK attaches to the cell membrane, the autophosphorylated tyrosine Tyr397 in the linker between the FERM and kinase domain (4) provides the binding site for the SH2 domain of Src kinase and leads to subsequent FAK activation. The following phosphorylation of tyrosine residues Tyr576 and Tyr577, which are located in the activation loop of FAK and shielded from the cytosol by the FERM domain, enhances the kinase activity of FAK in vitro (5).
Figure 1.
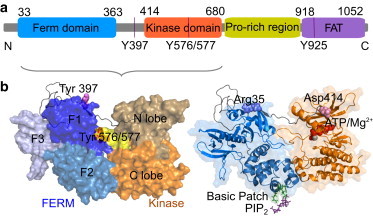
Structure and interactions of FAK. (a) Domain organization of FAK including phosphorylation sites. (b) Structure of FAK FERM and kinase domains and organization into subdomains F1–F3 in FERM and N- and C-lobes in the kinase domain. Primary phosphorylation sites (left), sites of FRET labels (Arg35 and Asp414), ATP/Mg2+, the basic patch, and PIP2 (right) are highlighted. To see this figure in color, go online.
Many stimuli have been reported to induce FAK activation, such as integrin signaling (6) and direct interactions with growth factor receptors (7–9). For the last decade, a key role of phosphatidylinsositol-4,5-bis-phosphate (PIP2) in these interactions has been emphasized (10,11). PIP2 is a lipid integral to the plasma membrane. It is also a pleiotropic ligand molecule, which modulates diverse biological processes, including signaling, intracellular vesicle traffic, and cellular motility (12). PIP2 can be locally generated in FAs by the enzyme phosphatidylinositol 4-phosphate 5-kinase type Iγ (PIP5KIγ), which adds the 5-phosphate to PI4P. It has been shown that PIP5KIγ is required for efficient FAK activation, providing strong support to the notion that PIP2 is a key mediator of the integrin-FAK signaling link (11). It was demonstrated that PIP2 interacts directly with the basic patch of the FERM domain (10), which leads to PIP2-induced activation of FAK in vitro (10) and in vivo (13). However, molecular details of the FAK-PIP2 interactions and mode of activation remain unclear.
To elucidate these, biochemical, structural, and fluorescence resonance energy transfer (FRET) experimental data have recently been employed (11). The experimental evidence presented therein suggests the binding of PIP2 to a basic patch of the FERM domain to induce conformational rearrangements resulting in a decreased FRET efficiency measured between the kinase N-lobe and FERM-F1. This conformational transition, which was found to be further modulated by ATP binding, promoted efficient FAK autophosphorylation of tyrosine Tyr397, but could not induce conformational changes required for autophosphorylation of the key tyrosine residues (Tyr576/577) in the active state of the kinase domain.
To provide direct insight into the underlying molecular mechanism of these changes at high spatial and temporal resolution, which has been inaccessible by these experiments, we conducted a series of molecular dynamics (MD) simulations followed by force distribution analysis (FDA) and principal component analysis (PCA). We studied the structure and dynamics of the FERM and kinase fragment of FAK (FK-FAK) in three systems (Fig. 1), namely: apo-FK; FAK bound to ATP (FK-ATP); and FAK bound to ATP and PIP2 (FK-ATP-PIP2). We propose an atomic detailed model of ATP and PIP2-bound FK-FAK, which is in agreement with previous structural and mutant data. We find ATP and PIP2 binding to induce distinct long-range conformational changes in the kinase and FERM domains, which are likely to allosterically control FAK activity. More specifically, ATP binding to the kinase active site induces a closure of the two kinase lobes, which is reversed by subsequent PIP2 binding to the FERM basic patch with structural consequences for the kinase-FERM interaction. The observed allosteric transitions are qualitatively in line with recent FRET measurements at the kinase-FERM interface (11). We identify an allosteric network of primarily electrostatic interactions spanning from the PIP2 binding site to the distant interface between the kinase N-lobe and the FERM F1-lobe, comprising candidate residues for mutagenesis to test our predicted mechanism.
