

Abstract
Langerin, a trimeric C-type lectin specifically expressed in Langerhans cells, has been reported to be a pathogen receptor through the recognition of glycan motifs by its three carbohydrate recognition domains (CRD). In the context of HIV-1 (human immunodeficiency virus-1) transmission, Langerhans cells of genital mucosa play a protective role by internalizing virions in Birbeck Granules (BG) for elimination. Langerin (Lg) is directly involved in virion binding and BG formation through its CRDs. However, nothing is known regarding the mechanism of langerin assembly underlying BG formation. We investigated at the molecular level the impact of two CRD mutations, W264R and F241L, on langerin structure, function, and BG assembly using a combination of biochemical and biophysical approaches. Although the W264R mutation causes CRD global unfolding, the F241L mutation does not affect the overall structure and gp120 (surface HIV-1 glycoprotein of 120 kDa) binding capacities of isolated Lg-CRD. In contrast, this mutation induces major functional and structural alterations of the whole trimeric langerin extracellular domain (Lg-ECD). As demonstrated by small-angle x-ray scattering comparative analysis of wild-type and mutant forms, the F241L mutation perturbs the oligomerization state and the global architecture of Lg-ECD. Correlatively, despite conserved intrinsic lectin activity of the CRD, avidity property of Lg-ECD is affected as shown by a marked decrease of gp120 binding. Beyond the change of residue itself, the F241L mutation induces relocation of the K200 side chain also located within the interface between protomers of trimeric Lg-ECD, thereby explaining the defective oligomerization of mutant Lg. We conclude that not only functional CRDs but also their correct spatial presentation are critical for BG formation as well as gp120 binding.
Introduction
Dendritic cells (DCs) are professional antigen-presenting cells that play a crucial role in the defense against pathogens. Invading pathogens are recognized by toll-like receptors and receptors such as C-type lectins expressed on the surface of DCs. DCs are then able to process and present antigens, initiating primary immune response and inducing memory. They can be divided into several subsets distinguishable by specific markers and notably proteins of the C-type lectin family. Indeed, Langerhans cells (LCs) and interstitial DCs can be identified by a large panel of C-type lectins such as langerin (Lg) and DC-SIGN (dendritic cell-specific ICAM-3 grabbing nonintegrin), respectively (1–3). These two lectins belong to the same family and present some strong similarity in sequence and structural organization. Moreover, despite different specificities, they exhibit some common general recognition properties toward high mannose glycans. Indeed, they are both involved in human immunodeficiency virus-1 (HIV-1) binding through specific recognition of the high mannose glycan present on the gp120 envelope protein of the virus (4). However, this shared property hides a drastically different impact in virus fate (5). Although DC-SIGN can be hijacked by HIV-1 to promote its transmission to T cells by DCs (1), langerin has been reported to protect LCs from HIV-1 infection and to promote viral degradation at low viral load (6).
At high viral load or in case of a reduced expression level, langerin is saturated and LCs can be infected and become efficient in HIV-1 transmission (6,7). Indeed, coinfection with herpes simplex virus type 2 induces HIV-1 susceptibility of LCs by abrogating langerin function (7). Located in the epidermis and stratified mucosal tissue, LCs are on the frontline for interaction with pathogens, and they have been suspected to be the initial HIV-1 targets after sexual exposure. Biological mechanisms by which HIV-1 is acquired and transmitted from mucosal site are still poorly understood. Indeed, the ability of LCs to switch from an anti-HIV-1-barrier status to a HIV-1-transmission mode, more specifically the role of langerin in this process is actively investigated.
The molecular mechanism by which langerin plays its protective role is far from being understood. Current knowledge is limited to the fact that langerin is able i), to bind HIV-1 particles and, ii), to promote their internalization into organelles specific to LCs, the Birbeck granules (BGs). Interestingly, langerin is also the central element required for the generation of these specific organelles. BGs are pentalamellar and zipped membranes defining tennis racket-like structures when their cytomembrane sandwiching structures (CMS) are fused to endocytic structures, i. e., clathrin-coated vesicles and early endosomes. Langerin accumulation is essential to generate this membrane architecture as illustrated by the induction of such CMS by langerin cDNA transfection into fibroblasts or even in melanoma cell lines (3,8). Langerin extracellular domain (ECD) contains a neck region responsible for its trimeric organization and a C-terminal carbohydrate recognition domain (CRD) responsible for the Ca2+-dependent lectin properties (9). In a previous study, we demonstrated the requirement of CRDs for BGs formation and suggested a macromolecular organization of langerin compatible with BG architecture (10).
The langerin gene has been shown to present nonsynonymous single nucleotide polymorphisms within the CRD coding region. The corresponding amino acid substitutions, when characterized, have been reported to alter the sugar binding activity or switch langerin specificity from one sugar to another (11,12). In contrast, to date only two point mutations leading to altered BG organization have been reported in langerin. These mutations are both located in the CRD and have been identified in human or in murine context (W264R or F244L, respectively) (13,14). Such mutations represent an opportunity to study the relationship between BG alteration and a specific modification of langerin properties. We undertook this study with the aim of gaining some insight into the structural basis of the langerin role in BG formation. We reconstituted these mutations in recombinant versions of langerin CRD (Lg-CRD) and langerin entire ECD (Lg-ECD) to evaluate their impact on lectin properties, notably the binding to HIV-1 envelope glycoprotein gp120.
After confirmation of the impact of mutations in BG structure, we characterized the effects of both mutations on isolated Lg-CRD and entire Lg-ECD in parallel. The W264R mutation is shown to cause global unfolding of the Lg-CRD. In stark contrast, the mutation of phenylalanine 241 into leucine in human Lg-CRD (corresponding to F244L in the murine gene) leaves functional and structural properties of Lg-CRD globally unchanged, although the whole Lg-ECD exhibits major functional and structural alterations. Indeed, the oligomerization state of Lg-ECDF241L is perturbed, and consequently its global architecture, as demonstrated by small-angle x-ray scattering (SAXS) comparative analysis of wild-type (WT) and mutant forms of recombinant Lg-ECD. Moreover, although the intrinsic lectin activity of the Lg-CRD is not impaired by the mutation, the avidity property of the oligomeric form is modified as shown by its weaker binding to gp120 glycan moiety. In this work, we establish a direct link between a mutation in the Lg-CRD and modification of Lg-ECD quaternary structure, loss of efficiency in gp120 recognition, and alteration of BG architecture.
