

Abstract
Protein kinase R (PKR) is activated by dsRNA produced during virus replication and plays a major role in the innate immunity response to virus infection. In response, viruses have evolved multiple strategies to evade PKR. Adenovirus virus-associated RNA-I (VAI) is a short, noncoding transcript that functions as an RNA decoy to sequester PKR in an inactive state. VAI consists of an apical stem-loop, a highly structured central domain, and a terminal stem. Chemical probing and mutagenesis demonstrate that the central domain is stabilized by a pseudoknot. A structural model of VAI was obtained from constraints derived from chemical probing and small angle x-ray scattering (SAXS) measurements. VAI adopts a flat, extended conformation with the apical and terminal stems emanating from a protuberance in the center. This model reveals how the apical stem and central domain assemble to produce an extended duplex that is precisely tuned to bind a single PKR monomer with high affinity, thereby inhibiting activation of PKR by viral dsRNA.
Introduction
Infection of mammalian cells by viruses triggers the innate immunity response by interaction of viral nucleic acids with the pattern recognition receptors RIG-I, MDA5, and TLR3 (1). These pathways converge in the expression of type 1 interferons. Secreted interferons induce several hundred genes, including key proteins involved in antiviral defense: PKR, RNase L, and MxA. Viruses have evolved diverse mechanisms to evade the innate immunity pathway (2). The crucial role of PKR in this pathway is highlighted by the large number of viruses that disable PKR to promote viral replication (3) and by the rapid evolution of PKR under selective pressure from viruses (4,5).
PKR contains two tandem dsRNA binding domains at the N-terminus and a C-terminal kinase domain connected by a long, unstructured linker. The enzyme is induced in a latent form and is activated by viral dsRNA to phosphorylate the translational initiation factor eIF2α, leading to arrest of viral protein synthesis in the host cell. PKR activation is mediated by dimerization of the kinase domains (6–8). A minimum of 30 to 33 bp of regular duplex RNA is required to bind two PKR monomers and activate the kinase (9,10), supporting the dimerization model. Secondary structure defects typically impede the ability of dsRNAs to activate PKR (11).
Adenovirus and Epstein-Barr virus each produce noncoding, highly structured RNAs that act as RNA decoys and sequester PKR but do not activate, thereby allowing viral replication to proceed (3). Adenovirus virus-associated RNA-I (VAI) contains ∼160 nt and accumulates to micromolar concentrations late in infection. Enzymatic probing measurements (12,13) reveal a conserved secondary structure consisting of three distinct domains: an apical stem, a highly structured central domain, and a terminal stem (Fig. 1 a). The apical stem is the primary PKR site, with some evidence for interaction with the central domain (14–17). Deletion of the entire terminal stem does not affect PKR binding or inhibitor potency (3,18,19). PKR binding stoichiometry and affinity are strongly modulated by divalent ion and a single monomer binds with high affinity to VAI in the presence of Mg2+ (19). It has been proposed that the central domain is stabilized by a pseudoknot (20,21). Recent small angle x-ray scattering (SAXS) measurements indicate that VAI adopts an extended conformation (22).
Figure 1.
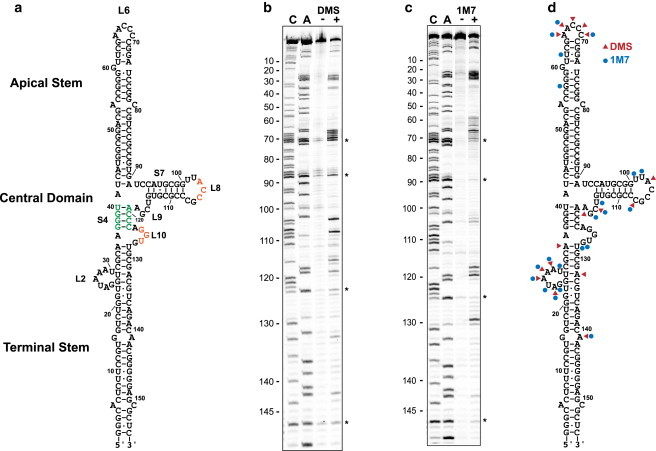
Structure probing of VAI. (a) Secondary structure of VAI. The complementary triplets in loops 8 and 10 are depicted in red, and the conserved tetrastem 4 is depicted in green. (b) DMS probing of VAI structure. The gel includes C and A sequencing ladders, a control lane without DMS (-), and a lane with DMS (+). Artifacts arising from reverse transcriptase pause sites are marked with an asterisk. (c) SHAPE probing of VAI structure. The gel includes C and A sequencing ladders, a control lane without SHAPE reagent (-) and a lane with 1M7 (+). Artifacts arising from reverse transcriptase pause sites are marked with an asterisk. (d) DMS and SHAPE reaction sites mapped onto the secondary structure diagram of VAI. To see this figure in color, go online.
In this study, we employ a combination of chemical probing, SAXS, and analytical ultracentrifugation to define a structural model of VAI. VAI is stabilized by a pseudoknot and adopts a flat, extended conformation with the apical and terminal stems emanating from a protuberance in the middle. The apical stem and central domain assemble to produce an extended duplex that is precisely tuned to bind a single PKR monomer with high affinity.
Materials and Methods
PKR was expressed and purified as previously described (23). RNAs were synthesized in vitro by T7 RNA polymerase using linearized plasmid templates and purified as previously described (17,19). VAI mutants were analyzed using the Kinefold (24) structure prediction algorithm to ensure that the mutations did not stabilize alternative folds within the central domain.