Materials and Methods
Molecular dynamics simulations and data analysis
For all simulations, the crystal structure (Protein Data Bank (PDB) 2J0J) of the FAK fragment comprising the FERM and kinase domain (residues 35–686) (3) was used as a starting structure. The flexible loops, including those of residues 362–394, 402, and 574–583, were not resolved and were added using MODBASE (14). The structure of FK-ATP was obtained by inserting ATP in the same binding pose resolved in the crystal structure of the FAK kinase domain (K-FAK) in complex with ANP (PDB 2J0L) (3). The initial geometry of the FK-ATP-PIP2 complex was obtained using a molecular docking procedure, with the putative PIP2 binding site assigned to the FERM basic patch (residues 216–222). Details of the docking and refinement procedure have been described previously in Goñi et al. (11). Partial atomic charges on the PIP2 molecule with a reduced lipid tail (C2-PIP2) were assigned using the RESP methodology (15–17) and obtained with the program RED (15,16), using HF/6-31G∗ as basis set. The total charge on the PIP2 headgroup was set to −5, which corresponds to the fully deprotonated state, because potential protons of PIP2 in its most common state with −4 as a total charge (18) are likely to be displaced by Lys/Arg interactions upon binding to FAK.
Molecular docking was performed with the UCSF DOCK6.5 suite (19), using the semirigid docking procedure and energy grid scoring in an implicit solvent. The grid spacing was 0.025 nm, and the grid included 1.2 nm beyond the FAK basic patch. The energy score was the sum of electrostatic and van der Waals contributions. In the course of the docking procedure, the PIP2 molecule was subjected to 2500 cycles of molecular-mechanical energy minimization. The number of maximum ligand orientations was 5000. The best-scoring 25 FK-ATP-PIP2 complexes were further analyzed by means of MD simulations. The best structure was chosen according to the favorable Coulombic interaction between the PIP2 headgroup and the basic patch in the FERM domain. The obtained docking pose was in line with alanine scanning results (10), and robust with regard to the PIP2 lipid tail length. Also, we previously could show that PIP2 allostery is mainly due to the charge neutralization of the basis patch, because a basic patch mutant gave highly similar force distribution patterns. We thus conclude that the PIP2-bound FAK structure is appropriate for the subsequent MD simulations and force analyses.
All MD simulations were carried out using GROMACS 4.0.5 (www.gromacs.org) (20). The OPLS all-atom force field (21) for the protein and the TIP4P water model (22) were used. The temperature was kept constant at T = 300 K by using velocity rescaling with a coupling time of 0.1 ps. The pressure was kept constant at 1 bar using an isotropic coupling to Parrinello-Rahman barostat with a coupling time of 0.1 ps (23). A cutoff of 1 nm was used for all nonbonded interactions. Long-range electrostatic interactions were treated with the particle-mesh Ewald (24) method using a grid spacing of 0.12 nm with cubic interpolation. All bonds between hydrogens and heavy atoms were constrained using the LINCS algorithm (25). An integration time step of 2 fs was used. During all simulations, the neighbor list was updated every 10 steps.
Each of the three structures, apo-FK, FK-ATP, and FK-ATP-PIP2, were immersed in a cubic TIP4P water box containing ∼200,000 atoms. Sodium and chloride counterions (0.1 M concentration) were added to neutralize the system. Before the MD simulations, all investigated systems were energy-minimized using the steepest descent method for 10,000 steps. This was followed by 2-ns MD simulations, during which position constraints were used on all protein atoms. Finally, each system was equilibrated during 200-ns MD simulations. The last 120-ns trajectories of each simulation have been used for analysis.
Subsequently, trajectories were analyzed by PCA (26) and FDA (27) as implemented in the software GROMACS. PCA was used to monitor the global motions within the kinase domain, and FDA was used to investigate the changes in internal forces of the molecules under external perturbation, i.e., ATP or PIP2 binding. During FDA, atomic pairwise forces as defined by the force field were calculated, and residue forces were obtained by vector summation of atomic forces according to
| (1) |
where i is an atom of residue ri, and j is an atom of residue rj, with ri and rj being different. Angle and dihedral forces were decomposed into pairwise forces between the three or four involved atoms, respectively. Forces from particle-mesh Ewald were ignored. We compared residue pairwise forces between apo-FK and FK-ATP, as well as between FK-ATP and FK-ATP-PIP2, respectively, averaged over 360 ns (three trajectories, from each the final 120 ns) for each state. These differences in forces between residue pairs measure the allosteric signal propagation upon binding of ATP or PIP2. Fig. 4 and Fig. S2, Fig. S3, and Fig. S4 in the Supporting Material show the largest continuous graphs of force changes at varying cutoff values (28). The punctual stress shown in Fig. S2 d, Fig. S3 d, and Fig. S4 d is defined as the sum of the absolute values of scalar pairwise forces acting on an atom i,
| (2) |
and serves as a simple measure for where pairwise forces accumulate (27).
