

Abstract
Inherited retinal diseases are uncommon pathologies and one of the most harmful causes of childhood and adult blindness. Leber congenital amaurosis (LCA) is the most severe kind of these diseases accounting for approximately 5% of the whole retinal dystrophies and 20% of the children that study on blind schools. Clinical ophthalmologic findings including severe vision loss, nystagmus and ERG abnormalities should be suspected through the first year of life in this group of patients. Phenotypic variability is found when LCA patients have a full ophthalmologic examination. However, a correct diagnosis may be carried out; the determination of ophthalmologic clues as light sensibility, night blindness, fundus pigmentation, among other, join with electroretinographics findings, optical coherence tomography, and new technologies as molecular gene testing may help to reach to a precise diagnosis. Several retinal clinical features in LCA may suggest a genetic or gene particular defect; thus genetic-molecular tools could directly corroborate the clinical diagnosis. Currently, approximately 20 genes have been associated to LCA. In this review, historical perspective, clinical ophthalmological findings, new molecular-genetics technologies, possible phenotype-genotypes correlations, and gene therapy for some LCA genes are described.
Keywords: Gene therapy, Leber congenital amaurosis, Retinal dystrophies, Childhood blindness
Core tip: Leber congenital amaurosis (LCA) is the most severe retinal dystrophy causing blindness before the age of 1 year. Clinical ophthalmological findings together with electroretinogram study, OCT imaging and retinal molecular-genetic technologies provide a precise diagnosis in these individuals. Gene-specific phenotypic features exist in LCA, and in this way is possible to predict the underlying genetic defect in some patients on the basis of ophthalmological clues. Clinical, molecular-genetics, phenotype-genotype and gene therapy aspects of LCA are described.
INTRODUCTION
Photoreceptor and retinal pigment epithelium dystrophies are inherited retinal disorders that result in severe visual impairment in childhood and adult years. They affect more than two million individuals worldwide[1,2] and are sorted according to the inheritance trait (autosomal recessive, autosomal dominant, X-linked or mitochondrial); however, these methods of subdividing retinal disease are unsatisfactory due to the fact that they do not always discern the etio-pathogenesis of retinal degeneration[3]. Over the last decade, new molecular-genetic technologies have classified the pathogenic nucleotide variants (mutations) in a more exact way; thus, at this moment, approximately 221 genes are known to cause these diseases[4]. Retinal degeneration phenotypes have variable expressivity that can range from mild retinal dysfunction (night blindness, color blindness) to poor vision and total blindness[5].
Leber congenital amaurosis (LCA) is the most severe and earliest form of the inherited retinal diseases that causes childhood blindness[6]. This dystrophy is a genetically heterogeneous recessive disease affecting 1 in 30000[7] to 1 in 81000[8] subjects; although in consanguineous populations or isolated communities may be more frequent[9]. LCA represents almost 5% of all retinal dystrophies and 20% of children with visual impaired in special schools[10]. Although LCA have a broad expression variability, some clinical features may be specific to individual genetic abnormalities, providing a useful means of determining which gene may be responsible, thus narrowing the number of genes that may need to be tested and thereby significantly reducing the involved cost[11]. To date, eighteen genes involved in LCA have been identified, which encodes proteins important in several retinal developmental and physiologic pathways[4]. Recently, therapeutic gene replacement trials for a specific form of human LCA have started, and represent the first example for inherited blindness treatment. In the following sections, we will describe the historical perspective, the clinical characteristics, the involved genes and their functions, the genotype-phenotype correlations, and the current gene therapy treatments for LCA.
HISTORICAL CLINICAL PERSPECTIVE
In 1869, Theodor Leber described a blind child with vision loss, wandering nystagmus, amaurotic pupils, and congenital retinitis pigmentosa (RP). These characteristics were present at or close birth. This German ophthalmologist classified the disease as a new group of pigmentary retinal dystrophy, or tapetoretinal degeneration[12].
In 1957, a reduced or non-recordable ERG was identified as an essential component in the LCA diagnosis[13]. In such report, congenital keratoconus/keratoglobus and cataracts were features associated with this dystrophy, which was named LCA[13,14]. At the same year, Alstrom et al[15] reported that in a study from Sweden 20% of blind children had LCA (heredoretinopathia congenitalitis monohybrid recessive autosomalis), which had predominantly an autosomal recessive inheritance. In 1963, Waardenburg et al[16] described intrafamilial expression variability and association with keratoconus and cataracts.
CLINICAL CHARACTERISTICS
An appropriate clinical evaluation and ophthalmological history, as well as the determination of suggestive retinal clues make a correct diagnosis of the early-onset childhood retinal dystrophies. The utilization of newer diagnostic tools as optical coherence tomography (OCT), join to electrophysiological test (ERG) support the diagnosis. However, at present, genetic-molecular testing is necessary to obtain a definitive diagnosis of retinal dystrophies through pathogenic variants identification[10].
LCA is characterized by at least three findings: severe and early visual impairment, sluggish or near-absent pupillary responses, and severely subnormal or non-detectable ERG[12,13,17]. In LCA patients the absent of fixation or oscillations of the eyes may be seen as early as 6 wk of life. Phenotypic variability on the retina can be identified; thus, fundus appearance ranged from normal or mild retinal involvement to macular coloboma or maculopathy, bone-spicule pigment migration, marbleized fundus, among others. Refractive errors as high hypermetropia, photoaversion (photophobia), nyctalopia and the oculodigital sign, are also commonly observed[17,18].
Visual function
Visual function and visual acuity (VA) are broadly variable, generally range from 20/200 to perception of light or inclusive no perception of it[18]; thus, the prognosis in these patients are complicated. The natural history of visual impairment has been divided into three types: a stable development in most of affected subjects, visual progressive decline, and an appreciable improvement in a minority[8]. In this way, VA, fundus appearance, and systemic findings were assessed in 55 patients with LCA. Twenty-two patients were seen for follow-up examinations (5 years). Seventeen (77%) patients were found to have stable VA, four (18%) had deterioration of VA, and one patient (5%) improvement[19]. Another longitudinal study carried out in 14 patients reported that 50% of them have invariable VA, 29% with VA deterioration, and 21% with visual improvement[20]. In a small series of nine LCA patients was showed VA stability was demonstrated in 55% of patients, while 11% and 33% demonstrated decline and improvement, respectively[21]. In summary, in all studies, 90 patients were examinated: 15%, 75%, and 10% of cases have showed deterioration, stability and improvement, respectively[8]. Patients with mutations in specific LCA genes have demonstrated distinctive VA among the different LCA subtypes. We will return to this point later on.
Ophthalmological features associated to LCA
Refraction defects are variable. Subjects with LCA most commonly are hyperopic[19,22,23]; although, they may also be highly myopic[24]. It has been proposed that an unusual emmetropic development may be caused by severe visual impairment[23]. Some children with LCA children are photophobic[24], whereas others LCA patients can have nyctalopia[25] and these symptoms may be gene-specific as we will soon see[2]. Franceschetti´s oculo digital sign, comprising eye poking, pressing, and rubbing is usual in LCA children; it is not pathognomonic for this retinal dystrophy and it can be found in other diseases[26,27]. Some LCA patients may present keratoconus and cataract, which exacerbate the poor vision of this pathology. Mutations in AIPL1 and CRB1 genes may be identified in these patients[28-31].
No ophthalmological features associated in LCA
Mental retardation was the most significant systemic association in LCA patients. It was reported in up to 52% on this disease[15,32-35]. As in most of these reports cerebral imaging studies were not performed, it seems that this figure is overestimated. In more recent studies where the brain was evaluated, numerous cases were found to have cerebral anomalies as cerebellar involvement (Joubert syndrome), thereby excluding LCA diagnosis[18].
Stereotypic movements and behavior (hand and rubbing movements, hair touching, facial grimaces, among others) are particularly marked in LCA[6].
Olfactory dysfunction has been described in some LCA patients (and carriers) due to mutations in the CEP290 gene[36].