Structure probing
For primer extension assays VAI was inserted into an RNA structure cassette (25). A primer complementary to the 3′ region of the cassette containing a 5′-amine was purchased from IDT (Coralville, IA) (5′-AmC6-GAACCGGACCGAAGCCCG-3′) and labeled with 6-carboxyltetramethylrhodamine (6-TAMRA) N-hydroxysuccinamidyl ester. 1-methyl-7-nitroisatoic anhydride (1M7) was synthesized using an established protocol (26). Before DMS modification, RNAs were annealed at 95°C for 3 min in 20 mM phosphate, 0.1 mM EDTA, pH 7.5 and snap-cooled on ice. 200 mM NaCl and 5 mM MgCl2 were added, and the RNAs were allowed to fold for 30 min at 37°C. RNAs were treated with 38.4 mM of DMS dissolved in ethanol for 15 min at room temperature, and the reactions were quenched by adding 51.8 mM β-mercaptoethanol. Before SHAPE analysis, RNAs were annealed at 95°C for 3 min in 40 mM HEPES, 0.1 mM EDTA, pH 8.0 and snap-cooled on ice. 200 mM NaCl and 5 mM MgCl2 were added, and the RNAs were allowed to fold for 30 min at 37°C. RNAs were treated with 150 mM of 1M7 dissolved in DMSO for 2 min at 37°C. Samples were ethanol precipitated, dried, and resuspended in 0.5 X TE (5 mM Tris-HCl, 0.5 mM EDTA), pH 8.0. Reverse transcription was performed as previously described (25). Samples were run on 8 M urea sequencing gels containing 8% to 12% acrylamide and immediately imaged on a Typhoon Trio (GE Life Sciences, Piscataway, NJ). Gel images were quantitated using SAFA (27). The data were normalized to the band corresponding to A65, which was uniformly reactive in all VAI constructs. Reactivities below 0.20 were considered unmodified.
Structural modeling
SHAPE reactivities were imported into RNAStructure (28), and secondary structures were determined using pseudo-free energy constraints (29) with default settings for the slope and intercept. Ten three-dimensional models of VAI were generated with RNAComposer (30) using the secondary structure constraints generated by RNAStructure. A pseudoknot between loops 8 and 10 was imposed where appropriate. These models were aligned to SAXS envelopes using SUPCOMB (31). The final models were chosen based on a low normalized spatial discrepancy (NSD) and adherence to the structure probing data.
Analytical ultracentrifugation
PKR binding to VAI was characterized using sedimentation velocity analytical ultracentrifugation as previously described (17,19). Before analytical ultracentrifugation, PKR and VAI constructs were buffer exchanged into 20 mM HEPES, pH 7.5, 200 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, and 0.1 mM TCEP using Biogel P6 spin columns and mixed at several ratios. The data were processed using DCDT+ (32) to produce normalized g(s∗) sedimentation velocity distribution functions. Dissociation constants were determined by global analysis with SEDANAL (33) using a 1:1 binding model. Hydrodynamic parameters were calculated using HYDROPRO (34).
Small angle x-ray scattering
SAXS data were collected at beamline X9 of the National Synchrotron Radiation Laboratory at Brookhaven National Laboratories. RNA samples at concentrations of 1, 2, and 4 mg/ml were buffer exchanged into 20 mM HEPES, pH 7.5, 200 mM NaCl, 0.1 mM EDTA, 0.1 mM TCEP, and 5% glycerol. Sample homogeneity was verified by sedimentation velocity analysis (see Fig. S1 in Supporting Material). MgCl2 was added directly before analysis. Samples were filtered through a 0.02 μm syringe filter (Anotop) and centrifuged for 5 min at 12,000 RPM immediately before analysis. During data collection, the samples were maintained at 20°C and flowed continuously through a capillary to prevent radiation damage. Analysis of successive frames confirms the absence of radiation damage.
Guinier analysis was performed using the low-q portion of the data where Rg•q ≤ 1.3. The p(r) pair distribution function was calculated using GNOM (35) with a maximum q corresponding to 8/Rg. DMax was determined by the minimum of Χ2 as this parameter was incremented. Ab initio bead models were generated using the data collected at 2 mg/ml by simulated annealing using DAMMIF (36). For each structure, 25 simulated annealing runs were performed and the resulting models were superimposed, averaged, and filtered using DAMAVER (37). The mean NSD was calculated for each ensemble: 0.81 ± 0.06 (VAI), 0.82 ± 0.03 (VAI + Mg2+), 0.86 ± 0.03 (L8 + Mg2+), and 0.77 ± 0.04 (ΔTS + Mg2+). Surfaces were calculated using pdb2vol from SITUS (38).
Results
Secondary structure of VAI
Several alternative secondary structures have been reported for VAI based on enzymatic and chemical structure probing and phylogenetic analyses (39,40). Therefore, we used both DMS and SHAPE probing to resolve the base pairing within VAI. The RNA was inserted into a cassette to facilitate analysis by primer extension (25). The pattern of chemical modifications observed in this study is the same as detected in the absence of the cassette (17). As expected, DMS reacts extensively with residues lying within loops 2, 6, and 9 (Fig. 1, b and d). Interestingly, most of the residues in loops 8 and 10 are protected, consistent with a tertiary interaction in the central domain (20,21). DMS also modifies A132, which lies just above loop 2, and A141, which corresponds to a G-A mismatch in the terminal stem.
Consistent with DMS probing, SHAPE measurements show modification within loops 2, 6, and 9, and protection in loops 8 and 10 (Fig. 1, c and d). Loop 2 is more reactive in SHAPE relative to DMS because 1M7 reacts with 2'-OH regardless of sequence where DMS selectively modifies adenine and cytosine nucleobases. As observed in DMS probing, A141 modification in SHAPE indicates distortion/flexibility at the G-A mismatch in the terminal stem. Interestingly, loop 6 is protected from 1M7 modification at positions C67-C70. These bases are modified by DMS indicating that this loop is not involved in tertiary interactions. SHAPE is more sensitive to local nucleotide dynamics than DMS probing (41) and the additional protection from 1M7 modification could be because of base stacking interactions within this loop. 1M7 also reacts with G57 in the apical stem. Although this residue lies within the duplex portion of the apical stem, modification in this region has been previously noted by enzymatic probing (40) suggesting that it may be dynamic or distorted.