Figure 4.
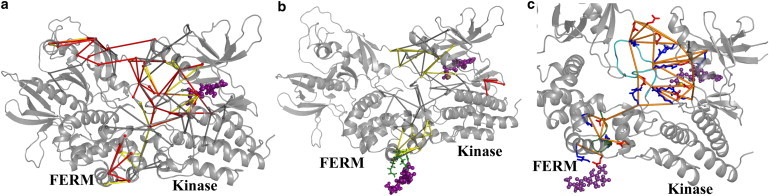
Force distribution upon ATP and PIP2 binding of FAK. (a) Differences of residue pairwise forces between apo-FK and FK-ATP and (b) FK-ATP and FK-ATP-PIP2. (Sticks) Network of force differences. Only networks of forces connecting more than five residues are shown. (Different colors of sticks indicate different cutoffs of forces, including red (350 pN), yellow (300 pN), and gray (200 pN).) The positions of ATP and PIP2 (purple) are shown at representative positions, as observed during the simulations. (c) The force network (orange) connects residue pairs with a change >250 pN upon ATP binding and a reverse change of >250 pN upon PIP2 binding (see red in Table S1 in the Supporting Material). Positively (blue) and negatively charged residues (red) are shown as sticks. (Cartoon) The activation loop (residues 569–584, dark green) and the loop 181–189 (cyan) are highlighted. To see this figure in color, go online.
Protein crystallization, data collection, and structure determination
The kinase domain of the avian FAK (sequence: 411–686) was expressed and purified following a protocol described in Lietha and Eck (29). K-FAK (6.7 mg/mL) was incubated with various ligands at 4°C for several hours. Low-affinity ligands were taken from a small-compound library with scaffolds targeting the ATP binding site (see Table S2 in the Supporting Material). They include bis-amino pyrimidine, bis-anilino imidazotriazine, and low-molecular-weight fluorinated compounds. It was found out later, when solving the crystal structure of the protein, that not all the ligands tested bound to the kinase domain; thus, we obtained several apo structures of the protein. Crystals were grown, mixing an equal volume of the protein-ligand mix with the crystallization condition (100 mM Tris, pH 8.5, 50–300 mM LiSO4, 18–30% PEG4000, 10 mM TCEP). Crystals were cryoprotected by a quick soak into the following cryosolution: 100 mM Tris pH 8.5, 200 mM LiSO4, 32% PEG4000, 10 mM TCEP, and 8% ethylene glycol; it was then flash-frozen.
Native data were collected on PXI-XS06 beamline at Swiss Light Source, Villigen, Switzerland and processed with the software XDS (30). The molecular replacement protocol in the software PHASER (31), was applied to provide an initial set of phases using the K-FAK model from PDB 2JKK. This was followed by an initial round of model building using ARP/wARP (32). Refinement was performed using the program REFMAC (33) and manual rebuilding was carried out with the software COOT (34). All refinement data is given in Table S2.
Results and Discussion
PIP2- and ATP-induced conformational changes of FAK
To characterize interactions among ATP, PIP2, and FAK at the atomistic level, we performed MD simulations of 1) a FAK fragment containing the FERM and kinase domains (apo-FK), 2) the same FAK fragment bound to ATP (FK-ATP), and 3) FAK bound to ATP and PIP2 (FK-ATP-PIP2) (Fig. 1).
The term “PIP2” here refers to a molecule with a shortened lipid tail containing only two carbon atoms (C2-PIP2). At pH7, PIP2 is most likely in a singly protonated state and carries the net-charge −4 (18). However, the proton bound to PIP2 is likely to be displaced upon binding to a protein (18). Accordingly, in the FK-ATP-PIP2 simulation system, a net charge of −5 has been assigned to the PIP2 headgroup.
In all simulations, comprising 0.6 μs for each state, the FERM domain remained associated with the kinase domain. However, ATP and PIP2 binding induced large-scale conformational changes at the interface of the N-lobe and F1 domain (Fig. 2). We first monitored the distance between Arg35 (the N-terminal part of the FERM domain) and Asp414 (the N-terminal part of the kinase domain), which were the labeling sites for FRET sensors in previous measurements (11), in the course of the MD simulations, in order to validate the computed conformational changes.
Figure 2.