DIFFERENTIAL DIAGNOSIS OF LCA
Some inherited retinal no syndromic diseases share similarities with LCA. Achromatopsia and congenital stationary night blindness may present poor eye fixation and nystagmus, which are similar to LCA, but they show normal retinal fundus. On the other hand, ocular albinism also have nystagmus and poor fixation; however, it has albinotic retinal fundus (absent retinal pigment and choroidal vessels visible) and foveal hypoplasia[8].
Complete achromatopsia or colorblindness is an autosomal recessive pathology that presents marked photophia-photoaversion and blepharospam, decreased VA, and inability to discriminate color. At night, they have a significantly improvement. Incomplete form of achromatopsia or blue-cone monochromacy is a congenital stationary cone dysfunction that presents the same symptoms, although it is less severe. In both subtypes of cone dystrophies ERG recordings have rod photoreceptor normal function, while cone function is absent or subnormal[8,18].
CSNB is a heterogeneous group of nonprogressive retinal pathologies that present reduction of VA, myopia, strabismus, and mainly impaired night vision. ERG report absence of rod function in the complete form of CSNB, while in the incomplete form there is a subnormal rod function response[8,18].
Since the sixth week of age albinism may be confused with LCA; however, several features present in albinism as hypopigmentation of the skin, hair and eyes, and a normal ERG make a differential diagnosis[8,18].
Some syndromic inherited disorders may present similar ocular characteristics to LCA. For this reason it is important search features as mental retardation, deafness, kidney disease (nephronophthisis), skeletal anomalies, cerebral and cerebellar anomalies, among others, which can be associated to retinal photoreceptor degeneration.
Bardet-Biedl syndrome (BBS) is an autosomal recessive disorder with a retinal degeneration that presents a rapid progress, characterized by poor vision and night blindness since the first decade of life and total blindness before the second decade. Nystagmus is extremely infrequent. Different forms of retinal dystrophy have been described, including a cone-rod dystrophy or rod-cone dystrophy, choroidal dystrophy, and so-called global severe retinal dystrophy[37]. Truncal obesity and diabetes mellitus, postaxial polydactyly, hypogonadism in males and genital anomalies in females, renal malformations, developmental delay/behavioral anomalies, ataxia, anosmia, cardiovascular anomalies, among others, can be seen in BBS[38]. Alström syndrome is an autosomal recessive disease similar as BBS that also affects severely vision since early ages; but they do not present polydactyly[39].
The neuronal-ceroid-lipofuscinosis are heterogeneous disorders characterized by intracellular storage of ceroid lipofuscin. Progressive vision loss, retinal degeneration, macular degeneration and optic atrophy cause blindness since the age of two years. Developmental delay/mental retardation, psychomotor degeneration, hypotonia, ataxia, seizures, spatiscity, and death are common characteristics seen in this syndrome. ERG shows early non-recordable responses[40,41].
Senior-Loken syndrome has 2 major features: cystic kidney known as nephronophthisis and early childhood-onset retinal degeneration (retinitis pigmentosa or LCA). Similar features join to cerebellar hypoplasia may be seen in Jourbert syndrome and Meckel syndrome, both diseases known as cerebello-oculo-renal syndromes. Anomalies in cilia proteins (which are essential for the normal development and function of a wide array of specialized tissues as retina, inner ear, kidney, and brain) have been associated in these entities. Thus, mutations in several genes related to centrosomal and ciliary function (as CEP290) can cause phenotypic heterogeneity, ranging from LCA to the syndromes above mentioned[42,43].
Refsum disease, neonatal adrenoleucodystrophy and Zellweger syndrome are peroxisomal disorders that have similar ocular LCA phenotype; however, the systemic cerebral, hepatic, and renal features dominate the phenotype, and the patients almost always suffers an early death[18].
MOLECULAR GENETICS OF LCA
LCA is a highly clinical and genetic heterogeneous disease that is inherited as an autosomal recessive trait in most of the affected. Recent advances in knowledge based in molecular genetics of the retina have allowed the improvement and widen of the clinical diagnosis. In this way, patients with LCA or childhood early retinal dystrophies are sooner identified, and new mutations and LCA genes are being discovered[12,15,44].
Identification of LCA genes
Mutations in least 22 genes (Table 1) have been identified in patients suffering from LCA, nonetheless in 30%-50% of LCA patients no genetic cause is confirmed (Table 1). The advent of new genotyping technologies such as DNA microarrays or next generation sequencing (NGS) offers the opportunity of discovering new LCA loci[4]. Currently, some old methods (linkage analysis and candidate gene approach) or newer methods (homozygosity/autozygosity mapping and NGS) are used for LCA genes identification.
Table 1.
Leber congenital amaurosis genes and phenotype-genotype correlations
| Locus name | Gene symbol | Chromosomal locus | Protein name | Protein function | % of LCA | LCA phenotype | Other retinal dystrophies | Mutations number |
| LCA1 | GUCY2D | 17p13.1 | Retinal guanylyl ciclase 1 | Hydrolysis cGMP | 6-21 | Marked poor vision, photophobia, hyperopia, nystagmus. Normal appearing fundus or mild granular pigmentary changes in periphery. OCT with significant thinning perifoveal | dCRD, dCD | 155 |
| LCA2 | RPE65 | 1p31.3-p31.2 | Retinal pigment epithelium protein 65 | Isomerohydrolase in vitamin A visual cycle | 3-16 | Night blindness, nystagmus, poor vision on the first year, transient vision improvement and later deterioration in their third to fifth decades. OCT: thinner retina in both central and perifoveolar areas | rRP, dRP, RPci | 138 |
| LCA3 | SPATA7 | 14q31.3 | Spermatogenesis-associated protein 7 | Possible vesicular transport | Appro- ximately 3 | Transient photophobia on the first year, but at three years old all patients had night blindness. Visual acuity at the end of the first decade remained stable. After, only hand motion and 20/200 can be seen. Fundus with typical appearance of RP rapidly progressive | rRP | 18 |
| LCA4 | AIPL1 | 17p13.1 | Aryl hydrocarbon interacting protein | Rod PDE chaperone | 5-10 | Keratoconus, cataract, and hyperopia. Variable night blindness or light sensibility. Poor vision. Fundus with bone spicules pigmentation and variable degree of maculopathy. OCT with reduced macular thickness | dCRD, rRP | 52 |
| LCA5 | LCA5 | 6q14 | Lebercilin | Ciliary functions | 1-2 | Severe reduced vision at, or near birth. Nystagmus and high hypermetropia. Visual acuity range between 0.20 to light perception. Extensible peripheral field loss. Fundus examination with widespread atrophy of the retina and RPE. Scattered white dots at RPE. Macula is normal most time, but in few patients may be seen macular coloboma. OCT: macular atrophy, disruption of retinal lamination and presence of hyporeflective well-circumscribed area in the outer nuclear layer, with a hyperreflective border (rossettes). Fundus autofluorescence shows hypofluorescence in the macula | No | 35 |
| LCA6 | RPGRIP1 | 14q11 | RP GTPase regulator-interacting protein 1 | Connecting cilium, disc morphogenesis | 4-6 | Severe loss of vision early in life. Acuity visual worse than 20/200. At the beggining normal retina is seen, then it progress to pigmentary retinopathy. OCT shows remaining photoreceptor in the fovea | rCRD | 82 |
| LCA7 | CRX | 19q13.3 | Cone-rod homeobox | Elongation of photoreceptor outer segment, photoreceptor development, phototransduction | 1%-3% | Severe vision impairment is expected early in life. Nystagmus and high hyperopia. Fundus grayish with clumping or dot-pigment deposits and macular coloboma-like defect. OCT shows macular atrophy without noticeable signal of the junction between inner segments and outer segments | dCRD, dRP dLCA and rLCA | 58 |