Materials and Methods
Transfection and expression of langerin constructs in COP5 cells
Murine fibroblastic COP5 cells, derived from C127 cell lines (15) (Health Protection Agency, catalogue No. 87091701, Porton, UK), were cultured in RPMI 1640 medium supplemented with 10% v/v heat-inactivated fetal bovine serum, 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazine ethane sulfonic acid), 2 mM L-glutamine, and 50 μM 2-mercaptoethanol and gentamicin (80 μg/mL). The cells were transfected using the GeneJuice Transfection Reagent (Merck, Fontenay Sous Bois, France) with cDNA encoding the various langerin expression constructs (all constructs used for WT and mutant langerin alike corresponded to full-length langerin spanning amino acids 1 to 328). Langerin expression was evaluated by FACS (fluorescence-activated cell sorting) analysis 48 h after transfection.
Flow cytometry analysis
COP5 transfected cells were incubated with antilangerin mAb (clone 808E10 or with antilangerin mAb (clone 310F7) for intracytoplasmic labeling (Dendritics, Lyon, France). MAbs were revealed with goat antimouse Ig-FITC conjugated (Becton Dickinson, Pont de Claix, France). Intracellular labeling was performed in the presence of permeabilization medium (0.3% w/v saponin, 2% w/v bovine serum albumin (BSA)). Negative controls were performed with an isotype IgG1 control. Fluorescence was analyzed with a FACScan flow-cytometer (Becton Dickinson).
Transmission electron microscopy
After washing, isolated COP5 fibroblasts transfected with langerin cDNA were fixed with 2% w/v glutaraldehyde in 100 mM cacodylate buffer at pH 7.4 for 24 h. After rinsing in 100 mM cacodylate buffer with 150 mM sucrose for 24 h, cells were processed for transmission electron microscopy. They were postfixed with an aqueous solution of 1% w/v osmium tetroxide in cacodylate buffer with sucrose and embedded in epoxy medium after dehydration through a graded series of ethanol. Ultrathin sections were stained with lead citrate and uranyl acetate and examined with a JEOL 1200EX electron microscope with acceleration voltage of 80 kV (Centre des Microstructures, Lyon University, France).
Cloning and expression of recombinant langerin domains
Lg-CRD and Lg-ECD, corresponding respectively to amino acids 188–328 and 68–328 of langerin, were expressed as previously described (10,16). Mutations in the Lg-CRD were introduced into the human langerin cDNA by polymerase chain reaction using synthetic oligonucleotides. The forward-primer sequence, used for F241L mutation, was 5′-GAGAGTGAGCAGGAGCTGCTGTATAAAACAGCGGG-3′. The forward-primer sequence, used for W264R human mutation, was 5′-AGGGATGGAAGGGGACCGCTCCTGGGTGGATGAC-3′. The changes were checked by DNA sequencing (Beckman Coulter Genomics, Grenoble, France).
Protein purification
Lg-CRDWT and Lg-CRDF241L were purified using the protocol already described in (10,16). All forms of Lg-ECD were expressed as inclusion bodies so that a refolding step was performed before the purification procedure. The pellet obtained from a 1 L culture was resuspended into 30 mL of buffer A (25 mM Tris pH 8.0 and 150 mM NaCl) with addition of one complete EDTA (ethylene-diaminetetraacetic acid)-free tablet (Roche Diagnostic, Meylan, France). Cells were lysed by a 4 min sonication with 2 s pulses separated by 10 s pauses. Inclusion bodies were isolated by centrifugation at 100,000 × g for 30 min, 4°C. They were resuspended into 30 mL of buffer A with addition of 2 M urea and 1% v/v X100 Triton and centrifuged at 100,000 × g for 30 min at 4°C. Pellets were washed in 30 mL of buffer A and centrifuged again at 100,000 × g for 30 min, 4°C. Inclusion bodies were then solubilized in 30 mL of buffer A with addition of 6 M Guanidine and 0.01% v/v β-mercapto-ethanol. Aggregates were discarded at 100,000 × g for 30 min, 4°C. The supernatant was diluted down to 2 mg/mL in the same buffer. The solution was then diluted five fold by flash-dilution into a buffer containing 25 mM Tris pH 8.0, 1.25 M NaCl, and 25 mM CaCl2. A first dialysis step was performed against 25 mM Tris pH 8.0 (1 volume against 7). The resulting solution was dialyzed against buffer B (25 mM Tris pH 8.0, 150 mM NaCl, and 4 mM CaCl2). For Lg-ECDs, purification was performed by affinity chromatography on a mannan-agarose column (Sigma, St-Quentin Fallavier, France) equilibrated with buffer B and eluted in buffer A supplemented with 1 mM EDTA (buffer C). The eluted protein was dialyzed against buffer B. For refolded Lg-CRDW264R, purification was performed as for Lg-CRDWT using the N-terminal Strep-tag II affinity tag (16).
Retardation assay
The functionality of Lg-CRD (WT and mutants) was tested as previously described (16) on a mannan-agarose column (11 mL, Sigma).
Crystallization, data collection, and processing
Lg-CRDF241L was crystallized by the hanging drop method with initial mixing of 1 μL of Lg-CRDF241L (5 mg/mL in buffer B) and 1 μL of reservoir solution (100 mM MES (2-(N-morpholino)ethanesulfonic acid) pH 6.0; 200 mM MgCl2 and 17% w/v PEG 3350 (poly ethylene glycol)). A Lg-CRDF241L crystal was cryoprotected by transfer into a solution composed of 100 mM MES pH 6.0, 200 mM MgCl2, and 35% w/v PEG 3350 and flash-frozen in liquid nitrogen. X-ray diffraction data were collected at ID14-2 beamline (ESRF, Grenoble, France). 360 images were recorded with an oscillation range of 0.5° and 10 s exposure time. Data were processed using the XDS program (17). The space group is P42, the same as that of Lg-CRDWT. The structure was solved by molecular replacement using the MolRep program (18) and Lg-CRDWT structure as model (10). The structure was refined alternating refinement cycles performed with the Refmac program (19) and manual construction steps using the Coot program (20). The atomic coordinates and structure factors of Lg-CRDF241L have been deposited in the Protein Data Bank. The attributed code is 4AK8.
Circular dichroism spectroscopy
A Jobin Yvon CD6 circular dichroism (CD) spectropolarimeter was used. Data were recorded at room temperature in a 0.1 cm cell with a wavelength step of 1 nm and an integration time of 8 s. Each spectrum is the average of three successive scans, after correction with buffer B. Protein concentration was 0.1 mg/mL for all samples. The formula used to calculate the ellipticity is (33 × ΔA × 100)/(l × Protein concentration/MW × (number of residues – 1)), with l = cell length (0.1 cm), (number of residues – 1) = 154, and MW = 17905.1 Da and 17871.1 Da for Lg-CRDWT and Lg-CRDF241L, respectively.