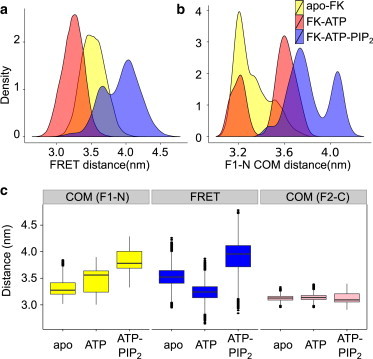
Conformational changes upon ATP/PIP2 binding measured by interdomain distances as observed in MD simulations of the three states of FK-FAK. (a) Distance distribution between the residues where FRET labels were inserted in the FERM and kinase domain (Fig. 1b). (b) Distance distribution between the centers of mass of the FERM F1 domain and the kinase N-lobe. (c) Average and deviations of the COM distance between the F1 lobe and the kinase N-lobe (yellow), the FRET distance (blue), and the COM distance between the FERM F2 subdomain and the kinase C-lobe (pink). To see this figure in color, go online.
We observed the shortest median FRET distance (3.2 nm) for FK-ATP, whereas the FRET distances of apo-FK and FK-ATP-PIP2 were ≥3.5 nm (Fig. 2 a). This trend is consistent with the experimental data, which gave a larger FRET efficiency for FK-ATP as for apo-FK and FK-ATP-PIP2 (11). However, we find the distances between the FRET labels do not reflect the interdomain distance. Namely, >50% of apo-FK center-of-mass (COM) distances between the N-lobe and F1 were in the range of 3.1–3.3 nm, which is shorter than the median COM distance of FK-ATP (3.5 nm) and FK-ATP-PIP2 (3.8 nm) (Fig. 2 b). Thus, binding of ATP to apo-FK leads to an apparent closure as measured by the FRET labels both in our simulations and in experiments; however, it hides an opening of the respective subdomains, the N-lobe and F1, relative to each other. In contrast, PIP2 binding to FK-ATP results in a partial opening of the FERM-kinase interface leading to increased distances of both the COM of the two domains and the respective FRET labels.
Accordingly, the addition of PIP2 to FAK can induce the switch from an inactive to an active kinase domain. We here refer to the allosteric changes as an opening and closing of the N-lobe and F1, as suggested by the COM measurements, even though the motions include a twisting (see below) and the N/F1 interface remains largely unaffected, including the burial of Tyr576/577. We also note that, in none of the three states, was full dissociation of the inhibitory FERM domain from the catalytic kinase domain observed. Instead, the distance between the FERM F2-lobe and the kinase C-lobe remained unchanged within our simulation timescale (Fig. 2 c, right), suggesting that the FRET sensors measure partial FAK activation only. The validation of the observed ATP- and PIP2-dependent dynamics in FAK by comparison to FRET experiments allows us to further analyze the underlying functional dynamics induced by these two ligands.
Detection of functional modes in FAK dynamics
To analyze the functional motions underlying the observed distance changes, which represent a rather coarse measure of the protein conformational changes, a PCA (26) of the kinase domain, excluding the activation loop, in the apo state was carried out. Differences between the protein’s conformational motions in its three states were observed by projecting the trajectories of the kinase domain in the FK-ATP and FK-ATP-PIP2 states onto the eigenvalues calculated for the apo state. We observed major differences of the protein dynamics after binding of ATP and then of PIP2, as reflected by the projection of the dynamics on the first two eigenvectors, which account for ∼45% of the overall protein motion (see Fig. S1). Here, eigenvector 1 (EV1) represents an open-to-closed motion (Fig. 3 b) and EV2 corresponds to the twisting motion of the FAK kinase domain (Fig. 3 c).
Figure 3.
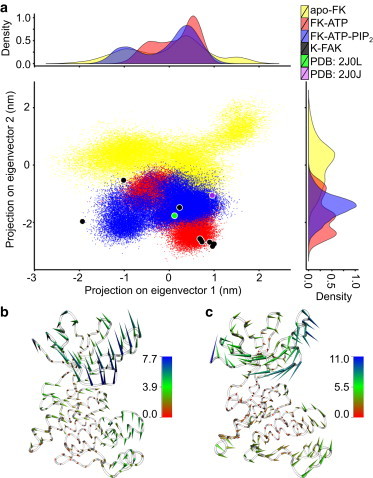
Conformational changes upon ATP/PIP2 binding measured by principal component analysis (PCA). (a) apo-FK (yellow), FK-ATP (red), FK-ATP-PIP2 (blue) during MD simulations, and crystal structures of staurosporine-bound FK-FAK (violet, PDB 2J0J), the ANP-bound K-FAK (green, PDB 2J0L), and apo K-FAK (black) were projected on the first two eigenvectors obtained from a PCA of the fluctuations of apo-FK Cα atoms. Distributions of the three states along the two apo eigenvectors are also shown. (b and c) Porcupine plot of the FAK kinase domain, illustrating the motion along the first (b) and second (c) eigenvectors (EV) obtained from PCA. Coloring is according to the deviation in eigenvalue between the two extreme structures along the respective EV. EV1 (b) describes an opening, EV2 (c) a twisting motion. To see this figure in color, go online.