| LCA 8 | CRB1 | 1q 31-32.1 | Crumbs homologue | Determining ad maintaining photoreceptor architectura | 9-13 | Nictalopia, nystagmus, keratoconus, corioretinal atrophy and nanophthalmos. Fundus with numular pigment clump, bone spicules and para-arteriolar preservation. Coloboma-like lesions and Coast like lesions | RPpa, rRP | |
| LCA9 | NMNAT1 | 1p36.22 | Nicotinamide nucleotide adenylyltransferase 1 | Rate-limiting enzyme NAD (+) biosynthesis | ---- | Severe form of retinal hereditary degeneration, mainly atrophic macular lesion. Macular pseudocoloboma. Retina´s remainder with pigmentary changes. Nistagmus and severe loss of vision (only light or hand movements perception) | ---- | 44 |
| LCA 10 | CEP290 | 12q21.32 | Centrosomal protein Cep290 | Ciliary function | 20 | Nystagmus, hyperopia, keratoconus and cataract. Photophobia. Light perception or no vision. Atrophic spot (dot-like) in RPE, intraretinal bone spicules in most patients. A striking tapetal reflex (specific intraretinal greyish and white marbled areas). Perifoveal thinning by OCT | Syndromes (Senior-Loken, Joubert, Meckel) | 187 |
| LCA 11 | IMPDH1 | 7q32.1 | Inosine 5´- monophosphate dehydrogensase 1 | De novo synthesis de guanine nucleotide | 8 | Nystagmus with no fixation to light. Retina showing diffuse RPE mottling. No pigmentary deposits | dRP | 18 |
| LCA12 | RD3 | 1q32.3 | Protein RD3 | Transcription and splicing. Suppress retinal membrane guanylate cyclase activity. Role in retinal maturation | < 1 | Night blindness, severe nystagmus. Initial refraction was hypermetropic and changed to myopic in the disease´s course. Severe impaired visual acuity. Attenuated vessels, salt and pepper aspect, and bone spicules are seen on fundus. Macular changes as hammer beaten appearance were note on the third decade of live. OCT reveal disorganization of all retinal layers | ---- | 9 |
| LCA13 | RDH12 | 14q23.3 | Retinol dehydrogensase 12 | Unusual dual specificity for all-trans-retinol and cis-retinols | 4-5 | Poor vision. Night blindness. Chorioretinopathy (reticular or fishnet pattern) with dense hyperpigmentation and bone spicules. There is little or no autofluorescence on the macula. SDOCT: severe macular thinning and loss of the foveal laminar architecture | dRP | 76 |
| LCA 14 | LRAT | 4q31.3 | Lecithinretinol acyltransferase | Esterification essential in vitamin A visual cycle | < 1 | Poor vision, nyctalopia, and visual field constriction since childhood. Peripheral RPE atrophy with little pigment migration into retina. Asteroid hyalosis occurs more frequently than RP (37% vs 3%). Reduced AF signal | rRP, EORD | 13 |
| LCA 15 | TULP1 | 6p21.3 | Tubby-like protein | Protein transport from the photoreceptor inner segment to the outer segment | ---- | Night blindness, nystagmus, moderately to severely limited visual field. Severely disturbed color vision. Fundoscopic findings are variable; pronounced maculopathy in older patients, pigmentary retinopathy in all patients. Pigmentary spicules also are variable affected | rRP | 47 |
| LCA 16 | KCNJ13 | 2q37 | Inwardly-rectifying potassium cannel subfamily J members | Maintaining resting membrane potential | ---- | Poor night vision, nystagmus. Cataract. Fundoscopy reveals considerable levels of pigments at RPE and a different configuration that the one seen on typical RP | dVRD | 6 |
| LCA 17 | GDF6 | 8q22.1 | Grow differentiation factor 6 | Codes for a widely expressed growth factor in the TGF-b pathway specifying the dorsal-ventral retinal axis | ---- | Ocular and skeletal features. Limited vision to detect hand motions | Klippel Feil syndrome, dominant microphthalmia | 15 |
| CABP4 | 11q13.1 | Calcium binding protein 4 | Modulate voltage dependent calcium channel | ---- | Poor vision, nystagmus, photophobia, poor visual acuity | rCSNB, rCRSD | 6 | |
| CNGA3 | 2q11.2 | Cone photoreceptor cGMP-gated cation channel alpha subunit | Important for normal vision and olphatory signaling transduction | ---- | LCA phenotype is not described | Colour blindness total, achromatopsia, cone dystrophy | 82 | |
| ALMS1 | 2p13.1 | ALMS1 | ALMS protein | ---- | LCA phenotype is not described | Alstrom syndrome | 129 | |
| IQCB1 | 3q21.1 | ---- | LCA phenotype is not described | Recessive Senior Loken syndrome | ||||
| MYO7A | 11q13.5 | ---- | LCA phenotype is not described | Usher syndrome, congenital deafness without RP | 304 |
dLCA: Dominant Leber congenital amaurosis; rLCA: Recessive Leber congenital amaurosis; dCRD: Dominant cone-rod dystrophy; dCD: Dominant con dystrophy; rRP: Recessive retinitis pigmentosa; dRP: Dominant retinitis pigmentosa; Rpci: Retinitis pigmentosa with choroidal involvement; PDE: Phosphodiesterase; recessive retinitis pigmentosa with para arteriolar preservation (rRPpa); EORD: Early onset retina dystrophies; AF: Autofluorescence; dVRD: Dominant viteroretinal degeneration; rCSNB: Recessive congenital stationary night blindness; CRSD: Recessive cod-rod synaptic disorder.
Linkage analysis has identified some LCA genes including AIPL1, GUCY2D and RDH12[45-49]. On the basis of retinal expression other group of genes related to LCA have also been found (RPE65, LRAT, CRB1, IMPDH1, CRX, and RPGRIP1)[50-56].
Recently, microarrays technology is utilized to identify mutations that had already been described in other retinal dystrophy reports. This method has the advantage that is relatively cheap and fast; although it has the disadvantage that new mutations will be missed and will not identified. Thus, at least one pathogenic mutation will be found in approximately 60% of the patients when this technology is used. Another method based in microarray technology named homozygosity mapping is important in autosomal recessive diseases where the same ancestral allele is present in affected patients. Thus, this genetic-method is used for detect homozygous mutations (the same mutation in both alleles). CEP 290, LCA5, IQCB1, and SPATA are some genes that have been identified by this microarray technology[57].
New mendelian syndromes, new disease genes discovered and even new mechanism of pathogenicity have been identified[58]. A combination of homo-zygosity mapping and/or exome sequencing have successfully identified mutations in novel LCA genes including KCNJ13[59], ALMS[60], CNGA3[60], MYO7A[60], BBS4[61], NMNAT1[62,63] and PRPH2[64].
Genes, function and genotype-phenotype correlation
LCA exhibits a wide intra-and interfamilial clinical heterogeneity. However, it has been recognized that in some instances the retinal phenotype can suggest the underlying molecular defect. This phenotype-genotype correlation may help to determine rapidly a responsible gene, thus decreasing both the number of genes to be analyzed and the cost of molecular analyses. In the next sections we describe some of the most frequently mutated LCA genes.
GUCY2D (LCA1, OMIM #204000): GUCY2D gene encodes a transmembrane protein termed guanylyl cyclase 1 which is specifically expressed in the retina and localized in the outer segment of the photoreceptors[65]. This enzyme is involved in the resynthesis of cGMP, a key step in the phototransduction recovery process. GUCY2D mutations accounts for 6%-21% (Table 1) of recessive LCA and up to 40% of dominant cone dystrophies[4,66]. Most GUCY2D mutations produce truncation of the protein and total loss of its function[67].
Patients with GUCY2D mutations had markedly poor vision early in life without an obvious subjective degree of progression and their visual acuity can range from 20/200 to light perception[68,69]. Patients have poor responses to visual stimuli, photophobia, preference for dim lights, hyperopia, nystagmus, and do not report night blindness[70]. Retinal fundus generally remains without abnormal findings throughout life. Patients can develop peripheral mild pigmentary changes, optic disc pallor and vascular attenuation[69]. Non-recordable ERGs are typical while OCT imaging shows a significant retinal thinning in the perifoveal area[69].