Thermal shift assay
An iQ5 real-time polymerase chain reaction detection system from BioRad was used. The fluorophore used for the unfolded protein detection was SYPRO orange. The temperature range was varied from 293 K to 368 K at a rate of 1 K/min. The stability of Lg-CRDWT, Lg-CRDF241L, and Lg-CRDW264R at 0.5 mg/mL was determined in 100 mM HEPES pH 7.0, 500 mM NaCl, 4 mM CaCl2, 2 μL of 50× diluted commercial stock of SYPRO orange.
Cross-linking coupled to matrix-assisted laser desorption-ionization time-of-flight mass spectrometry
The WT and mutant Lg-ECDs were cross-linked by ethylene glycol bis(succinimidylsuccinate) (EGS). Proteins were dialyzed against 25 mM MES pH 6.0, 150 mM NaCl, and 4 mM CaCl2 to eliminate the Tris-HCl that is incompatible with the use of EGS. Proteins at 0.5 mg/mL were incubated with 5 mM EGS for 2 h at 4°C and the reaction was stopped by addition of Tris-HCl. Matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) spectra were acquired with a Bruker Daltonics Ultraflex III mass spectrometer. Native and cross-linked Lg-ECDWT and Lg-ECDF241L were serially diluted in 0.1% v/v TFA (trifluoroacetic acid) and mixed on the MALDI probe with an equal volume of sinapinic acid matrix (saturated in acetonitrile/water/TFA acid 50/50/0.1, v/v/v). The TOF analyzer was set in the linear mode for protein detection. Data were analyzed and smoothed using Bruker FlexAnalysis software.
Surface plasmon resonance
All experiments were performed on a Biacore 3000 using a functionalized CM4 chip and the corresponding reagents from Biacore. Two flow-cells were activated by 50 μL of an EDC/NHS mixture (ethyl(dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide) carbodiimide). Flow-cell one was blocked with 50 μL of ethanolamine and used as a control surface. Flow-cell two was functionalized at a level of 514 RU (Resonance units) with a 10 μg/mL solution of gp120YU2 (a kind gift from F. Véas and A. Méchulam) (21) in 10 mM sodium acetate pH 4.0 buffer. Increasing amounts of Lg-CRD and Lg-ECD (both WT and mutant) were injected at 20 μL/min flow rate. Lg-CRDs sensorgrams were fitted with Langmuir equation using the BIAeval software. The running buffer was buffer B supplemented with 0.005% v/v P20 surfactant. Each protein was diluted into the running buffer. 20 μL of 50 mM EDTA pH 8 were injected to regenerate the chip.
SAXS
SAXS experiments were carried out on the SWING beamline at the SOLEIL synchrotron radiation facility (Saint-Aubin, France). The sample to detector (Aviex CCD) distance was set to 1820 mm, allowing useful data collection over the momentum transfer range 0.007 Å−1 < q < 0.5 Å−1 with q = 4πsinθ/λ, where 2θ is the scattering angle and λ the wavelength of the x-rays (λ = 1.0 Å). Preliminary experiments showed that langerin solutions always contain a large fraction of aggregates. To separate aggregates from isolated proteins, SAXS data were collected on solutions eluting from the online size-exclusion high-performance liquid chromatography (SE-HPLC) column available on SWING and directly connected to the SAXS measuring cell (22). Flow rate was 150 μL/min, frame duration was 2 s, and the dead time between frames 0.5 s. For each frame, the protein concentration (between 0.5 and 1 mg/mL at the top of elution peak) was estimated from ultraviolet absorption at 280 nm using a spectrometer located immediately upstream of the SAXS measuring cell. The absorbance at 280 nm E0.1% (mg.ml−1) = 1.91 for the ECD was calculated from the sequence using the program ProtParam (http://www.expasy.org/). Selected identical frames corresponding to the main elution peak were averaged. A large number of frames were collected before the void volume and averaged to account for buffer scattering, which was subsequently subtracted from the averaged protein signal. The scattered intensities were put on an absolute scale using water scattering (23). Data analysis and model calculations were performed using programs from the ATSAS package (24). For clarity, P(r) profiles have been scaled to I(0), so that the total area under the curve is 1, but have not been scaled to MM.
Results and Discussion
Impact of langerin mutations on BG structure
Langerin gene transfection in COP5 fibroblasts leads to the formation of CMS typical of BGs (3). Mutation-induced alteration of Birbeck granule structure was reported for mutation F244L in the murine langerin gene transfected-COP5 (14) and for mutation W264R isolated from a healthy person whose LCs lack BG altogether (13). Considering the difference in gene origin and cellular environment of these two reported mutations, we decided to analyze those two mutations within the same genetic and cellular context, i.e., the human gene transfected in the previously described COP5 fibroblast model.
First, using flow cytometry, we controlled the cell surface expression of langerin on COP5 transfected cells using the mAb anti-langerin clone 808E10 directed against a Lg-CRD epitope, and intracytoplasmic expression using the mAb anti-langerin clone 310F7, which recognizes an intracytoplasmic epitope (see Fig. S1 in the Supporting Material). Although transfection level reaches only 12% to 20%, expression of langerin is clearly observed using the two mAbs except for the mutation W264R where mAb 808E10 labeling on cell surface is not observed, probably due to an epitope disappearance, as suggested by a global unfolding of the CRD in this mutant (see below).
Typical CMS were observed following transfection of the WT human langerin gene (Fig. 1, A and B). Phenylalanine 244 in murine langerin is conserved in humans as phenylalanine 241 as shown by sequence alignment (14). The transfection of Lg-ECDF241L gene leads to the formation of superimposed and thickened membranes structures different from typical CMS of BG, as previously described with the mouse gene (Fig. 1, D–E). We also confirm the total loss of BG associated with W264R mutation already reported (Fig. 1 F).
Figure 1.
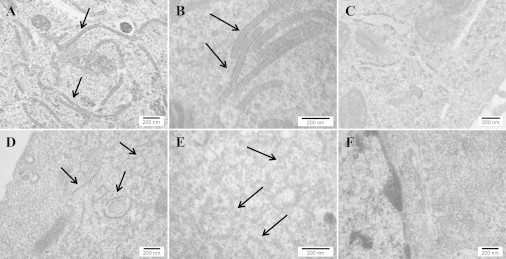
Electron microscopy pictures of transfected COP5 cells. (A, B, and D–F) Cells transfected with: langerinWT gene (A and B), langerinF241L gene (D and E), and langerinW264R gene (F). (C) Cells not transfected. The characteristic BG structure is shown in (A and B). Arrows point to CMS (observed with langerinWT) or superimposed and thickened membranes (observed with langerinF241L). Comparison of (B) with (E) shows the structural modification of CMS caused by the F241L mutation (substitution of the double layers with an inner striation with short, curved, and irregularly thickened double layers without inner electron-dense bodies), whereas panel (F) shows the loss of CMS with the W264R mutation.