As can be seen in Fig. 3 a, apo-FK samples a broad range of conformations along both the open-to-close interdomain motion and the intradomain twisting motion. ATP, which is located between the kinase N- and C-lobe, restricted the open-close transition and constrained the structure of the kinase domain. PIP2 binding to the basic patch in the FERM domain, in turn, released some of these constraints, and the kinase domain was found in a more open conformation (negative eigenvalues of EV1), in agreement with the observed increases in distance (Fig. 2 a). We also projected available x-ray structures onto the two collective coordinates. We note, however, that a direct comparison is not straightforward, because most experimental structures lack the FERM domain. Crystal structures of the FK-FAK in the presence of staurosporine (violet) and the ANP-bound K-FAK (green) (3) have previously been experimentally resolved and lie well within the dynamics observed in our MD simulations. Interestingly, the FERM-inhibited structure (PDB:2J0J) features a more closed state along EV1 as compared to the ANP-bound structure (PDB:2J0L), either due to the presence of the FERM domain or ligands at the ATP-binding site added during the crystallization protocol (3).
For comparison, we solved a number of structures of the true apo-state of the kinase in absence of the autoinhibitory domain or any ligand bound to the active site (black). These structures reproduce the large variability of the apo state along EV1 seen in our simulations. Along EV2, surprisingly, they cover the conformational space observed in our MD simulations for FK-ATP and FK-ATP-PIP2, rather than apo-FK. A possible interpretation is that the FERM domain causes a twist in the kinase domain, which vanishes in the kinase structure alone and similarly decreases when weakening the FERM-kinase interface upon ATP/PIP2 binding. Apparently, the presence of the FERM domain in our simulations gives rise to this shift in conformational space. Structural data of the apo-state of the FERM-kinase construct would be required to further resolve this.
ATP- and PIP2-induced allostery of FAK
We observed in our MD simulations that the binding of ATP and PIP2 induced large-scale conformational changes in the FAK kinase domain and at the domain interface. However, both the PIP2 and ATP binding sites are located quite far (≥1.5 nm) from the N/F1 interface and the autophosphorylation site. Thus, it is unclear how exactly these two small molecules induce conformational changes at such distant sites. To elucidate the molecular nature of the allosteric pathway connecting the involved sites, FDA was performed. FDA allows us to investigate changes in internal pairwise forces under external perturbations such as ligand binding. During FDA, atomic pairwise forces were calculated, and forces between amino-acid residues were obtained by vector summation of atomic forces (27). Matrices of interresidue forces were averaged over all trajectories for each state (apo-FK, FK-ATP, and FK-ATP-PIP2). Analyzing changes in pairwise forces instead of coordinates has the advantage of tracking changes in relatively stiff regions such as the protein core, and thus is not biased toward large-amplitude motions such as those detected by PCA (Fig. 3).
Initially, the differences of interresidue forces in FAK upon binding of ATP were calculated to reveal the allosteric pathway underlying the ATP-induced FERM/kinase domain closure we observed experimentally and computationally. The resulting network of differences in pairwise residue forces is shown in Fig. 4 a as sticks connecting the respective residues. To reduce the noise, only the connected graph with more than five residues is displayed as vertices (28). ATP binding leads to a network of forces propagating from the active site to the activation loop and also spanning the FERM F1 domain. Apparently, ATP and Mg2+ act as an electrostatic lock by interacting with the adjacent charged residues, resulting in the observed long-range FERM/kinase closure motion and loss in interlobe flexibility within the kinase domain (Fig. 3). The force network originates from the ATP/Mg2+ binding site, because a high force cutoff restricts the network to this region (see Fig. S2 a). With decreasing force cutoff, in turn, the signal also reaches the PIP2 binding site in the FERM F2 lobe and the domain-interface between the FERM F1 lobe and kinase N-lobe via the activation loop (see Fig. S2), but without apparent structural changes at the FERM F2 subdomain and kinase C-lobe interface (Fig. 2).