The DMS and SHAPE data were jointly used as input for the RNAStructure algorithm (28,29) to produce the secondary structure shown in Fig. 1 a. This model generally agrees with one of the secondary structure models of VAI but differs in the apical stem where we detect an alternative pairing and a shorter loop 6, as previously suggested (40).
Central domain tertiary interactions
Loops 8 and 10 contain complementary ACC and GGU triplet sequences that have the potential to base pair and form a pseudoknot. Replacement of the ACC triplet with UAA in the L8 construct greatly enhances SHAPE modification of residues in loop 8 and remarkably, also induces in extensive modification of loop 10 (Fig. 2). Quantitative analysis reveals reactivities greater than 0.5 in these loops (Fig. 2 d), and several nucleotides in loop 10 exhibit SHAPE reactivities greater than 1.5. Mutation of GGU to CCA in L10 also greatly enhances modification in both loops 8 and 10. These data indicate tertiary interactions between these regions. Sedimentation velocity measurements were used to assess the consequences of disruption of this interaction on the affinity of PKR binding to VAI. Titration of wild-type VAI with PKR induces an increase in the sedimentation coefficient of the RNA, consistent with formation of a 1:1 complex (Fig. 2 c), and global analysis of these data reveal a Kd of 334 nM (Fig. 2 d). The L8 mutation weakens PKR binding affinity by approximately sixfold, indicating a significant disruption. The L10 mutation is less detrimental, causing a 2.5-fold reduction in binding affinity.
Figure 2.
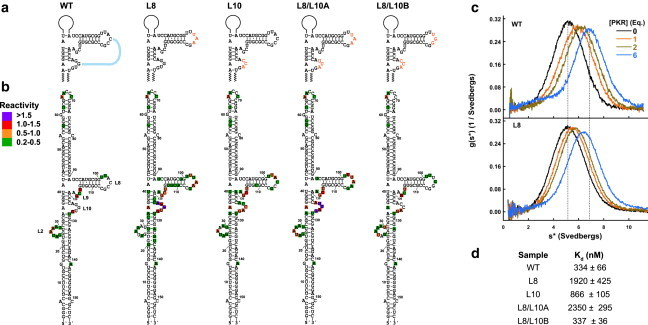
Effect of central domain mutations on VAI structure and PKR binding. (a) Secondary structure of VAI indicating central domain mutations. (b) SHAPE reactivity of central domain mutations. Reactivities were quantitated using SAFA and normalized to the band corresponding to A65. The reactivities are indicated in color scale indicated in the legend. (c) Sedimentation velocity titration of PKR binding to WT (top) and L8 (bottom) VAI. The data were processed using DCDT+ (32) to produce normalized g(s∗) sedimentation velocity distribution functions. The shift in the peak maximum upon addition of 6 eq. of PKR to WT VAI is indicated by the dotted lines. The magnitude of the shift is decreased for L8 because of a reduced binding affinity. (d) Dissociation constants for PKR interaction with central domain mutants determined by global analysis using SEDANAL (33). To see this figure in color, go online.
To test whether reconstitution of base pairing between loops 8 and 10 is sufficient to recover the pseudoknot in VAI, we made two additional sets of mutations: L8/L10A and L8/L10B (Fig. 2 a). L8/L10A contains the same sequence in loop 8 as L8, but with a complementary UUA triplet in loop 10. L8/L10B contains the same mutation in loop 10 as L10, but with a complementary UGG triplet in loop 8, thus swapping bases between loops 8 and 10. The SHAPE reactivity pattern of L8/L10A is very similar to the L8 mutant and the PKR binding affinity remains about as weak as L8. Thus, mutation of the ACC triplet to UAA in loop 8 cannot be rescued by compensatory A-U base pairing. In contrast, the SHAPE modification pattern and PKR binding affinity of L8/L10B are close to wild-type VAI. Therefore, the identity, but not the orientation, of the base pairs between loops 8 and 10 is important for maintenance of the tertiary structure within the central domain.
The loop 8 and loop 10 mutants all show similar modification within loop 2. However, L8 shows modification in several nucleotides at the periphery of this loop (G20 and U31), but with low intensity. Additional nucleotides extending even further from loop 2 are modified but the intensities are below the limit of quantitation (0.2 reactivity) used in this study. L8 also shows modification of nucleotides on the complementary strand near loop 2 (A132, G134, and U135). The lower intensity of modifications in this region may indicate a mixture of open and closed conformations. The L2 mutant was prepared to examine the consequences of expansion of loop 2 from positions 20 to 29 (Fig. S2). As predicted, L2 shows extended modification within loop 2 similar to that observed in L8 but does not exhibit the enhanced modification in loops 8 and 10 found in L8. The Kd for PKR binding to L2 is 322 ± 35 nM, which is similar to wild-type. Therefore, opening of loop 2 does not otherwise alter the tertiary structure or function of VAI.
Because alterations of the triplet in loop 8 produced more dramatic reduction in PKR binding affinity than in loop 10, we prepared additional mutations in this region. The A103U substitution induces fourfold reduction in PKR binding affinity. However, loop 8 remains protected from SHAPE modification with a slight increase in modification of loop 10 relative to the wild-type (Fig. 3). A103U also shows enhanced modification near loop 2 at A132, G134, and U135, similar to the L8 construct (Fig. 2 b). The C105A and C104A/C105A mutations do not significantly alter PKR binding affinity but result in significant modification in loops 8 and 10, indicating disruption of the native tertiary structure. In fact, both of these mutants contain several reactivities within loop 8 and 10 greater than 1.5. Thus, perturbation of tertiary structure in the central domain does not necessarily correlate with changes in PKR interactions.
Figure 3.