These two mutations are located within the CRD reinforcing the essential role of this domain in BG formation as already suggested (10). To understand the effect of these mutations from a molecular point of view, different recombinant CRDs containing these mutations, as well as full-length ECDs, have been produced and characterized.
Characterization of mutation effects on langerin CRD
Langerin WT and mutants CRD production and purification
For the Lg-CRDs purification step, we used the Strep-tag II as described in (16). The soluble expression of the Lg-CRD, in the Escherichia coli periplasm, is a first indication of a proper folding of Lg-CRDs. This is confirmed by the similar elution profiles of Lg-CRDwt and Lg-CRDF241L of the soluble fraction. In contrast, Lg-CRDW264R was not present in the soluble fraction and formed inclusion bodies. As reported for CRDs of other C-type lectin such as DC-SIGN (25,26), a refolding step yielded a soluble form of Lg-CRDW264R.
Mannan binding properties of mutants
Functionality of Lg-CRDF241L was evaluated with the WT as positive control, using the delayed elution of proteins on a mannan-agarose column as a marker of their affinity for the resin (10). BSA, used as negative control, eluted in the void volume of the column, whereas Lg-CRDF241L eluted with a delay comparable to that observed with Lg-CRDwt (Fig. 2 A). The F241L mutation does not significantly affect the capacity of Lg-CRD to interact with the mannan. In contrast, Lg-CRDW264R eluted in the void volume as the negative control, suggesting that despite the isolation of a soluble form after refolding, this one is not functional.
Figure 2.
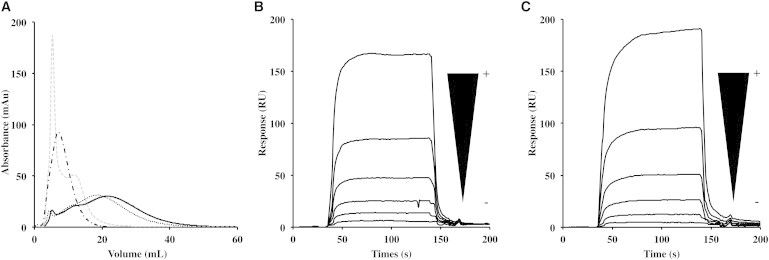
Functional characterization of Lg-CRDs. (A) Delayed elution of Lg-CRDs and BSA in Mannan-Agarose affinity column. BSA, used as a negative control (dotted-dashed line), Lg-CRDWT (solid line), Lg-CRDF241L (dotted line), and Lg-CRDW264R (gray dashed line). (B and C) Sensorgrams of the interaction of Lg-CRDs with gp120. B: Lg-CRDWT. C: Lg-CRDF241L. Both proteins were injected in series of twofold dilutions over the concentration range [12.5 μM, 400 μM].
Analysis of gp120 binding properties
Binding of Lg-CRDs to the envelope glycoprotein gp120 of HIV-1 has been studied using surface plasmon resonance (Fig. 2, B and C). We observed very similar interaction signals for both Lg-CRDWT and Lg-CRDF241L. The CRD harboring the F241L mutation appears to be fully functional as regards gp120 recognition.
Structural integrity of WT and mutants Lg-CRD
The secondary structures of the three Lg-CRDs have been compared using CD spectroscopy (Fig. 3 A). We observed a perfect superposition of the Lg-CRDF241L and Lg-CRDwt spectra indicative of identical secondary structures. In contrast, the CD pattern of Lg-CRDW264R was markedly different from that of Lg-CRDWT.
Figure 3.
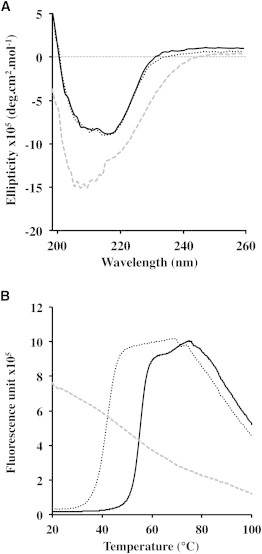
Structural characterization of Lg-CRDs. (A) CD spectra of Lg-CRDWT (solid line), Lg-CRDF241L (dotted line), and Lg-CRDW264R (gray dashed line). (B) Thermal shift assay of Lg-CRDWT (solid line), Lg-CRDF241L (dotted line), and Lg-CRDW264R (gray dashed line) as a function of temperature.
Structural stability has been investigated using thermal shift assay. Misfolding of Lg-CRDW264R is confirmed by the strong fluorescence signal observed at 20°C indicating that hydrophobic regions are already exposed to the solvent (Fig. 3 B). The fluorescence decrease observed at higher temperature is due to protein aggregation. In view of the location of W264 within a central hydrophobic core of the protein, it could be anticipated that its replacement by an arginine would cause the observed Lg-CRD global unfolding (Fig. S2). Although Lg-CRDF241L behaves as a well-folded protein, with a clear melting transition, it exhibits a Tm 10°C lower than that of the WT protein (Fig. 3 B). This reduced stability is the first noticeable effect of the mutation.
X-ray structure of Lg-CRDF241L
To explain the stability decrease in F241L mutant, we crystallized and solved the structure of Lg-CRDF241L (see Table S1 for data collection and structure statistics). The superimposition of Lg-CRDF241L structure over that of Lg-CRDwt structure, previously solved by us and others (10,27), shows that the replacement of the phenylalanine by a leucine causes the loss of a π orbital stacking between the F241 and the F202 from α2 helix and β1 strand (Fig. 4, A–C). This interaction may play a role in anchoring the first N-terminal β-sheet onto the rest of the Lg-CRD and its loss may explain the reduced thermal stability of the F241L mutant reported previously. Moreover, the mutation induces a large movement of K200 side chain that fills the space left empty by the loss of the phenylalanine ring (Fig. 4 C). As a result, K200 establishes a new hydrogen bond with T245 in Lg-CRDF241L. This is accompanied by the displacement of three water molecules, one of which is substituted by the amine group of K200. The net contribution of this additional H-bond to the stability is lower than the initial hydrophobic cluster centered on F241. H-bond being more sensitive to temperature increase, this explains the lower stability achieved. However, these modifications are essentially local, do not significantly impair the recognition properties of gp120, and do not provide an easy explanation for the deep disorganization of the Birbeck granule superstructure (Fig. 1).
Figure 4.
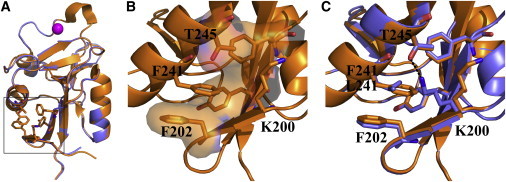
Crystallographic structures of Lg-CRDWT (orange) and Lg-CRDF241L (blue). (A) Superimposition of both structures. (B) Focus on F241 and F202 π-orbital stacking in Lg-CRDWT structure. (C) Same focus as C with both superimposed structures.