RPE65 (LCA2, OMIM #204100): RPE65 encodes a retinal pigment epithelium-specific 65-Kd protein[65], which forms a complex with LRAT to act as the isomerohydrolase in the process of visual pigment regeneration. RPE65 is highly expressed in the RPE cells[71]. High RPE65 concentrations have demonstrated by immunocytochemistry and immunoblotting in the central area of the retina. LCA2 patients show an evident early cone loss, although residual cone preserve structure and function that may be present for many years in humans, supporting the existence of alternative pathways for cone survival[18]. RPE65 mutations account for 6%-16% of LCA cases and for approximately 2% of recessive RP cases[72]. Mutations in the RPE65 gene are associated to severe visual loss; shortly after birth or in the first few years of life, affected child is noted to be less visually responsive than normal child[72].
In LCA2 patients, night blindness is a common characteristic and for that reason they prefer being in well-illuminated environments. Patients typically have nystagmus and poor vision since the first year of life. Improvement of visual function over the first few years of life can occur and vision may be relatively fair through teenage years, with later deterioration as patients reach their third to fifth decades[2,11]. Myopia and cataract are frequently associated[69]. The fundus appears normal at birth, but several abnormalities can progressively develop. Most patients present optic disc pallor, vascular attenuation and pigmentary retinal changes. Bull’s eye maculopathy can be present at early stages while geographic atrophic lesions, diffuse hypopigmentation (dot-like changes) and pigment clumping can be observed at late stages (Figure 1). SD-OCT show 1-3 retinal layer with progressive disorganized lamellar structures. Patients with RPE65 have significantly thinner retinas in both the central and perifoveal areas[69].
Figure 1.
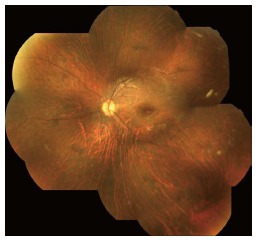
Patient´s fundus with RPE65 mutations.
AIPL1 (LCA4, OMIM #604393): AIPL1 gene encodes the aryl hydrocarbon receptor protein-like 1, which is involved in nuclear transport or chaperone activity for rod phosphodiesterase (PDE)[4]. This protein is expressed early during human development in the central and peripheral retina, coinciding with rod and cone development; in contrast, AIPL1 expression is restricted to rod photoreceptors in the adult retina[73]. Mutations in AIPL1 resulting in blinding diseases can be classified into three categories: Class I and class II changes (missense and stop mutations) results in LCA (5%-10% of recessive LCA cases), while class III mutations (small in-frame deletions) originate dominant forms of cone dystrophy (CORD5) or juvenile retinitis pigmentosa (RP13)[18,74].
The phenotype of LCA patients due to AIPL1 mutations is relatively severe and is characterized by maculopathy and pigmentary retinopathy since young ages[31,75]. Nystagmus is observed in all patients starting at birth or at early infancy[75]. Keratoconus (26%-30%) and cataract (26%-60%) are commonly associated ocular findings[31,76] while hyperopia is the most common refraction error[2,76]. In addition, light gazing, night blindness, and photophobia can also be present[2,76]. Patients show markedly decreased visual acuities since early ages, ranging from 20/600 to no light perception associated with severe visual fields loss, and extinguished ERGs responses[76]. Fundus examination can vary from normal fundus to a salt-and-pepper retinal dystrophy and no apparent macular involvement in young patients; in contrast, older patients can exhibit typical features of retinitis pigmentosa as mild retinal vessel attenuation, bone spicule pigmentation, nummular pigmentation, and pale optic disc in combination with macular anomalies that can vary from mild foveal atrophy to macular coloboma. Autofluorescence (AF) imaging in AIPL1 young patients demonstrates mild generalized reduction in AF at the posterior pole and a relatively preserved macular AF, which demonstrate some degree of structurally intact photoreceptors and retinal pigment epithelial cells[77]. OCT imaging indicates a reduction in macular thickness in all patients (with salt-and-pepper dystrophy or RP-like lesions) with retinal lamellar structures partially retained, displaying 3 retinal layers with preservation of the outer nuclear layer and photoreceptor inner/outer segment juncture[76].
CRB1 (LCA8, OMIM #613835): The CRB1 gene encodes a transmembrane protein named crumbs homolog 1 that is expressed in brain and retina and plays a central role in determining and maintaining the apico-basal cell polarity and adherens junction in embryonic epithelia[18,66]. CRB1-associated LCA is suggested to be caused by a developmental defect in the retina, since LCA patients with mutations in CRB1 have a thickened retina and lack of distinctive layering, resembling an immature retina[78]. Mutations in this gene cause multiple retinal phenotypes including LCA, retinitis pigmentosa, early onset rod-cone dystrophy, and cone-rod dystrophy[4]. The frequency of LCA8 varies considerably between populations in different geographic regions and ranges from 17% in Spain to 0% in India[79,80].
Nystagmus is a common finding and visual acuities vary widely; thus the majority of patients retain walk-around vision in their first decade, during which median visual acuity is approximately 20/200. Later, visual acuity deteriorates with advancing age[29]. However, some reports have described VA as good as 20/40[2]. Approximately half of patients also have high hypermetropia[2,81]. Night blindness is present in the majority of affected; however, decrease of central vision and photophobia may present in some cases[81]. Keratoconus, occurring in 14%-70% of cases, contributes to visual deficiency in patients with LCA8[80]. Patients carrying CRB1 mutations have shown to be predisposed to chorioretinal atrophy[82] and nanophthalmos[83]. The fundus retinal hallmark of LCA8 is nummular pigment clumps (Figure 2), which are found in more than half of the patients. Although these feature are typical of CRB1 mutations, it is not pathognomonic and has also been associated with mutations in other retinal dystrophy genes as NR2E3[82], NRL[84] or TULP1[85]. Bujakowska et al[81] reported two patterns of fundus pigmentation in their cohort: a typical bone spicule-shaped pigment migration within the peripheral retinal, and clumped pigmentary changes of nummular appearance at the level of the RPE[81]. Preservation of the para-arteriolar retinal pigment epithelium, retinal telangiectasia with exudation (Coat’s-like vasculopathy), and macular lesions as coloboma-like lesions in macular or cystoids edema may also be seen[81,84]. SD-OCTs in patients with CRB1 mutations show increased retinal thickness and loss of the outer limiting membrane[84,85].
Figure 2.
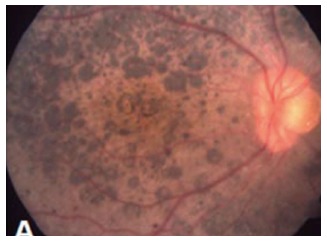
Patient´s fundus with CRB1 mutations.
CEP290 (LCA 10, OMIM #611755): CEP290 gene encodes the centrosomal protein 290 which is a centrosomal protein with a probable ciliary function and expressed at high levels in the photoreceptor connecting cilium. This protein putatively interacts with the protein retinitis pigmentosa GTPase regulator and also with nephrocystin-S, which is mutated in nephronophthisis type 5[66]. CEP290 mutations account for 6%-22% of non-syndromic LCA, depending on the population studied[18].