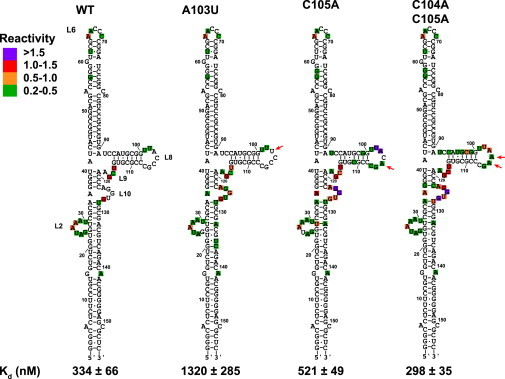
Effects of loop 8 mutations on VAI structure and PKR binding. The locations of the mutations are indicated by red arrows. The SHAPE reactivities were quantitated and displayed as indicated in Fig. 2. The dissociation constants were determined by sedimentation velocity analysis. To see this figure in color, go online.
The pattern of SHAPE modification observed in VAI is preserved in the ΔTS construct that lacks loop 2 as well as the rest of the terminal stem (see Fig. S3). Both loops 8 and 10 are protected in wild-type ΔTS but are extensively modified in ΔTS L8. As observed in full length VAI, the A103U substitution results in only mild enhancement in SHAPE reactivity in loop 10. Thus, the interaction between loops 8 and 10 is preserved in the absence of loop 2 and the terminal stem. Unlike full length VAI, 1M7 modifies nucleotides 84-86 in ΔTS and in the ΔTS mutants. However, these bases lie within a region of regular duplex and the complementary bases are unmodified. Interestingly, the L8 and A103U mutations do not strongly affect PKR binding to the ΔTS construct, with Kd values of 155 ± 24 nM and 229 ± 34 nM, respectively. Therefore, deletion of loop 2 and the terminal stem eliminates the effect of central domain mutations on PKR interactions.
The central domain of VAI contains a highly conserved tetrastem 4. Disruption of this stem abrogates PKR inhibition in vivo and compensatory mutations partially restore function (20). We determined the effects of compensatory base pair substitutions in the central region of this helix on VAI structure. Substitution of the two central G-C base pairs with A-U, individually (S4A and S4B) or together (S4AB), does not cause a significant structural change in the central domain (see Fig. S4). S4B and S4AB both had additional low-level reactivities in nucleotides 123 to 125, and S4B also had a low modification at A103. However, these are subtle differences and do not imply a large structural change as seen with several of the loop 8 and loop 10 mutants. Thus, the strong sequence conservation within stem 4 is not correlated with structural changes. The PKR binding affinity is not substantially altered in either S4A or S4AB but is approximately threefold reduced in S4B, providing another example of a mutation in the central domain that preserves tertiary structure while inhibiting PKR binding.
Structural model of VAI
The residue-level constraints from SHAPE and long-distance constraints derived from SAXS analysis were combined to develop a structural model of VAI. Fig. 4 a shows SAXS scattering curves for VAI in the presence and absence of Mg2+. The curves become flat in the low-q range, and the Guinier plots are linear (inset), indicating that the samples are monodisperse and homogeneous. The radius of gyration (Rg) of VAI obtained from Guinier analysis is independent of concentration from 1 to 4 mg/ml (see Table S1), indicating the absence of self-association and Kratky plots of q2•I vs. q exhibit a clear maximum and decrease at high q, as predicted for a well-ordered macromolecule (for an example, see Fig. S6).
Figure 4.
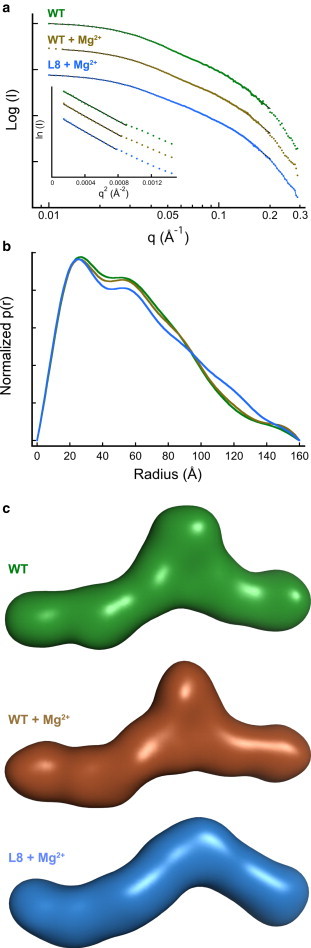
SAXS analysis of VAI. (a) SAXS scattering curves for WT (green), WT + Mg2+ (brown), and L8 + Mg2+ (blue). The solid black lines show the fits of the data to p(r) distributions. All samples are at 2.0 mg/ml. Inset: Guinier analysis giving Rg values of 44.8 ± 1.0 Å (WT), 45.1 ± 0.1 Å (WT + Mg2+), and 45.9 ± 0.7 Å (L8 + Mg2+). The data are vertically offset for clarity. (b) p(r) distance distribution functions. The distributions were produced using GNOM (35) with a maximum q corresponding to 8/Rg. (c) Ab initio models generated by simulated annealing using DAMMIF (36). Twenty-five models were superimposed, averaged, and filtered using DAMAVER (37). The bead models were converted to surface representations using SITUS (38). To see this figure in color, go online.
Addition of divalent ion results in an insignificant change in Rg of VAI from 45.7 ± 1.1 Å (average of three concentrations) to 47.2 ± 0.2 Å, confirming the absence of a substantial structural change induced by Mg2+ (17,19). The p(r) pair distribution function for VAI shows a characteristic maximum at ∼25 Å, corresponding to the approximate diameter of an A-form RNA duplex, a shoulder near 55 Å, and a maximum dimension (Dmax) of 160 Å (Fig. 4 b). The distributions for VAI in the presence and absence of Mg2+ are nearly superimposable. The triplet substitution in L8 induces a slight increase in Rg to 48.4 ± 0.1 Å in the presence of divalent ion and a concomitant enhancement of the contribution of longer distances scattering pairs in the p(r) curve. However, the maximum dimension is not altered. Thus, disruption of the tertiary interaction between loops 8 and 10 measurably perturbs the global structure of VAI.