Feinberg et al. (28) published a structure of a trimeric truncated form of the langerin ECD. In this trimer, F241 is found at the interface between neighboring monomers so that its mutation is likely to destabilize interactions within the trimer. To clarify this issue, we decided to study this mutation in the context of the whole ECD.
Characterization of F241L mutation effects on Lg-ECD
Lg-ECDWT and Lg-ECDF241L production and purification
Recombinant Lg-ECD is produced in inclusion bodies in bacteria and must therefore be refolded before protein purification, whereupon functional oligomers are sorted by affinity purification onto mannan agarose column. Both Lg-ECDWT and Lg-ECDF241L bound to the column and required EDTA for elution in contrast to the CRDs that were just delayed. This is due to an avidity effect, thereby revealing the existence of oligomeric forms for each protein (data not shown).
Alteration of gp120 binding properties of Lg-ECDF241L
Titration experiments of both Lg-ECDWT and Lg-ECDF241L were performed on an immobilized gp120 surface as described previously for Lg-CRDs. Both Lg-ECDs are able to recognize high mannose glycans from gp120 but Lg-ECDF241L required the use of much higher concentrations to reach a similar response level to that of the WT as shown in Fig. 5. Data analysis yielded a ratio of 7 between the apparent affinities of the two Lg-ECDs (Fig. S3). Because Lg-CRDF241L affinity was slightly better, if at all different, from that of Lg-CRDWT, the weaker affinity of Lg-ECDF241L implies a reduction in the avidity of the mutant, due to the modification of the Lg-CRDs arrangement or even of the oligomeric state of the protein.
Figure 5.
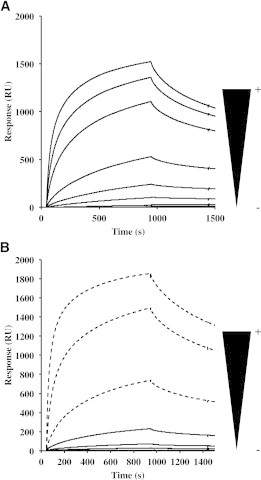
Sensorgrams of the interaction of Lg-ECDs with gp120. (A) Lg-ECDWT; concentrations used are 5, 10, 50, 100, 200, 500, 750, and 1000 nM. (B) Lg-ECDF241L; black lines correspond to concentrations of 62.5, 250, 500, and 1000 nM and dotted lines correspond to additional concentrations used, 2, 4, and 6.6 μM.
Cross-linking coupled to MALDI-MS of Lg-ECDWT and Lg-ECDF241L
To address this question, we first performed cross-linking experiments coupled with mass spectrometry (MS). Although results show the expected trimer of WT protein, a dimer is clearly observed for Lg-ECDF241L (Fig. 6). This prompted us to undertake a SAXS study to investigate into more detail the oligomeric state and conformation of both Lg-ECDs.
Figure 6.
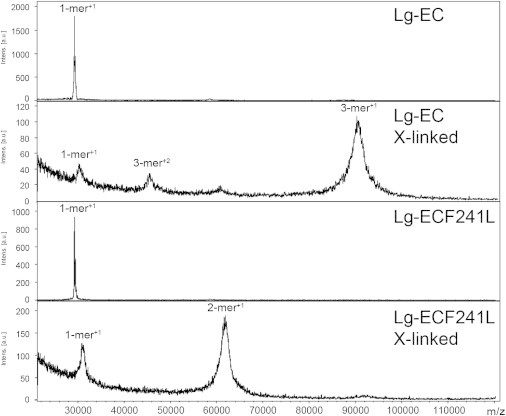
Oligomeric state of Lg-ECD assessed by cross-linking and MALDI-MS analysis. Top to bottom panel correspond to MALDI-MS spectra of Lg-ECDWT, Lg-ECDWT cross-linked with EGS, Lg-ECDF241L, Lg-ECDF241L cross-linked with EGS, respectively.
SAXS analysis of Lg-ECDWT and Lg-ECDF241L
Preliminary experiments showed that concentrated langerin solutions always contain a large fraction of aggregates. To separate aggregates from the isolated proteins, SAXS data were collected on solutions eluting from the online SE-HPLC column directly connected to the SAXS measuring cell on the SWING beamline at the Synchrotron SOLEIL (22). Elution profiles measured with the ultraviolet spectrometer (λ = 280 nm) are displayed in Fig. 7 for both WT and mutant proteins. In the case of WT protein the profile exhibits a single narrow peak beyond those corresponding to aggregates. In the case of the mutant protein, a clear shoulder is seen at the elution volume of the WT protein, whereas the main elution peak is markedly shifted toward longer elution times.
Figure 7.
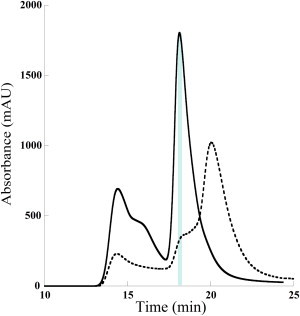
SAXS-coupled SE-HPLC elution profiles of Lg-ECDs. Lg-ECDWT (continuous line) and Lg-ECDF241L (dashed line). The light blue rectangle corresponds to the elution of the WT protein.
Lg-ECDWT
Scattering patterns recorded around the maximum of the elution peak are identical, so that 20 frames could be averaged to yield a scattering curve amenable to further analysis. All structural parameters derived from the SAXS data are given in Table 1, Guinier plots are shown in Fig. S4, whereas P(r) profiles are shown in Fig. 8. The estimate of the molecular mass derived from the I(0)/c value is in good agreement with the molecular mass of a trimer.
Table 1.
Structural parameters derived from SAXS data
| Molecule | Rg Guinier (Å)a | Rg P(r) (Å) | Dmax (Å) | c (mg/ml) | MM (kDa) from I(0)b | MM(kDa) from volumec | calculated MM (kDa)d |
|---|---|---|---|---|---|---|---|
| Lg-ECDWT | 55.6 ± 1 | 58.4 ± 1 | 210 | 0.71 | 88.8 | 92.4 | 88.2 |
| Lg-ECDF241L, trimer | 55.9 ± 1 | 55.6 ± 1 | 200 | 0.20 | 92.7 | 87.8 | 88.1 |
| Lg-ECDF241L, dimer | 41.8 ± 1 | 42.9 ± 1 | 140 | 0.49 | 58.9 | 55.6 | 58.7 |
| Lg-ECD model | 65.2e | 69.3 | 230 | 88.2 |
Rg obtained with the Guinier approximation in the range 0.4 < qRg < 1.