Additional PIP2 binding resulted in a pronounced increase in the distance between the FERM F1 lobe and the kinase N-lobe, which is even more eye-striking than the ATP-dependent allostery, given the remote location of the PIP2 binding site at the FERM F2 lobe. According to the force network depicted in Fig. 4 b and Fig. S3, PIP2 binding to ATP-bound FAK results in a spatially connected force network, which spans the F2/C-lobe interface and the ATP binding site all the way up to the linker. The PIP2 headgroup is very negatively charged. The total charge on the whole molecule varies from −5 to −4, depending on the PIP2 protonation state (18). Thus, binding of PIP2 to the basic patch in the FERM domain causes substantial changes in the protein electrostatic potential at the F2/C-lobe interface, such that PIP2 binding indirectly adjusts the salt-bridge interactions in this region (11). These structural adaptions then further distribute into the kinase N-lobe and adjacent linker, according to our FDA.
Forces induced by PIP2 are generally lower than those from ATP, such that this allostery was only detected at a lower cutoff of 200 pN. A possible reason is that PIP2 resides at the well-structured FERM domain, in which both force transmission and structural changes are much less pronounced than in the susceptible kinase domain to which ATP binds. Indeed, increasing the cutoff for PIP2-induced forces to 350 pN results in a single and very small force network at the dynamic kinase interlobe region domain (see Fig. S3 a), a site overlapping with the hinge of the opening-closure motion instead of the PIP2 binding site, where the perturbation originates.
While the force networks due to ATP and PIP2 binding largely overlap and involve similar residue pairs (see Fig. S2 and Fig. S3), their binding results in opposing distance changes between the FERM F1 and kinase N lobes (Fig. 2). We analyzed the residue interaction network within FAK, which gives rise to the observed force networks, to find a possible explanation for the distinct structural closure and opening motion induced by ATP and PIP2, respectively. A remarkable number of residue pairs within the allosteric network show a significant force change upon ATP binding (>250 pN), which is largely rescued by PIP2 binding (>250 pN, marked red in Table S1 and shown in Fig. 4 c). These ATP-induced but PIP2-reversed force alterations are thus likely to reflect the motions probed by FRET, which follow the same trend. Within this set of residue pairs reversely affected by ATP and PIP2, one or both residues in each of the specifically perturbed interactions resides in the activation loop (residues 569–584) or in the loop 181–189. The former typically plays a pivotal role in allosteric regulation of kinases. The latter is located at the domain interface close to the FERM basic patch, and has been observed to be disordered in a charge-neutralizing basic patch mutant of FAK meant to mimic PIP2 binding (11).
Our PIP2-dependent alterations of residue interactions within this loop are possible explanations for the order-to-disorder transition observed in the crystal structures of wild-type and mutant. Remarkably, all but one of the PIP2-affected pairwise interactions within the kinase-FERM construct are saltbridges that reversibly swap partners; we argue this to be the molecular basis for the observed dramatic change in the relative positioning of the lobes within the kinase domain as well as of the kinase and its autoinhibitory FERM domain as observed in MD simulations here and FRET experiments previously (11).
Conclusions
We here studied the dynamic allostery involved in PIP2-mediated activation of FK-FAK. Based on the results obtained from MD simulations, PCA, and FDA that was performed for the three different protein systems, we concluded that the highly charged ATP/Mg2+ acts as a hub for electrostatic interactions with charged residues in the gap between kinase N- and C-lobe. These interactions restrict the twisting-motional mode of the kinase domain and tie the correlated motions of the N- and C-lobes. Binding of highly negatively charged PIP2 at the basic patch of the FERM domain leads to the rearrangement of saltbridges between the α-helix of the basic patch (residues 216–222), and another α-helix at the FERM F2- and kinase C-lobe interface (residues 181–189). This results in a dominolike cascade of changes in saltbridges of the kinase domain, spanning from the interface between the FERM F2- and kinase C-lobe through the activation loop to the interface between the FERM F1- and kinase N-lobe. The results of MD, PCA, and FDA not only describe the molecular mechanism of PIP2-induced FAK activation, but also allow us to predict mutations, which might be critical for the FAK activation mechanism.
Based on our analysis, we predict mutations at R184 and R597 to critically interfere with ATP and PIP2-dependent allostery. As observed in our results, we can conclude that the action of PIP2 is mainly of an electrostatic nature, by changing the electrostatic interactions between basic patch with its nearby α-helix and inducing a rearrangement at F2/N-lobe interface. We believe that the other basic patch interaction partners, such as C-Met (35) or the FAT domain, might allosterically effect FAK dynamics in a similar fashion—a hypothesis straightforwardly testable by experiments.