Ab initio bead models were constructed from the SAXS data using a simulated annealing procedure (DAMMIF) (36). This approach has been shown to provide accurate low-resolution RNA structures (42–45). VAI has a central bulged region flanked by a short arm and a longer, kinked arm (Fig. 4 c). The structure is almost planar, as is commonly observed in smaller RNAs (46). The width of the arms (25 to 30 Å) corresponds to an A-form RNA duplex. Mg2+ does not significantly affect the shape of VAI. Indeed, superposition of structural models derived from eight independent SAXS measurements performed in the presence and absence of divalent ion reveals a remarkably consistent overall shape with a maximum normalized spatial discrepancy (NSD) of only 0.536 (Fig. S5). The longer arm is ∼75 Å, consistent with the length of RNA duplex present in either the terminal or apical stem.
We assigned these features to the domains of VAI based on SAXS analysis of ΔTS VAI. SHAPE demonstrates that the structures of the central domain and apical stems are preserved in ΔTS (Fig. S3). This RNA is well folded and homogeneous (Fig. 5). As expected, both Rg and Dmax of the deletion construct are reduced relative to VAI (Table S1). As in the case of VAI, these structural parameters are not significantly changed in the presence of Mg2+ (data not shown). The ab initio model of ΔTS contains a kinked, helical region and a wide domain but lacks the shorter helix (Fig. 5 c). The bulge present in VAI is also less prominent in ΔTS. Thus, the kinked helix in VAI corresponds to the apical stem and the short helix is the terminal stem. Although the central domain resides in the middle of the VAI, the reduction of the size of the bulge in ΔTS indicates a contribution from the terminal stem. The atomic models described below support these assignments.
Figure 5.
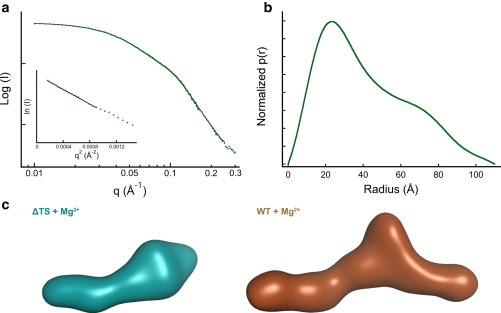
SAXS analysis of ΔTS VAI. (a) SAXS scattering curves of ΔTS VAI. The solid black line shows the fit of the data to a p(r) distribution. Inset: Guinier analysis giving Rg = 32.7 ± 1.1 Å. (c) Ab initio model of ΔTS compared to WT VAI. The bead models were generated by simulated annealing using DAMMIF (36). Twenty-five models were superimposed, averaged, and filtered using DAMAVER (37). The models were converted to surface representations using SITUS (38). To see this figure in color, go online.
Atomic structures of VAI, ΔTS, and L8 were predicted based on the experimental secondary structure constraints from SHAPE analysis using a fragment assembly approach (30). The VAI and ΔTS models incorporate the loop 8-loop 10 interaction. The VAI model aligns reasonably well with the bead model, with NSD = 0.884 (Fig. 6 a). However, the scattering predicted from the atomic model of VAI does not superimpose with the experimental scattering, giving rise to an elevated χ2 (Fig. S7). We believe that this discrepancy is primarily because of the kink in the apical stem of the ab initio bead model that is not present in the atomic model. Deletion of the pseudoknot constraint resulted in VAI models that did not fit as well, with NSD of ∼0.93 to 0.95. The alignment of the atomic model confirms our assignment of the VAI domains. Interestingly, the protuberance in the middle of the structure is mainly associated with loop 2. The central domain itself lies below this protuberance and adopts a compact structure. Stem 7 extends out from the three-helix junction and bends sharply to bring loops 8 and 10 in proximity to base pair. In the VAI model predicted by RNAComposer C104-G125 and C105-G124 are paired, but A103-U126 does not pair (Fig. 6 c). At the three-way junction, coaxial stacking of the conserved stem 4 extends the apical stem. The validity of this model is supported by hydrodynamic calculations. The sedimentation coefficient predicted from the atomic model is s20,w = 5.53 S, which agrees well with measured values of 5.55 to 5.59 S. The atomic model for ΔTS is similar to VAI and fits the SAXS envelope very well, with NSD = 0.837. The central domain occupies the wider portion of the SAXS envelope. However, the orientation of the model with respect to rotation around the helical axis is not well defined. As in the case of VAI itself, there is good agreement between the predicted (s20,w = 4.78 S) and measured (s20,w = 4.59 S) sedimentation coefficients.
Figure 6.
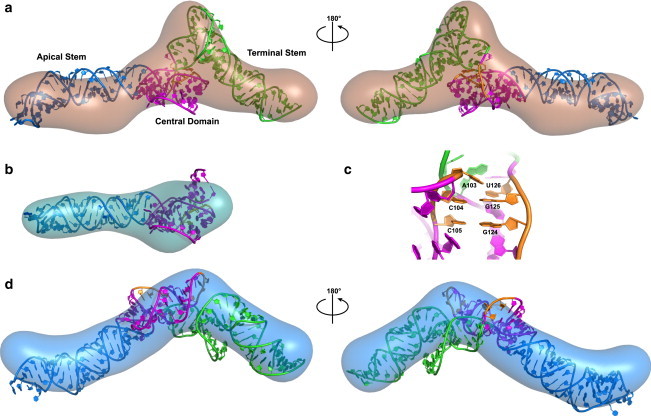
Overlay of atomic models with ab initio SAXS models: (a) VAI, (b) ΔTS, (c) expanded view of the pseudoknot in VAI, and (d) L8 mutant. Atomic models were generated with RNAComposer (30) using the secondary structure constraints derived from SHAPE analysis. Models are color coded by domain: apical stem (blue), central domain (purple), and terminal stem (green). The pseudoknot is colored gold. The atomic models were aligned with the SAXS envelopes using SUPCOMB (31). To see this figure in color, go online.