Molar mass M obtained from the I(0) value using the partial specific volume 0.732 cm3.g−1 calculated with the program SEDNTERP available at http://sednterp.unh.edu/.
Molar mass M obtained from the whole I(q) curve (qmax = 0.3Å) using the macromolecule volume and the SAXS-MoW method available at http://www.if.sc.usp.br/∼saxs/.
The calculated masses were derived from the sequence.
The Guinier Rg value is identical to the P(r) derived value when calculated over the experimentally inaccessible q-range q Rg <0.14.
Figure 8.
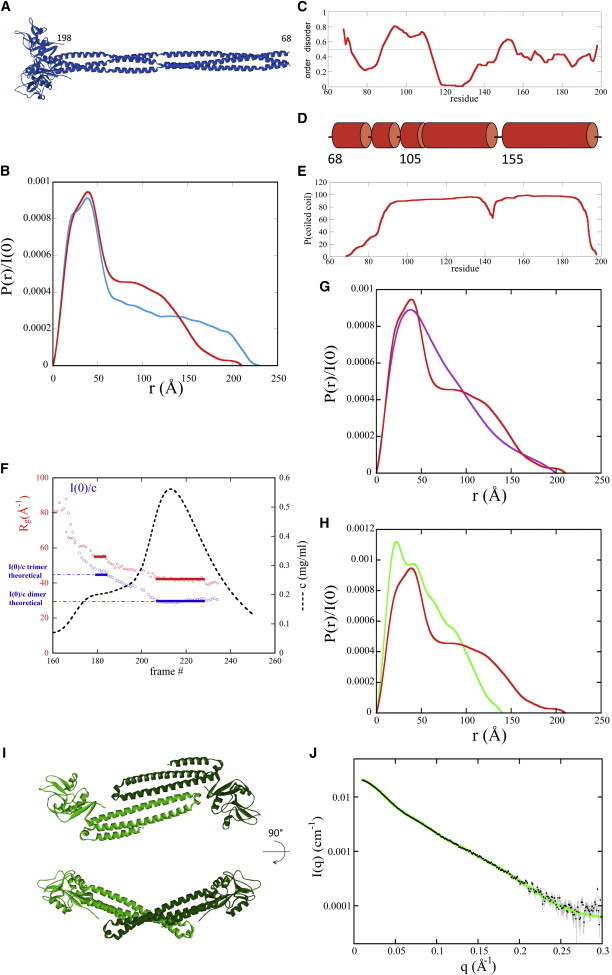
SAXS analysis. (A) Model of Lg-ECDWT trimer from (30) after incorporation of the trimeric head and proximal coiled-coil region from 3KQG. (B) Comparison of the distance distribution function of Lg-ECDWT model shown in A (cyan line), with that derived from the experimental curve for Lg-ECDWT (red curve). (C) Disorder prediction using PondR (30) in the range 68–198 (neck). (D) Prediction of secondary structure in the same range. Only helices are predicted. (E) Prediction of coiled coil using Logicoil (31). (F) Variation along the SEC elution profile of the protein concentration c (black dashed line), radius of gyration Rg (red diamonds), and I(0)/c (blue squares). Each frame corresponds to a 2 s SAXS pattern. (G) Comparison of the distance distribution functions of the WT trimer (red line) and of the trimer of F241L mutant (purple line). (H) Comparison of the distance distribution functions of the WT trimer (red line) and of the dimer of F241L mutant (green line). (I) Two 90° rotation views of a typical model obtained with SASREF for the dimer of F241L mutant. (J) Black dots: SAXS experimental data for the dimer of F241L mutant with error bars (gray lines); for q < 0.17 Å−1 error bars are smaller than the black dots; green line: calculated curve using CRYSOL from the model shown in Fig. 8I.
Previously, we proposed a structural model of the Lg-ECD based on the results of hydrodynamic studies (10). We modified this model by incorporating the crystal structure of the trimeric head of langerin containing the CRD and one coiled-coil repeat (Protein Data Bank 3KQG) (28,29) (Fig. 8 A). We calculated the scattering pattern I(q) of the resulting model together with the corresponding distance distribution function P(r). Comparison of the Rg and Dmax values of the model with experimental values (Table 1) and of its P(r) with the experimental one (Fig. 8 B) clearly indicate that the protein neck in solution does not adopt the straight structure long of ∼200 Å proposed in our initial model and is markedly shorter. Most striking is the drop around 140 Å observed on the experimental profile crossing the calculated profile that, in contrast, exhibits a slowly decreasing plateau until beyond 200 Å. The major peak centered around 30 to 40 Å is very similar in both profiles, suggesting that the trimeric CRD head adopts in solution a conformation similar or even identical to that observed in the crystal.
These observations prompted us to question one of the assumptions behind our model, namely that the neck adopted an extended threefold coiled-coil conformation. We thus investigated the predictions of several coiled-coil prediction programs including the recently available Logicoil program (31). We also used several order/disorder analysis and structure predictions programs. The results are presented in a synthetic way in Fig. 8, C–E. The PondR profile (Fig. 8 C) shows that the neck is predicted to be highly structured with two localized exceptions around residue number 100 and 150, respectively. PsiPred (Fig. 8 D) predicts mostly α-helical structures with a few interruptions in the N-terminal region and close to the central stretch pointed out in PondR. The prediction of the program Logicoil is shown in Fig. 8 E. The neck is essentially predicted to adopt a coiled-coil conformation with two exceptions in the N-terminal region and just upstream from residue 150. The SAXS pattern contains too little structural information to determine a unique model for langerin conformation in solution. However, combining the SAXS pattern with the results of the various sequence analysis programs mentioned previously may guide what amounts to an educated guess. In view of its speculative character, this attempt is briefly presented in the Supporting Material (Fig. S5).
In conclusion, the conformation of the ectodomain of langerin in solution is certainly very different from the shape of DC-SIGN ectodomain. Indeed, DC-SIGN neck that adopts a rigid, straight coiled-coil arrangement ending with the CRD tetramer is based on the existence of conserved heptad repeats making for a very regular coiled-coil (26,32). Langerin neck sequence is not as strictly periodic as that of DC-SIGN neck and exhibits two regions predicted to be unstructured and that are potential sources of fragility. However, it is likely that, in the context of full-length langerin, membrane anchoring significantly constrains these less-ordered regions.
Lg-ECDF241L
The elution profile shown in Fig. 7 exhibits a pronounced shoulder at an elution time similar to that of the WT trimer followed by the main peak. Each scattering pattern along the elution profile was individually analyzed. Fig. 8 F presents the variation along the elution profile of the protein concentration c (measured using OD at 280 nm), the Rg, and I(0)/c that is proportional to the mass of the scattering object. I(0)/c values show that the scattering species corresponding to the shoulder and the top of the elution peak are compatible with trimers and dimers respectively.