Phosphorylation assays showed that PIP2 can enhance FAK autophosphorylation (11). Our allosteric network originating from PIP2 indeed spans the protein all the way up to the loop in which the Y397 autophosphorylation site is located (see Fig. S4). We, however, could not detect any significant change in structure or dynamics of Y397 itself, which would explain the PIP2-dependent regulation of FAK. This might be due to the limited submicrosecond timescale of our MD simulations. As previously observed for other proteins (36,37), forces transmit through proteins on timescales much shorter than those of large-scale conformational transitions, suggesting FDA to be a versatile method to detect allostery in proteins on standard MD timescales.
The activation loop is critically involved in the detected allosteric pathway. Interestingly, we observed an overall larger root-mean-square fluctuation of this loop in FK-ATP-PIP2, while most residues in this loop were restricted in FK-ATP (Fig. 5). The ATP binding strongly reduces the flexibility of the activation loop, including the major phosphorylation site Y576/577, but the further PIP2 binding released this constraint. We propose that the experimental observations of the increase in Src phosphorylation of Y576/577 in presence of PIP2 (11) can be explained by a less compact protein conformation adopted by FAK, involving an increased flexibility of the activation loop upon PIP2 binding.
Figure 5.
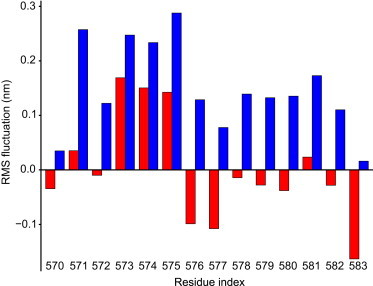
Ligand-induced changes in the activation loop at the FERM-kinase interface, where the major phosphorylation site Y576/577 is located. Differences in root-mean-square fluctuations to the apo-state (red) for FK-ATP and (blue) for FK-ATP-PIP2. To see this figure in color, go online.
In none of the simulations performed herein could we observe the dissociation of FERM and kinase domains. That is consistent with the experimental data reported (11), which shows that PIP2 is not sufficient to induce catalytic turnover of FAK, but Src recruitment for subsequent Tyr566/567 phosphorylation is additionally required. External mechanical force might act as another factor contributing to FAK activation. For several focal adhesion proteins it has been shown that stretching forces can regulate focal adhesion maturation and signaling (38–40), and several studies describe FAK as a key tension sensor in focal adhesions (41–43). FAK’s FAT domain binds to actin filaments via paxillin (44), and FAK’s FERM domain binds to the membrane via PIP2 (10,11). It is therefore feasible that stretching forces at focal adhesion sites propagate through these attachment points to FAK and enhance FAK activation. We have shown that PIP2 and ATP binding are important first steps in FAK allosteric control. However, further experimental and computational studies are required to fully decipher the conformational plasticity and multidimensional activation landscape of this fascinating molecular signaling hub.
Acknowledgments
We thank Bogdan Costescu and Maxime Louet for helpful discussions. Additionally, we thank Andrea Lin for her help with the crystallization of FAK kinase and the Swiss Light Source beamline staff for their support.
Funding from the Klaus Tschira Stiftung, the Federal Ministry of Education and Research SysTec program, and the Center for Modelling and Simulation in the Biosciences at Heidelberg University is gratefully acknowledged.