Finally, we generated an atomic model of the L8 mutant. Based on the structure-probing results, the loop 8–loop 10 interactions were not enforced and the size of loop 2 was increased. Overall, the best fitting model agrees with SAXS results, with an NSD = 0.915. In the resulting model, the orientation of the three-way junction is altered such that stem 7 points toward the protuberance and the terminal stem points downward (Fig. 6 d). Thus, the protuberance is formed from stem 7 rather than the large loop 2, consistent with the reduced size of this feature in the SAXS envelope. Also consistent with the SAXS results, the L8 mutation does not alter the maximum dimension of the atomic model.
Discussion
PKR binds to dsRNAs as short as 10 to 15 bp but activation requires longer (> 30 to 33 bp) stretches of duplex (9,10). PKR also interacts with highly structured single-stranded RNAs that contain bulges, loops, pseudoknots, and single-stranded tails. It is believed that activation by such RNAs containing complex secondary and tertiary structures is mediated by assembly of long double-stranded elements by dimerization, coaxial stacking, mimicry of canonical A-form dsRNA, and inclusion of symmetrical defects (11). However, the structural principles by which RNA inhibitors of PKR mediate high-affinity binding without activating the kinase are not as well understood.
Using a combination of chemical probing, SAXS and structure prediction algorithms, we have developed a three-dimensional structural model for VAI that is consistent with previous biochemical data and provides insights in the mechanism by which this complex RNA interacts with PKR. Chemical probing data indicates that the central domain is stabilized by a pseudoknot between loops 8 and 10. In other adenovirus serotypes, nucleotides within loop 10 are also protected from single-stranded nucleases and phylogenetic and secondary structure analysis indicates that the loop 8-loop 10 interaction is conserved across all adenovirus VAI RNAs (39). The RNA adopts a flat, extended conformation with the apical and terminal stems emanating from a protuberance in the middle that is comprised of the central domain and a large, single-stranded loop. A key feature in the structure is coaxial stacking of the apical stem onto stem 4 in the central domain.
Recently, Conn and coworkers also detected a pseudoknot in the central domain where the tertiary structure is stabilized at low pH, low temperature, and in the presence of Mg2+ (21). In contrast, we find that the SAXS structure (Fig. 4) and reactivity in loops 8 and 10 (data not shown) are not sensitive to divalent ion. A recent SAXS analysis of VAI (22) reported hydrodynamic parameters similar to those reported in this study. The overall shape of VAI is similar to that shown in Fig. 4 but the domains were not assigned. The shape of ΔTS deduced by Dzananovic et al. (22) is quite different from our model, and their alignment of an atomic model for ΔTS within the SAXS envelope results in an orientation of the 5′- and 3′-ends that is incompatible with the structure of full-length VAI.
Our detailed analyses of PKR interactions with VAI (17,19) supports a model whereby VAI functions as an inhibitor by binding a single monomer, thereby preventing dimerization of PKR on the RNA and subsequent activation. VAI contains two stems of ∼20 bp, each of which should be capable of binding a PKR. Indeed, when expressed as isolated domains, the terminal stem (17) and apical stem (17,47) each bind one PKR with weak affinity. In the context of full-length VAI, the portion of the terminal stem proximal to the central domain is occluded by stem 7 (Fig. 6 a), which would impede its ability to bind PKR. In contrast, the apical stem is accessible to interaction with PKR. Footprinting and affinity cleavage studies indicate that the apical stem represents the primary binding site for PKR (14–16). The structure also rationalizes the observation that the apical stem and central domain cooperatively interact to form a high-affinity PKR binding site (17). As expected for a nonsequence-specific interaction, the affinity of PKR binding increases with the length of dsRNA (48–50). In the atomic model, stem 4 in the central can coaxially stack onto the apical stem, resulting in an effectively longer duplex. Consistent with this model, affinity cleavage reveals that stem 4 in the central domain is a secondary PKR binding site (15).
The native tertiary structure in the central domain is not required for high-affinity PKR binding. Some mutations that disrupt the pseudoknot—C105A, C104A/C105A—do not substantially alter PKR binding. This observation is consistent with our proposal that that the high-affinity PKR binding site is formed from the conserved stem 4 and the apical stem. This motif is predicted to be largely unaffected by disruption of the pseudoknot (Fig. 6 d). Alternative functions for the tertiary structure in the central domain may include protecting VAI from cleavage or regulating the interaction of VAI structures or sequences with RNA binding proteins other than PKR (21). In this regard, VAI is reported to serve as a substrate for dicer (51,52) and to function as an activator of oligoadenylate synthetase (53,54).
Several mutations within the central domain significantly inhibit PKR binding without inducing substantial structural rearrangements. The A103U substitution in loop 8 reduces binding without affecting the loop 8-loop 10 interactions. It also inhibits binding in the context of L8 and L8/L10A, where the pseudoknot is disrupted. However, wild-type PKR binding affinity is recovered in L8/L10B where a complementary adenosine is provided in loop 10. Because PKR binding is not sensitive to the presence of the loop 8-loop 10 pseudoknot, it appears unlikely that A103 is directly recognized by PKR. Instead, we propose that mutations at this position result in a conformational change in another region of VAI that impedes PKR binding. A reasonable candidate is the large, single-stranded loop 2. This model is supported by the observations that the SHAPE reactivity of this loop is perturbed by the L8 and A103U mutations and SAXS indicates a substantial rearrangement in loop 2 in the L8 construct. Also, these mutations do not affect PKR binding in the context of the terminal stem deletion construct ΔTS, where loop 2 is not present (see Fig. S3).