Identical frames within the shoulder and around the top of the peak (5 and 18 frames, respectively) were averaged to improve the statistical quality for further analysis. Average patterns of the mutant trimer and dimer are shown in Figs. S6 and 8 J, respectively.
The trimer of the mutant protein is different from that of WT langerin as indicated by the smaller Rg value and by the profile of the distance distribution function that is not compatible with a neck following a head domain (Fig. 8 G).
The distance distribution function of the dimer is very different from that of WT langerin (Fig. 8 H). The maximal extension is much shorter, indicating that the dimer does not adopt a conformation analog to that of the WT trimer with a head part followed by an extended dimeric coiled coil. However, the asymmetric P(r) is characteristic of an elongated object. The value of the dimer maximal extension is 140 Å, much shorter than the length of one helix from the neck in straight conformation (∼200 Å). Therefore, helices have to fold over, possibly interacting with one another.
We have submitted the neck sequence of langerin to the program Robetta (33). The first resulting model showed an almost entirely helical chain folded over in three contiguous segments in an intrachain coiled-coil-like arrangement. Combining this model with a model of the monomer structure of the mutant Lg-CRD we obtain a full-length model of langerin chain. We then ran the rigid-body modeling program SASREF (34) to build a symmetric dimer (the twofold symmetry condition is imposed a priori). 9 out of 10 resulting dimer models were very similar. A typical one that gave an excellent fit to the data with a χ value of 1.2 (Fig. 8 J) is shown in Fig. 8 I. We propose this model, which combines totally independent informations (the neck sequence fed to Robetta and our SEC-SAXS data), although we are well aware that other models are possible. Indeed, the 10th model obtained with SASREF showed a head-to-head interaction through the domain top faces (as opposed to side interactions as in the WT trimeric head) with a slightly worse fit to the data. We are also aware that this is not a model for the full-length molecule anchored in the membrane because, in this case, only parallel interactions are possible.
In the WT trimer, the Lg-CRD surface containing the F241 residue lies within the interface between two protomers (Fig. 9, C and D), where the bottom of one CRD (containing F241) interacts with the C-terminal end of the coiled-coil neck of the neighboring monomer (Fig. 9 D). Fig. 9 B, where the major part of Lg-CRDs has been omitted for the sake of clarity, shows that the F241 residue from each protomer directly interacts with Q197 of the neighboring protomer. Beyond this direct interaction between protomers, the bulky side chain of F241 is located in the center of the CRD/CRD interface (Fig. 9 D) so that the F241L mutation, together with the already mentioned relocation of K200 (Fig. 4 C) modifies the Lg-CRD surface involved in this contact (Fig. 9 D). Our data globally suggest that phenylalanine in position 241 is crucial for the interface stability required for the formation of the WT-like trimer that cannot be ensured by the sole neck domain. Actually, analysis of coiled-coil propensity using the Logicoil program that is tuned toward discrimination between various oligomerization states predicts a trimeric coiled-coil for the distal (N-proximal) part of the neck (residues 100–140) contrasting with a favored dimeric coiled-coil for the CRD proximal part (residues 150–190), the two being separated by a short less-ordered region also predicted by other programs. We surmise that the mutant Lg-ECD trimer is associated by the N-terminal trimeric coiled-coil, although most likely presenting significant disorder in the rest of the chains, with a mixture of noninteracting chains. The sheer number and heterogeneity of the likely conformations in solution make any modeling attempt a meaningless undertaking.
Figure 9.
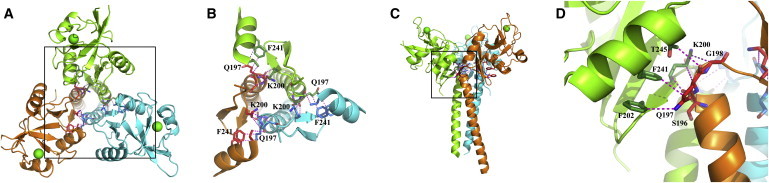
F241 and K200 are central residues of the interface between CRD of one protomer and C-terminal neck end of the neighboring one. (A). Top view. (B) Top view with a zoom over the square in panel A. (C) Lateral view. (D) Lateral view with a zoom over the square in (C).
In langerin, trimer stabilization appears to result from synergistic contributions from both CRD and neck domains. The apparently minor perturbation associated with the substitution of the phenyl ring by the aliphatic side chain of leucine and with lysine 200 reorientation is enough to shift the balance toward the dimeric coiled coil as observed by MALDI-MS study of cross-linked ECDs and by SAXS. The solutions of mutant Lg-ECD are thus highly heterogeneous both in terms of oligomerization states and of conformations.
Conclusions
In a previous study, we proposed a molecular mechanism for the membrane zippering leading to BG formation, in which CRDs at the tip of elongated neck domains within facing membranes could assemble and form a higher order structure at the center of the CMS (10). This proposal was mainly supported by electron microscopy observations, notably the disappearance of CMS in COP5 transfected with langerin lacking the entire carbohydrate domain. In this study, we showed that this DC-SIGN-like representation of the molecule, with a rigid neck, is not strictly supported by our observation of the ECD in solution that suggests a more flexible structure. However, we must add that in vivo, the array of full-length langerin molecules anchored in the membrane may give rise to intermolecular interactions stabilizing the trimer conformation. We also studied the structural consequences of the rather conservative substitution of hydrophobic leucine for hydrophobic phenylalanine and showed that they are locally subtle but have far-reaching functional implications. Lg-CRDF241L is still properly folded and fully functional, structural changes are few and local but located at an interface between neighboring CRDs. This interaction is perturbed enough to switch the global balance between trimer and dimer formation in solution, creating significant heterogeneities and finally leading to the deep alteration of CMS organization and to the assembly of irregularly thickened membrane structures observed in COP5 transfected cells (Fig. 1 E). The spatial arrangement of CRDs within the trimeric head of langerin is thus an absolute requirement for a proper BG architecture. Finally, in addition to BG formation, the affinity of Lg-ECDF241L for high mannose glycoconjugates in gp120 is severely reduced, showing that the specific spatial arrangement of CRDs is also key to the optimal recognition of the HIV-1 envelope glycoprotein.
Acknowledgments
We are very grateful to P. Roblin for expert assistance during measurements at the SWING beamline (Synchrotron Soleil) and to J. Pérez for insightful discussions. We thank Dr. Francisco Véas and Alain Méchulam for the kind gift of gp120 YU2. We also acknowledge the Centre des Microstructures from Lyon University for access to electron microscopes.