Although there is some inter-intrafamilial variability, patients carrying CEP290 mutations exhibit a relatively homogeneous distinctive phenotype[86]. Most of the patients present roving eye movements or nystagmus with sluggishly papillary reactions since early childhood. Eye poking, enophthalmos, hyperopia, keratoconus and juvenile cataract are frequently observed[86]. Photophobia is present later in life, and night blindness has sometimes been described. Most patients have severe visual deficiency and visual acuity is either light perception or no vision at all from birth. In some patients visual capability may be equal to or more than hand motion and most of them are able to record progressive deterioration of visual acuity in succeeding years[86]. Fundoscopic findings are variable, from no changes to small (dot-like), well defined, atrophic spot at the level of the RPE to more pronounced RPE atrophy with intraretinal bone spicule-like pigmentation and a preserved macular region. Pasadhika et al[69] described LCA 10 patients with various degrees of macular changes, from a blunted foveal reflex to bull’s eye maculopathy and optic disc drusen. In several patients, a striking tapetal reflex consisting of intraretinal greyish white marbled areas, more noticeable in the younger patients, is observed[86-88]. The small atrophic spots at the RPE layer and tapetal reflex-like changes seem to be specific of CEP290-associated LCA, since they have not been described in other forms of LCA. A distinct yellow scleral rim and pseudopapillary edema are also suggestive of this specific type of LCA[86]. Retinal SD-OCT analysis of patients with CEP290 mutations shows segmentation into only three layers. ONL was notably preserved at the central macular area, but was thinner from the perifoveal area to the periphery. Such preservation tended to decline with an increase in the patient’s age. Photoreceptor inner/outer segment was poorly defined in the central macula, but was invisible in the periphery. Moreover, cyst-like macular lesions can be identified in up to 43% of LCA 10 patients[69].
RDH12 (LCA 13, OMIM #612712): Retinol dehydrogenase 12 (RDH12) gene encodes a RDH12 protein that is a photoreceptor-specific enzyme involved in all-trans-and cis-retinol transformation[66]. RDH12 is expressed in the mouse and human photoreceptor inner segments and ONL[18]. Most RDH12 mutations result in reduced expression and activity of the retinal dehydrogenase 12 enzyme, which in turns disrupts the cycle of synthesis of the visual pigment chromophore, 11-cis-retinal, from 11-trans-retinal[66]. RDH12 mutations account for 4%-5% of recessive LCA, but may also cause other phenotypes as progressive rod-cone dystrophy, macular atrophy, and early-onset RP[4].
LCA 12 patients exhibit a homogeneous clinical picture characterized by poor visual function in early life, a transition period with visual improvement, and a progressive period of decline in visual function due to rod and cone degeneration[89]. At young ages (4-6 years), patients have visual acuities in the range of 40/100 to 50/100, and visual fields are relatively well preserved; however, an age-related progressive loss of visual acuity is evident, with values from 10/100 to light perception on people at ages above 20 years. In younger subjects, residual rod and cone ERGs responses are recordable, with preferential preservation of cone function documented by use of multifocal ERG recordings; however, both rod and cone ERG are undetectable in patients older than 20 years[90]. Night blindness is the predominant symptom of this type of LCA[2]. This subjective symptom together with visual field constriction may not be frequently reported at time of presentation, but may be commonly recognized later in the disease progression[91]. Most LCA 12 patients have no photophobia while mild posterior subcapsular cataract and mild hyperopia can occur[90]. A marked pigmentary retinopathy, ranging from mild RPE atrophy and mid peripheral hyperpigmentation to severe chorioretinopahty (reticular or fishnet pattern) with dense hyperpigmentation has been associated to this form of the disease[90]. Bone spiculae are almost always present (Figure 3)[90]. There is little or no autofluorescence at macula in severe disease. SD-OCT imaging demonstrates severe macular thinning as well as excavation and loss of the foveal laminar architecture[91].
Figure 3.

Patient´s fundus with RDH12 mutations.
Gene therapy in LCA
Recent advances in the knowledge of the genes and the pathophysiology associated with mutations in those genes, has opened a new era of mechanism-based molecular therapeutics in ophthalmology. Immune privilege, small organ size, easy access and compartmentalization, as well as contralateral control, make the eye a perfect organ for gene therapy treatment[92,93]. Thus, to date, several independent phase I/II clinical trials for inherited retinal dystrophies including LCA2 (RPE65), choroideremia (CHM), Usher syndrome 1b (MYO7A), Stargardt disease (ABCA4), Leber hereditary optic neuropathy (ND4), and autosomal recessive retinitis pigmentosa (MERTK) are being studied[93].
Human Therapy: RPE65: Recently, three independent clinical trials of gene replacement therapy for LCA (specifically by RPE65 gene) are carrying out to evaluate safety and efficacy[94-96]. Clinical assessment of these LCA2 patients analyzed ERG results, pupillary light reflex, nystagmus, and fundus abnormalities. Patients were treated in the most affected eye with a unique retinal injection of adenovirus vector (AAV2) carrying RPE65 gene. A safety assessment has not showed the presence of serious adverse event in all trials. The reports of short-term follow-up of these trials have demonstrated an improvement in selected measures of vision, including best-corrected visual acuity, kinetic visual field, nystagmus testing, pupilarry light reflex, microperimetry, dark-adapted perimetry, and dark-adapted full-field sensitivity testing[97]. Soon, the third clinical trial phase of RPE65 replacement in these patients, will start.
Animal models in other LCA genes: Preclinical promise: In several genetic-molecular studies, GUCY2D is the most common LCA gene affected. A normal retinal fundus evaluation and relatively preserved rod-cone architecture make this group of LCA patients aspirants for gene therapy treatment. GUCYKO mouse containing recombinant AAV-GUCY2D gene has restored and preserved cone function during long time[98,99]. A recently study carried out in non-human primate demonstrated a persistent function in both rod and cone cells, mainly in foveal, perifoveal and parafoveal photoreceptors. These results suggested that GUCYD2 gene therapy replacement could be a good treatment for LCA patients[100].
Other LCA genes considered for gene therapy are AIPL1 and CEP290. Recently, a study developed in hypomorphic and null function mouse model shown long time rescue of rod-cone photoreceptors. Thus, these results showed that CEP290 gene therapy could be applied to patients with partial or complete loss of gene function, mainly when intervention occurs early in life[101]. Another recent study, using self-complementary viral vector has achieved functional vision rescue for this rapid retinal dystrophy[102]. CEP290 mutations may maintain cone cell structure in macular region, mainly in the fovea; in this way, this group of patients are a perfect target for a new gene therapy. Recently, Burnight et al[103] built a viral vector containing the CEP290 gene and they demonstrated in cells culture that some mutated cells developed cilia after this therapy. Thus, this gene therapy rescued the ciliogenesis anomaly, and could be effective affected subjects[103].
CONCLUSION
Current technology, such as gene testing, OCT or autofluorescence imaging studies together with the knowledge of the ocular-phenotype features of distinct LCA diseases has increased the individual diagnosis of this retinal dystrophy. This will lead to better prognosis and treatment options for LCA patients. Currently, gene therapy for RPE65-LCA2 patients is a fact, and new similar emerging therapies will be soon available. Thus, the knowledge of genotype-phenotype is necessary for a better patient management.
Footnotes
P- Reviewer: Parmeggiani F S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 29, 2014
First decision: September 16, 2014
Article in press: November 19, 2014
References
- 1.Sundaram V, Moore AT, Ali RR, Bainbridge JW. Retinal dystrophies and gene therapy. Eur J Pediatr. 2012;171:757–765. doi: 10.1007/s00431-011-1615-2. [DOI] [PubMed] [Google Scholar]
- 2.Traboulsi EI. The Marshall M. Parks memorial lecture: making sense of early-onset childhood retinal dystrophies--the clinical phenotype of Leber congenital amaurosis. Br J Ophthalmol. 2010;94:1281–1287. doi: 10.1136/bjo.2009.165654. [DOI] [PubMed] [Google Scholar]
- 3.Moradi P, Moore AT. Molecular genetics of infantile-onset retinal dystrophies. Eye (Lond) 2007;21:1344–1351. doi: 10.1038/sj.eye.6702843. [DOI] [PubMed] [Google Scholar]
- 4.Daiger SP, Sullivan LS, Bowne SJ. The University of Texas Health Science Center, Houston, Texas. Available from: http: // www.retnet.org.