Conn and coworkers also found that PKR binding and inhibition by VAI are not dependent on the native tertiary structure within the central domain of VAI (21). Binding and PKR inhibition were not affected upon complete removal of loop 10 and substantial reduction in affinity required larger truncations within the 5′-half of the central domain. However, these studies were performed with a truncated RNA construct lacking the terminal stem (analogous to ΔTS) with a six-nucleotide deletion in the apical stem. Our analysis indicates that the effects of central domain mutations on PKR binding to ΔTS do not reflect the behavior of intact VAI.
Our study reveals that VAI has evolved to bind PKR with maximal affinity while avoiding activation of the kinase. As expected for a nonsequence-specific protein-nucleic acid interaction, PKR binding affinity and stoichiometry increase with the length of duplex RNA (9,48,49). The isolated apical stem-loop binds PKR fairly weakly (Kd = 1.7 μM) (17). Extension of the duplex region to 26 bp by stacking of stem 4 in VAI enhances PKR affinity by sixfold. Further enhancement of PKR binding to VAI by extension of the lattice is precluded because dsRNAs of 30 to 33 bp or longer induce activation (9,10). Given this limitation in binding affinity, accumulation of VAI to micromolar concentrations in the cell late in infection ensures that it effectively competes with activating viral RNAs to prevent kinase activation. The strategy employed by VAI may extend to other RNA inhibitors of PKR (3). The EBER-1 RNA produced by Epstein-Barr virus also inhibits PKR and has complex secondary structure that may fold to yield a similar length PKR binding duplex.
Author Contributions
K. L. F. performed biophysical and biochemical experiments, generated atomic models, and simulated hydrodynamic parameters. C. J. W., K. L. F., and J. L. C. collected SAXS data, and C. J. W. and J. L. C. analyzed it. K. L. F. and J. L. C. wrote the manuscript, and all authors reviewed it.
Acknowledgments
This work was supported by grant number AI-53615 from the NIH to J. L. C. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. We thank Drs. Marc Allaire and Lin Yang from NSLS X9 for beamline access and technical support. We also thank Drs. Mark Peczuh and Jaideep Saha for assistance in the synthesis of 1M7 for SHAPE analyses. The plasmid cassette for structure probing was a generous gift of Dr. Graeme Conn.
Supporting Material
Supporting Citations
References (55–58) appear in the Supporting Material.
References
- 1.Gürtler C., Bowie A.G. Innate immune detection of microbial nucleic acids. Trends Microbiol. 2013;21:413–420. doi: 10.1016/j.tim.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langland J.O., Cameron J.M., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 4.Rothenburg S., Seo E.J., Dittmar K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 2009;16:63–70. doi: 10.1038/nsmb.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elde N.C., Child S.J., Malik H.S. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature. 2009;457:485–489. doi: 10.1038/nature07529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole J.L. Activation of PKR: an open and shut case? Trends Biochem. Sci. 2007;32:57–62. doi: 10.1016/j.tibs.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dar A.C., Dever T.E., Sicheri F. Higher-order substrate recognition of eIF2α by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 8.Dey M., Cao C., Dever T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 9.Manche L., Green S.R., Mathews M.B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemaire P.A., Anderson E., Cole J.L. Mechanism of PKR activation by dsRNA. J. Mol. Biol. 2008;381:351–360. doi: 10.1016/j.jmb.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nallagatla S.R., Toroney R., Bevilacqua P.C. Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr. Opin. Struct. Biol. 2011;21:119–127. doi: 10.1016/j.sbi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mellits K.H., Kostura M., Mathews M.B. Interaction of adenovirus VA RNAl with the protein kinase DAI: nonequivalence of binding and function. Cell. 1990;61:843–852. doi: 10.1016/0092-8674(90)90194-j. [DOI] [PubMed] [Google Scholar]
- 13.Furtado M.R., Subramanian S., Thimmappaya B. Functional dissection of adenovirus VAI RNA. J. Virol. 1989;63:3423–3434. doi: 10.1128/jvi.63.8.3423-3434.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke P.A., Mathews M.B. Interactions between the double-stranded RNA binding motif and RNA: definition of the binding site for the interferon-induced protein kinase DAI (PKR) on adenovirus VA RNA. RNA. 1995;1:7–20. [PMC free article] [PubMed] [Google Scholar]
- 15.Spanggord R.J., Beal P.A. Selective binding by the RNA binding domain of PKR revealed by affinity cleavage. Biochemistry. 2001;40:4272–4280. doi: 10.1021/bi002512w. [DOI] [PubMed] [Google Scholar]
- 16.Spanggord R.J., Vuyisich M., Beal P.A. Identification of binding sites for both dsRBMs of PKR on kinase-activating and kinase-inhibiting RNA ligands. Biochemistry. 2002;41:4511–4520. doi: 10.1021/bi0120594. [DOI] [PubMed] [Google Scholar]
- 17.Launer-Felty K., Cole J.L. Domain interactions in adenovirus VAI RNA mediate high-affinity PKR binding. J. Mol. Biol. 2014;426:1285–1295. doi: 10.1016/j.jmb.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahid A.M., Coventry V.K., Conn G.L. Systematic deletion of the adenovirus-associated RNAI terminal stem reveals a surprisingly active RNA inhibitor of double-stranded RNA-activated protein kinase. J. Biol. Chem. 2008;283:17485–17493. doi: 10.1074/jbc.M802300200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Launer-Felty K., Wong C.J., Cole J.L. Magnesium-dependent interaction of PKR with adenovirus VAI. J. Mol. Biol. 2010;402:638–644. doi: 10.1016/j.jmb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Y., Mathews M.B. Secondary and tertiary structure in the central domain of adenovirus type 2 VA RNA I. RNA. 1996;2:937–951. [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson J.L., Vachon V.K., Conn G.L. Dissection of the adenoviral VA RNAI central domain structure reveals minimum requirements for RNA-mediated inhibition of PKR. J. Biol. Chem. 2014;289:23233–23245. doi: 10.1074/jbc.M114.550046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dzananovic E., Patel T.R., McKenna S.A. Solution conformation of adenovirus virus associated RNA-I and its interaction with PKR. J. Struct. Biol. 2014;185:48–57. doi: 10.1016/j.jsb.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Anderson E., Pierre-Louis W.S., Cole J.L. Heparin activates PKR by inducing dimerization. J. Mol. Biol. 2011;413:973–984. doi: 10.1016/j.jmb.2011.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xayaphoummine A., Bucher T., Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005;33:W605–W610. doi: 10.1093/nar/gki447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkinson K.A., Merino E.J., Weeks K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006;1:1610–1616. doi: 10.1038/nprot.2006.249. [DOI] [PubMed] [Google Scholar]