Supporting Material
References
- 1.Zamir E., Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci. 2001;114:3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 2.Hildebrand J.D., Schaller M.D., Parsons J.T. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J. Cell Biol. 1993;123:993–1005. doi: 10.1083/jcb.123.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lietha D., Cai X., Eck M.J. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schaller M.D., Hildebrand J.D., Parsons J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calalb M.B., Polte T.R., Hanks S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol. Cell. Biol. 1995;15:954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.David F.S., Zage P.E., Marcantonio E.E. Integrins interact with focal adhesions through multiple distinct pathways. J. Cell. Physiol. 1999;181:74–82. doi: 10.1002/(SICI)1097-4652(199910)181:1<74::AID-JCP8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 7.Chen S.Y., Chen H.C. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell. Biol. 2006;26:5155–5167. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plaza-Menacho I., Morandi A., Isacke C.M. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J. Biol. Chem. 2011;286:17292–17302. doi: 10.1074/jbc.M110.168500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieg D.J., Hauck C.R., Schlaepfer D.D. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 10.Cai X., Lietha D., Schaller M.D. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol. Cell. Biol. 2008;28:201–214. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goñi G.M., Epifano C., Lietha D. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc. Natl. Acad. Sci. USA. 2014;111:E3177–E3186. doi: 10.1073/pnas.1317022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thapa N., Anderson R.A. PIP2 signaling, an integrator of cell polarity and vesicle trafficking in directionally migrating cells. Cell Adhes. Migr. 2012;6:409–412. doi: 10.4161/cam.21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linseman D.A., Sorensen S.D., Fisher S.K. Attenuation of focal adhesion kinase signaling following depletion of agonist-sensitive pools of phosphatidylinositol 4,5-bisphosphate. J. Neurochem. 1999;73:1933–1944. [PubMed] [Google Scholar]
- 14.Pieper U., Eswar N., Sali A. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004;32:D217–D222. doi: 10.1093/nar/gkh095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanquelef E., Simon S., Dupradeau F.Y. R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011;39:W511–W517. doi: 10.1093/nar/gkr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dupradeau F.Y., Pigache A., Cieplak P. The R.E.D. tools: advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010;12:7821–7839. doi: 10.1039/c0cp00111b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frisch M.J., Trucks G.W., Fox D.J. Gaussian Inc.; Wallingford, CT: 2009. GAUSSIAN 09. [Google Scholar]
- 18.McLaughlin S., Wang J., Murray D. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 19.Lang P.T., Brozell S.R., Kuntz I.D. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen W.L., Tirado-Rives J. Computer-aided design of non-nucleoside inhibitors of HIV-1 reverse transcriptase. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1016/j.bmcl.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 22.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 23.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182. [Google Scholar]
- 24.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 25.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 26.Balsera M.A., Wriggers W., Schulten K. Principal component analysis and long time protein dynamics. J. Phys. Chem. 1996;100:2567–2572. [Google Scholar]
- 27.Costescu B.I., Gräter F. Time-resolved force distribution analysis. BMC Biophys. 2013;6:5. doi: 10.1186/2046-1682-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seifert C., Gräter F. Force distribution reveals signal transduction in E. coli Hsp90. Biophys. J. 2012;103:2195–2202. doi: 10.1016/j.bpj.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lietha D., Eck M.J. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS ONE. 2008;3:e3800. doi: 10.1371/journal.pone.0003800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCoy A.J., Grosse-Kunstleve R.W., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langer G., Cohen S.X., Perrakis A. Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Emsley P., Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Chen T.H., Chan P.C., Chen H.C. Phosphorylation of focal adhesion kinase on tyrosine 194 by Met leads to its activation through relief of autoinhibition. Oncogene. 2011;30:153–166. doi: 10.1038/onc.2010.398. [DOI] [PubMed] [Google Scholar]
- 36.Palmai Z., Seifert C., Balog E. An allosteric signaling pathway of human 3-phosphoglycerate kinase from force distribution analysis. PLOS Comput. Biol. 2014;10:e1003444. doi: 10.1371/journal.pcbi.1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young H.T., Edwards S.A., Gräter F. How fast does a signal propagate through proteins? PLoS ONE. 2013;8:e64746. doi: 10.1371/journal.pone.0064746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.del Rio A., Perez-Jimenez R., Sheetz M.P. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323:638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grashoff C., Hoffman B.D., Schwartz M.A. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawada Y., Tamada M., Sheetz M.P. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 2006;127:1015–1026. doi: 10.1016/j.cell.2006.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yano Y., Geibel J., Sumpio B.E. Tyrosine phosphorylation of pp125FAK and paxillin in aortic endothelial cells induced by mechanical strain. Am. J. Physiol. 1996;271:C635–C649. doi: 10.1152/ajpcell.1996.271.2.C635. [DOI] [PubMed] [Google Scholar]
- 42.Wang H.B., Dembo M., Wang Y. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl. Acad. Sci. USA. 2001;98:11295–11300. doi: 10.1073/pnas.201201198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang D., Mehta D., Gunst S.J. Mechanosensitive tyrosine phosphorylation of paxillin and focal adhesion kinase in tracheal smooth muscle. Am. J. Physiol. 1999;276:C250–C258. doi: 10.1152/ajpcell.1999.276.1.C250. [DOI] [PubMed] [Google Scholar]
- 44.Schmalzigaug R., Garron M.L., Premont R.T. GIT1 utilizes a focal adhesion targeting-homology domain to bind paxillin. Cell. Signal. 2007;19:1733–1744. doi: 10.1016/j.cellsig.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.