- 5.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–375. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Perrault I, Rozet JM, Gerber S, Ghazi I, Leowski C, Ducroq D, Souied E, Dufier JL, Munnich A, Kaplan J. Leber congenital amaurosis. Mol Genet Metab. 1999;68:200–208. doi: 10.1006/mgme.1999.2906. [DOI] [PubMed] [Google Scholar]
- 7.Stone EM. Leber congenital amaurosis-a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007;144:791–811. doi: 10.1016/j.ajo.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 8.Koenekoop RK. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol. 2007;49:379–398. doi: 10.1016/j.survophthal.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Sherwin JC, Hewitt AW, Ruddle JB, Mackey DA. Genetic isolates in ophthalmic diseases. Ophthalmic Genet. 2008;29:149–161. doi: 10.1080/13816810802334341. [DOI] [PubMed] [Google Scholar]
- 10.Koenekoop RK, Lopez I, den Hollander AI, Allikmets R, Cremers FP. Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions. Clin Experiment Ophthalmol. 2007;35:473–485. doi: 10.1111/j.1442-9071.2007.01534.x. [DOI] [PubMed] [Google Scholar]
- 11.Yzer S, Wolpert K, Borman AD, Tsang S. Gene-specific phenotypes and mechanism-based treatments in early-onset retinal dystrophies. Retinal Physician 2012. Available from: http: // www.retinalphysician.com. [Google Scholar]
- 12.Leber T. Uber retinitis pigmentosa und angeborene amaurose. Graefes Arch Clin Exp Ophthalmol. 1869;15:1–25. [Google Scholar]
- 13.FranceschettI A, Dieterle P. [Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia] Confin Neurol. 1954;14:184–186. [PubMed] [Google Scholar]
- 14.Waardenburg PJ. Does agenesis or dysgenesis neuroepithelialis retinae, whether or not related to keratoglobus, exist? Ophthalmologica. 1957;133:454–460. doi: 10.1159/000305543. [DOI] [PubMed] [Google Scholar]
- 15.Alstrom CH, Olson O. Heredo-retinopathia congenitalis monohybrida recessiva autosomalis. Hereditas. 1957;43:1–178. [Google Scholar]
- 16.Waardenburg PJ, Schappert-Kimmijser J. On various recessive biotypes of Leber congenital amaurosis. Acta Ophthalmol. 1963;41:317–320. doi: 10.1111/j.1755-3768.1963.tb02444.x. [DOI] [PubMed] [Google Scholar]
- 17.Chung DC, Traboulsi EI. Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J AAPOS. 2009;13:587–592. doi: 10.1016/j.jaapos.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 18.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Heher KL, Traboulsi EI, Maumenee IH. The natural history of Leber’s congenital amaurosis. Age-related findings in 35 patients. Ophthalmology. 1992;99:241–245. doi: 10.1016/s0161-6420(92)31985-2. [DOI] [PubMed] [Google Scholar]
- 20.Fulton AB, Hansen RM, Mayer DL. Vision in Leber congenital amaurosis. Arch Ophthalmol. 1996;114:698–703. doi: 10.1001/archopht.1996.01100130690009. [DOI] [PubMed] [Google Scholar]
- 21.Brecelj J, Stirn-Kranjc B. ERG and VEP follow-up study in children with Leber’s congenital amaurosis. Eye (Lond) 1999;13(Pt 1):47–54. doi: 10.1038/eye.1999.10. [DOI] [PubMed] [Google Scholar]
- 22.Dagi LR, Leys MJ, Hansen RM, Fulton AB. Hyperopia in complicated Leber’s congenital amaurosis. Arch Ophthalmol. 1990;108:709–712. doi: 10.1001/archopht.1990.01070070095043. [DOI] [PubMed] [Google Scholar]
- 23.Wagner RS, Caputo AR, Nelson LB, Zanoni D. High hyperopia in Leber’s congenital amaurosis. Arch Ophthalmol. 1985;103:1507–1509. doi: 10.1001/archopht.1985.01050100083024. [DOI] [PubMed] [Google Scholar]
- 24.Harris EW. Leber’s congenital amaurosis and RPE65. Int Ophthalmol Clin. 2001;41:73–82. doi: 10.1097/00004397-200101000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Lorenz B, Gyürüs P, Preising M, Bremser D, Gu S, Andrassi M, Gerth C, Gal A. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci. 2000;41:2735–2742. [PubMed] [Google Scholar]
- 26.Fazzi E, Signorini SG, Scelsa B, Bova SM, Lanzi G. Leber’s congenital amaurosis: an update. Eur J Paediatr Neurol. 2003;7:13–22. doi: 10.1016/s1090-3798(02)00135-6. [DOI] [PubMed] [Google Scholar]
- 27.Jan JE, Good WV, Freeman RD, Espezel H. Eye-poking. Dev Med Child Neurol. 1994;36:321–325. doi: 10.1111/j.1469-8749.1994.tb11852.x. [DOI] [PubMed] [Google Scholar]
- 28.Hameed A, Khaliq S, Ismail M, Anwar K, Ebenezer ND, Jordan T, Mehdi SQ, Payne AM, Bhattacharya SS. A novel locus for Leber congenital amaurosis (LCA4) with anterior keratoconus mapping to chromosome 17p13. Invest Ophthalmol Vis Sci. 2000;41:629–633. [PubMed] [Google Scholar]
- 29.Ehrenberg M, Pierce EA, Cox GF, Fulton AB. CRB1: one gene, many phenotypes. Semin Ophthalmol. 2013;28:397–405. doi: 10.3109/08820538.2013.825277. [DOI] [PubMed] [Google Scholar]
- 30.McMahon TT, Kim LS, Fishman GA, Stone EM, Zhao XC, Yee RW, Malicki J. CRB1 gene mutations are associated with keratoconus in patients with leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2009;50:3185–3187. doi: 10.1167/iovs.08-2886. [DOI] [PubMed] [Google Scholar]
- 31.Dharmaraj S, Leroy BP, Sohocki MM, Koenekoop RK, Perrault I, Anwar K, Khaliq S, Devi RS, Birch DG, De Pool E, et al. The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch Ophthalmol. 2004;122:1029–1037. doi: 10.1001/archopht.122.7.1029. [DOI] [PubMed] [Google Scholar]
- 32.Schappert-Kimmijser J, Henkes HE, Bosch J. Amaurosis congenital (Leber) Arch Ophthalmol. 1959;61:211–218. doi: 10.1001/archopht.1959.00940090213003. [DOI] [PubMed] [Google Scholar]
- 33.Dekaban AS. Mental retardation and neurologic involvement in patients with congenital retinal blindness. Dev Med Child Neurol. 1972;14:436–444. doi: 10.1111/j.1469-8749.1972.tb02616.x. [DOI] [PubMed] [Google Scholar]
- 34.Vaizey MJ, Sanders MD, Wybar KC, Wilson J. Neurological abnormalities in congenital amaurosis of Leber. Review of 30 cases. Arch Dis Child. 1977;52:399–402. doi: 10.1136/adc.52.5.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuil J, Meire FM, Delleman JW. Mental retardation in amaurosis congenita of Leber. Neuropediatrics. 1998;29:294–297. doi: 10.1055/s-2007-973580. [DOI] [PubMed] [Google Scholar]
- 36.McEwen DP, Koenekoop RK, Khanna H, Jenkins PM, Lopez I, Swaroop A, Martens JR. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc Natl Acad Sci USA. 2007;104:15917–15922. doi: 10.1073/pnas.0704140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.M'hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of bardet-biedl syndrome. Mol Syndromol. 2014;5:51–56. doi: 10.1159/000357054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437–446. [PMC free article] [PubMed] [Google Scholar]
- 39.Joy T, Cao H, Black G, Malik R, Charlton-Menys V, Hegele RA, Durrington PN. Alstrom syndrome (OMIM 203800): a case report and literature review. Orphanet J Rare Dis. 2007;2:49. doi: 10.1186/1750-1172-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preising MN, Lorenz B. [Genetics of neuronal ceroidlipofuscinoses. Aspects of genetic counseling] Ophthalmologe. 2010;107:612–615. doi: 10.1007/s00347-009-2107-x. [DOI] [PubMed] [Google Scholar]
- 41.Bennett MJ, Rakheja D. The neuronal ceroid-lipofuscinoses. Dev Disabil Res Rev. 2013;17:254–259. doi: 10.1002/ddrr.1118. [DOI] [PubMed] [Google Scholar]