This article is dedicated to our beloved colleague, Richard Kahn who passed away in October 2011. We miss the sound of his footsteps along the corridor, a physical expression of the intensity of his thoughts. It was an honor to know him.
Footnotes
Richard Kahn was deceased in 2011.
Contributor Information
Dominique Durand, Email: dominique.durand@u-psud.fr.
Franck Fieschi, Email: franck.fieschi@ibs.fr.
Supporting Material
References
- 1.Geijtenbeek T.B., Kwon D.S., van Kooyk Y. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 2.Valladeau J., Duvert-Frances V., Saeland S. The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface. Eur. J. Immunol. 1999;29:2695–2704. doi: 10.1002/(SICI)1521-4141(199909)29:09<2695::AID-IMMU2695>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 3.Valladeau J., Ravel O., Saeland S. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12:71–81. doi: 10.1016/s1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- 4.Turville S.G., Cameron P.U., Cunningham A.L. Diversity of receptors binding HIV on dendritic cell subsets. Nat. Immunol. 2002;3:975–983. doi: 10.1038/ni841. [DOI] [PubMed] [Google Scholar]
- 5.de Witte L., Nabatov A., Geijtenbeek T.B. Distinct roles for DC-SIGN+-dendritic cells and Langerhans cells in HIV-1 transmission. Trends Mol. Med. 2008;14:12–19. doi: 10.1016/j.molmed.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 6.de Witte L., Nabatov A., Geijtenbeek T.B. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat. Med. 2007;13:367–371. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 7.de Jong M.A., Geijtenbeek T.B. Langerhans cells in innate defense against pathogens. Trends Immunol. 2010;31:452–459. doi: 10.1016/j.it.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 8.McDermott R., Bausinger H., Hanau D. Reproduction of Langerin/CD207 traffic and Birbeck granule formation in a human cell line model. J. Invest. Dermatol. 2004;123:72–77. doi: 10.1111/j.0022-202X.2004.22728.x. [DOI] [PubMed] [Google Scholar]
- 9.Stambach N.S., Taylor M.E. Characterization of carbohydrate recognition by langerin, a C-type lectin of Langerhans cells. Glycobiology. 2003;13:401–410. doi: 10.1093/glycob/cwg045. [DOI] [PubMed] [Google Scholar]
- 10.Thépaut M., Valladeau J., Fieschi F. Structural studies of langerin and Birbeck granule: a macromolecular organization model. Biochemistry. 2009;48:2684–2698. doi: 10.1021/bi802151w. [DOI] [PubMed] [Google Scholar]
- 11.Ward E.M., Stambach N.S., Taylor M.E. Polymorphisms in human langerin affect stability and sugar binding activity. J. Biol. Chem. 2006;281:15450–15456. doi: 10.1074/jbc.M511502200. [DOI] [PubMed] [Google Scholar]
- 12.Feinberg H., Rowntree T.J., Taylor M.E. Common polymorphisms in human langerin change specificity for glycan ligands. J. Biol. Chem. 2013;288:36762–36771. doi: 10.1074/jbc.M113.528000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verdijk P., Dijkman R., Tensen C.P. A lack of Birbeck granules in Langerhans cells is associated with a naturally occurring point mutation in the human Langerin gene. J. Invest. Dermatol. 2005;124:714–717. doi: 10.1111/j.0022-202X.2005.23645.x. [DOI] [PubMed] [Google Scholar]
- 14.Valladeau J., Clair-Moninot V., Saeland S. Identification of mouse langerin/CD207 in Langerhans cells and some dendritic cells of lymphoid tissues. J. Immunol. 2002;168:782–792. doi: 10.4049/jimmunol.168.2.782. [DOI] [PubMed] [Google Scholar]
- 15.Tyndall C., La Mantia G., Kamen R. A region of the polyoma virus genome between the replication origin and late protein coding sequences is required in cis for both early gene expression and viral DNA replication. Nucleic Acids Res. 1981;9:6231–6250. doi: 10.1093/nar/9.23.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thépaut M., Vivès C., Fieschi F. Overproduction, purification and preliminary crystallographic analysis of the carbohydrate-recognition domain of human langerin. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2008;64:115–118. doi: 10.1107/S1744309108001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 18.Vagin A., Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Mechulam A., Cerutti M., Veas F. Highly conserved beta16/beta17 beta-hairpin structure in human immunodeficiency virus type 1 YU2 gp120 is critical for CCR5 binding. J. Mol. Med. 2005;83:542–552. doi: 10.1007/s00109-005-0673-1. [DOI] [PubMed] [Google Scholar]
- 22.David G., Pérez J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Cryst. 2009;42:892–900. [Google Scholar]
- 23.Orthaber D., Bergmann A., Glatter O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J. Appl. Cryst. 2000;33:218–225. [Google Scholar]
- 24.Petoukhov M.V., Franke D., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell D.A., Fadden A.J., Drickamer K. A novel mechanism of carbohydrate recognition by the C-type lectins DC-SIGN and DC-SIGNR. Subunit organization and binding to multivalent ligands. J. Biol. Chem. 2001;276:28939–28945. doi: 10.1074/jbc.M104565200. [DOI] [PubMed] [Google Scholar]
- 26.Tabarani G., Thépaut M., Fieschi F. DC-SIGN neck domain is a pH-sensor controlling oligomerization: SAXS and hydrodynamic studies of extracellular domain. J. Biol. Chem. 2009;284:21229–21240. doi: 10.1074/jbc.M109.021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chatwell L., Holla A., Skerra A. The carbohydrate recognition domain of Langerin reveals high structural similarity with the one of DC-SIGN but an additional, calcium-independent sugar-binding site. Mol. Immunol. 2008;45:1981–1994. doi: 10.1016/j.molimm.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 28.Feinberg H., Powlesland A.S., Weis W.I. Trimeric structure of langerin. J. Biol. Chem. 2010;285:13285–13293. doi: 10.1074/jbc.M109.086058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chabrol E., Nurisso A., Fieschi F. Glycosaminoglycans are interactants of Langerin: comparison with gp120 highlights an unexpected calcium-independent binding mode. PLoS ONE. 2012;7:e50722. doi: 10.1371/journal.pone.0050722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romero P., Obradovic Z., Dunker A.K. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Vincent T.L., Green P.J., Woolfson D.N. LOGICOIL—multi-state prediction of coiled-coil oligomeric state. Bioinformatics. 2013;29:69–76. doi: 10.1093/bioinformatics/bts648. [DOI] [PubMed] [Google Scholar]
- 32.Feinberg H., Tso C.K., Weis W.I. Segmented helical structure of the neck region of the glycan-binding receptor DC-SIGNR. J. Mol. Biol. 2009;394:613–620. doi: 10.1016/j.jmb.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raman S., Vernon R., Baker D. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petoukhov M.V., Svergun D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.