- 26.Mortimer S.A., Weeks K.M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. 2007;129:4144–4145. doi: 10.1021/ja0704028. [DOI] [PubMed] [Google Scholar]
- 27.Das R., Laederach A., Altman R.B. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA. 2005;11:344–354. doi: 10.1261/rna.7214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuter J.S., Mathews D.H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Low J.T., Weeks K.M. SHAPE-directed RNA secondary structure prediction. Methods. 2010;52:150–158. doi: 10.1016/j.ymeth.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popenda M., Szachniuk M., Adamiak R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012;40:e112. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kozin M.B., Svergun D.I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001;34:33–41. [Google Scholar]
- 32.Philo J.S. Improved methods for fitting sedimentation coefficient distributions derived by time-derivative techniques. Anal. Biochem. 2006;354:238–246. doi: 10.1016/j.ab.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 33.Stafford W.F., Sherwood P.J. Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys. Chem. 2004;108:231–243. doi: 10.1016/j.bpc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 34.García De La Torre J., Huertas M.L., Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semenyuk A.V., Svergun D.I. GNOM—a program package for small-angle scattering data processing. J. Appl. Crystallogr. 1991;24:537–540. [Google Scholar]
- 36.Franke D., Svergun D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volkov V.V., Svergun D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wriggers W. Conventions and workflows for using Situs. Acta Crystallogr. D Biol. Crystallogr. 2012;68:344–351. doi: 10.1107/S0907444911049791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma Y., Mathews M.B. Structure, function, and evolution of adenovirus-associated RNA: a phylogenetic approach. J. Virol. 1996;70:5083–5099. doi: 10.1128/jvi.70.8.5083-5099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wahid A.M., Coventry V.K., Conn G.L. The PKR-binding domain of adenovirus VA RNAI exists as a mixture of two functionally non-equivalent structures. Nucleic Acids Res. 2009;37:5830–5837. doi: 10.1093/nar/gkp595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weeks K.M. Advances in RNA structure analysis by chemical probing. Curr. Opin. Struct. Biol. 2010;20:295–304. doi: 10.1016/j.sbi.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burke J.E., Butcher S.E. Nucleic acid structure characterization by small angle X-ray scattering (SAXS) In: Beaucage Serge L., editor. Current Protocols in Nucleic Acid Chemistry. John Wiley & Sons, Inc.; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rambo R.P., Tainer J.A. Improving small-angle x-ray scattering data for structural analyses of the RNA world. RNA. 2010;16:638–646. doi: 10.1261/rna.1946310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang X., Wang J., Wang Y.X. An unusual topological structure of the HIV-1 Rev response element. Cell. 2013;155:594–605. doi: 10.1016/j.cell.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipfert J., Chu V.B., Doniach S. Low-resolution models for nucleic acids from small-angle x-ray scattering with applications to electrostatic modeling. J. Appl. Crystallogr. 2007;40:s229–s234. [Google Scholar]
- 46.Reiter N.J., Chan C.W., Mondragón A. Emerging structural themes in large RNA molecules. Curr. Opin. Struct. Biol. 2011;21:319–326. doi: 10.1016/j.sbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKenna S.A., Kim I., Puglisi J.D. Uncoupling of RNA binding and PKR kinase activation by viral inhibitor RNAs. J. Mol. Biol. 2006;358:1270–1285. doi: 10.1016/j.jmb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Ucci J.W., Kobayashi Y., Cole J.L. Mechanism of interaction of the double-stranded RNA (dsRNA) binding domain of protein kinase R with short dsRNA sequences. Biochemistry. 2007;46:55–65. doi: 10.1021/bi061531o. [DOI] [PubMed] [Google Scholar]
- 49.Husain B., Mukerji I., Cole J.L. Analysis of high-affinity binding of protein kinase R to double-stranded RNA. Biochemistry. 2012;51:8764–8770. doi: 10.1021/bi301226h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmedt C., Green S.R., Mathews M.B. Functional characterization of the RNA-binding domain and motif of the double-stranded RNA-dependent protein kinase DAI (PKR) J. Mol. Biol. 1995;249:29–44. doi: 10.1006/jmbi.1995.0278. [DOI] [PubMed] [Google Scholar]
- 51.Andersson M.G., Haasnoot P.C.J., Akusjärvi G. Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol. 2005;79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu S., Cullen B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004;78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desai S.Y., Patel R.C., Thimmapaya B. Activation of interferon-inducible 2′-5′ oligoadenylate synthetase by adenoviral VAI RNA. J. Biol. Chem. 1995;270:3454–3461. doi: 10.1074/jbc.270.7.3454. [DOI] [PubMed] [Google Scholar]
- 54.Meng H., Deo S., McKenna S.A. Regulation of the interferon-inducible 2′-5′-oligoadenylate synthetases by adenovirus VA(I) RNA. J. Mol. Biol. 2012;422:635–649. doi: 10.1016/j.jmb.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 55.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Svergun D., Barberato C., Koch M.H.J. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995;28:768–773. [Google Scholar]
- 57.Trabuco L.G., Villa E., Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petoukhov M.V., Franke D., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.