- 42.Ronquillo CC, Bernstein PS, Baehr W. Senior-Løken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 2012;75:88–97. doi: 10.1016/j.visres.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stone EM, Cideciyan AV, Aleman TS, Scheetz TE, Sumaroka A, Ehlinger MA, Schwartz SB, Fishman GA, Traboulsi EI, Lam BL, et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch Ophthalmol. 2011;129:81–87. doi: 10.1001/archophthalmol.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed E, Loewenstein J. Leber congenital amaurosis: disease, genetics and therapy. Semin Ophthalmol. 2010;23:39–43. doi: 10.1080/08820530701745215. [DOI] [PubMed] [Google Scholar]
- 45.Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H, Châtelin S, Souied E, Ghazi I, Leowski C, et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat Genet. 1996;14:461–464. doi: 10.1038/ng1296-461. [DOI] [PubMed] [Google Scholar]
- 46.Sohocki MM, Bowne SJ, Sullivan LS, Blackshaw S, Cepko CL, Payne AM, Bhattacharya SS, Khaliq S, Qasim Mehdi S, Birch DG, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet. 2000;24:79–83. doi: 10.1038/71732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Janecke AR, Thompson DA, Utermann G, Becker C, Hübner CA, Schmid E, McHenry CL, Nair AR, Rüschendorf F, Heckenlively J, et al. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 48.Stockton DW, Lewis RA, Abboud EB, Al-Rajhi A, Jabak M, Anderson KL, Lupski JR. A novel locus for Leber congenital amaurosis on chromosome 14q24. Hum Genet. 1998;103:328–333. doi: 10.1007/s004390050825. [DOI] [PubMed] [Google Scholar]
- 49.Keen TJ, Mohamed MD, McKibbin M, Rashid Y, Jafri H, Maumenee IH, Inglehearn CF. Identification of a locus (LCA9) for Leber’s congenital amaurosis on chromosome 1p36. Eur J Hum Genet. 2003;11:420–423. doi: 10.1038/sj.ejhg.5200981. [DOI] [PubMed] [Google Scholar]
- 50.Zhu M, Zhao S. Candidate gene identification approach: progress and challenges. Int J Biol Sci. 2007;3:420–427. doi: 10.7150/ijbs.3.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, Gal A. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. 2001;28:123–124. doi: 10.1038/88828. [DOI] [PubMed] [Google Scholar]
- 52.Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 53.Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T, Andréasson S, Berson EL. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68:1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lotery AJ, Jacobson SG, Fishman GA, Weleber RG, Fulton AB, Namperumalsamy P, Héon E, Levin AV, Grover S, Rosenow JR, Kopp KK, Sheffield VC, Stone EM. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol. 2001;119:415–420. doi: 10.1001/archopht.119.3.415. [DOI] [PubMed] [Google Scholar]
- 55.Freund CL, Wang QL, Chen S, Muskat BL, Wiles CD, Sheffield VC, Jacobson SG, McInnes RR, Zack DJ, Stone EM. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet. 1998;18:311–312. doi: 10.1038/ng0498-311. [DOI] [PubMed] [Google Scholar]
- 56.Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Littink KW, den Hollander AI, Cremers FP, Collin RW. The power of homozygosity mapping: discovery of new genetic defects in patients with retinal dystrophy. Adv Exp Med Biol. 2012;723:345–351. doi: 10.1007/978-1-4614-0631-0_45. [DOI] [PubMed] [Google Scholar]
- 58.Schnekenberg RP, Németh AH. Next-generation sequencing in childhood disorders. Arch Dis Child. 2014;99:284–290. doi: 10.1136/archdischild-2012-302881. [DOI] [PubMed] [Google Scholar]
- 59.Sergouniotis PI, Davidson AE, Mackay DS, Li Z, Yang X, Plagnol V, Moore AT, Webster AR. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am J Hum Genet. 2011;89:183–190. doi: 10.1016/j.ajhg.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Wang H, Cao M, Li Z, Chen X, Patenia C, Gore A, Abboud EB, Al-Rajhi AA, Lewis RA, et al. Whole-exome sequencing identifies ALMS1, IQCB1, CNGA3, and MYO7A mutations in patients with Leber congenital amaurosis. Hum Mutat. 2011;32:1450–1459. doi: 10.1002/humu.21587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang H, Chen X, Dudinsky L, Patenia C, Chen Y, Li Y, Wei Y, Abboud EB, Al-Rajhi AA, Lewis RA, et al. Exome capture sequencing identifies a novel mutation in BBS4. Mol Vis. 2011;17:3529–3540. [PMC free article] [PubMed] [Google Scholar]
- 62.Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44:1035–1039. doi: 10.1038/ng.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012;44:1040–1045. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X, Wang H, Sun V, Tuan HF, Keser V, Wang K, Ren H, Lopez I, Zaneveld JE, Siddiqui S, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50:674–688. doi: 10.1136/jmedgenet-2013-101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. Ensembl 2014. Nucleic Acids Research. 2014;42:D749–D755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weleber RG, Francis PJ, Trzupek KM, Beattie C. Leber Congenital Amaurosis.Pagon RA, Adam MP, Ardinger HH, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1298/ [PubMed] [Google Scholar]
- 67.Rozet JM, Perrault I, Gerber S, Hanein S, Barbet F, Ducroq D, Souied E, Munnich A, Kaplan J. Complete abolition of the retinal-specific guanylyl cyclase (retGC-1) catalytic ability consistently leads to leber congenital amaurosis (LCA) Invest Ophthalmol Vis Sci. 2001;42:1190–1192. [PubMed] [Google Scholar]
- 68.Milam AH, Barakat MR, Gupta N, Rose L, Aleman TS, Pianta MJ, Cideciyan AV, Sheffield VC, Stone EM, Jacobson SG. Clinicopathologic effects of mutant GUCY2D in Leber congenital amaurosis. Ophthalmology. 2003;110:549–558. doi: 10.1016/S0161-6420(02)01757-8. [DOI] [PubMed] [Google Scholar]
- 69.Pasadhika S, Fishman GA, Stone EM, Lindeman M, Zelkha R, Lopez I, Koenekoop RK, Shahidi M. Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2010;51:2608–2614. doi: 10.1167/iovs.09-3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jacobson SG, Cideciyan AV, Peshenko IV, Sumaroka A, Olshevskaya EV, Cao L, Schwartz SB, Roman AJ, Olivares MB, Sadigh S, et al. Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum Mol Genet. 2013;22:168–183. doi: 10.1093/hmg/dds421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Redmond TM. Focus on Molecules: RPE65, the visual cycle retinol isomerase. Exp Eye Res. 2009;88:846–847. doi: 10.1016/j.exer.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bowne SJ, Humphries MM, Sullivan LS, Kenna PF, Tam LC, Kiang AS, Campbell M, Weinstock GM, Koboldt DC, Ding L, et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;19:1074–1081. doi: 10.1038/ejhg.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van der Spuy J, Kim JH, Yu YS, Szel A, Luthert PJ, Clark BJ, Cheetham ME. The expression of the Leber congenital amaurosis protein AIPL1 coincides with rod and cone photoreceptor development. Invest Ophthalmol Vis Sci. 2003;44:5396–5403. doi: 10.1167/iovs.03-0686. [DOI] [PubMed] [Google Scholar]
- 74.Kolandaivelu S, Ramamurthy V. AIPL1 protein and its indispensable role in cone photoreceptor function and survival. Adv Exp Med Biol. 2014;801:43–48. doi: 10.1007/978-1-4614-3209-8_6. [DOI] [PubMed] [Google Scholar]
- 75.Tan MH, Mackay DS, Cowing J, Tran HV, Smith AJ, Wright GA, Dev-Borman A, Henderson RH, Moradi P, Russell-Eggitt I, et al. Leber congenital amaurosis associated with AIPL1: challenges in ascribing disease causation, clinical findings, and implications for gene therapy. PLoS One. 2012;7:e32330. doi: 10.1371/journal.pone.0032330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Testa F, Surace EM, Rossi S, Marrocco E, Gargiulo A, Di Iorio V, Ziviello C, Nesti A, Fecarotta S, Bacci ML, et al. Evaluation of Italian patients with leber congenital amaurosis due to AIPL1 mutations highlights the potential applicability of gene therapy. Invest Ophthalmol Vis Sci. 2011;52:5618–5624. doi: 10.1167/iovs.10-6543. [DOI] [PubMed] [Google Scholar]
- 77.Pennesi ME, Stover NB, Stone EM, Chiang PW, Weleber RG. Residual electroretinograms in young Leber congenital amaurosis patients with mutations of AIPL1. Invest Ophthalmol Vis Sci. 2011;52:8166–8173. doi: 10.1167/iovs.11-8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jacobson SG, Cideciyan AV, Aleman TS, Pianta MJ, Sumaroka A, Schwartz SB, Smilko EE, Milam AH, Sheffield VC, Stone EM. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet. 2003;12:1073–1078. doi: 10.1093/hmg/ddg117. [DOI] [PubMed] [Google Scholar]
- 79.Vallespin E, Cantalapiedra D, Riveiro-Alvarez R, Wilke R, Aguirre-Lamban J, Avila-Fernandez A, Lopez-Martinez MA, Gimenez A, Trujillo-Tiebas MJ, Ramos C, et al. Mutation screening of 299 Spanish families with retinal dystrophies by Leber congenital amaurosis genotyping microarray. Invest Ophthalmol Vis Sci. 2007;48:5653–5661. doi: 10.1167/iovs.07-0007. [DOI] [PubMed] [Google Scholar]
- 80.Sundaresan P, Vijayalakshmi P, Thompson S, Ko AC, Fingert JH, Stone EM. Mutations that are a common cause of Leber congenital amaurosis in northern America are rare in southern India. Mol Vis. 2009;15:1781–1787. [PMC free article] [PubMed] [Google Scholar]
- 81.Bujakowska K, Audo I, Mohand-Saïd S, Lancelot ME, Antonio A, Germain A, Léveillard T, Letexier M, Saraiva JP, Lonjou C, et al. CRB1 mutations in inherited retinal dystrophies. Hum Mutat. 2012;33:306–315. doi: 10.1002/humu.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McKay GJ, Clarke S, Davis JA, Simpson DA, Silvestri G. Pigmented paravenous chorioretinal atrophy is associated with a mutation within the crumbs homolog 1 (CRB1) gene. Invest Ophthalmol Vis Sci. 2005;46:322–328. doi: 10.1167/iovs.04-0734. [DOI] [PubMed] [Google Scholar]
- 83.Zenteno JC, Buentello-Volante B, Ayala-Ramirez R, Villanueva-Mendoza C. Homozygosity mapping identifies the Crumbs homologue 1 (Crb1) gene as responsible for a recessive syndrome of retinitis pigmentosa and nanophthalmos. Am J Med Genet A. 2011;155A:1001–1006. doi: 10.1002/ajmg.a.33862. [DOI] [PubMed] [Google Scholar]
- 84.Simonelli F, Ziviello C, Testa F, Rossi S, Fazzi E, Bianchi PE, Fossarello M, Signorini S, Bertone C, Galantuomo S, et al. Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci. 2007;48:4284–4290. doi: 10.1167/iovs.07-0068. [DOI] [PubMed] [Google Scholar]
- 85.Henderson RH, Mackay DS, Li Z, Moradi P, Sergouniotis P, Russell-Eggitt I, Thompson DA, Robson AG, Holder GE, Webster AR, et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br J Ophthalmol. 2011;95:811–817. doi: 10.1136/bjo.2010.186882. [DOI] [PubMed] [Google Scholar]
- 86.Yzer S, Hollander AI, Lopez I, Pott JW, de Faber JT, Cremers FP, Koenekoop RK, van den Born LI. Ocular and extra-ocular features of patients with Leber congenital amaurosis and mutations in CEP290. Mol Vis. 2012;18:412–425. [PMC free article] [PubMed] [Google Scholar]
- 87.Littink KW, Pott JW, Collin RW, Kroes HY, Verheij JB, Blokland EA, de Castro Miró M, Hoyng CB, Klaver CC, Koenekoop RK, et al. A novel nonsense mutation in CEP290 induces exon skipping and leads to a relatively mild retinal phenotype. Invest Ophthalmol Vis Sci. 2010;51:3646–3652. doi: 10.1167/iovs.09-5074. [DOI] [PubMed] [Google Scholar]
- 88.Perrault I, Delphin N, Hanein S, Gerber S, Dufier JL, Roche O, Defoort-Dhellemmes S, Dollfus H, Fazzi E, Munnich A, et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- 89.Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H, Hamel C, Dufier JL, Munnich A, Kaplan J, et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am J Hum Genet. 2004;75:639–646. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schuster A, Janecke AR, Wilke R, Schmid E, Thompson DA, Utermann G, Wissinger B, Zrenner E, Gal A. The phenotype of early-onset retinal degeneration in persons with RDH12 mutations. Invest Ophthalmol Vis Sci. 2007;48:1824–1831. doi: 10.1167/iovs.06-0628. [DOI] [PubMed] [Google Scholar]
- 91.Mackay DS, Dev Borman A, Moradi P, Henderson RH, Li Z, Wright GA, Waseem N, Gandra M, Thompson DA, Bhattacharya SS, et al. RDH12 retinopathy: novel mutations and phenotypic description. Mol Vis. 2011;17:2706–2716. [PMC free article] [PubMed] [Google Scholar]
- 92.Liu MM, Tuo J, Chan CC. Republished review: Gene therapy for ocular diseases. Postgrad Med J. 2011;87:487–495. doi: 10.1136/pgmj.2009.174912rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013;21:509–519. doi: 10.1038/mt.2012.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maguire AM, Simonelli F, Pierce EA, Pugh EN, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 96.Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, Banfi S, Surace EM, Sun J, Acerra C, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. 2013;120:1283–1291. doi: 10.1016/j.ophtha.2012.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boye SE, Boye SL, Pang J, Ryals R, Everhart D, Umino Y, Neeley AW, Besharse J, Barlow R, Hauswirth WW. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS One. 2010;5:e11306. doi: 10.1371/journal.pone.0011306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mihelec M, Pearson RA, Robbie SJ, Buch PK, Azam SA, Bainbridge JW, Smith AJ, Ali RR. Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum Gene Ther. 2011;22:1179–1190. doi: 10.1089/hum.2011.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baehr W, Karan S, Maeda T, Luo DG, Li S, Bronson JD, Watt CB, Yau KW, Frederick JM, Palczewski K. The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J Biol Chem. 2007;282:8837–8847. doi: 10.1074/jbc.M610369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tan MH, Smith AJ, Pawlyk B, Xu X, Liu X, Bainbridge JB, Basche M, McIntosh J, Tran HV, Nathwani A, et al. Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum Mol Genet. 2009;18:2099–2114. doi: 10.1093/hmg/ddp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ku CA, Chiodo VA, Boye SL, Goldberg AF, Li T, Hauswirth WW, Ramamurthy V. Gene therapy using self-complementary Y733F capsid mutant AAV2/8 restores vision in a model of early onset Leber congenital amaurosis. Hum Mol Genet. 2011;20:4569–4581. doi: 10.1093/hmg/ddr391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Burnight ER, Wiley LA, Drack AV, Braun TA, Anfinson KR, Kaalberg EE, Halder JA, Affatigato LM, Mullins RF, Stone EM, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014;21:662–672. doi: 10.1038/gt.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]