

Abstract
Capicua (CIC) has been implicated in pathogenesis of spinocerebellar ataxia type 1 and cancer in mammals; however, the in vivo physiological functions of CIC remain largely unknown. Here we show that Cic hypomorphic (Cic-L-/-) mice have impaired bile acid (BA) homeostasis associated with induction of proinflammatory cytokines. We discovered that several drug metabolism and BA transporter genes were down-regulated in Cic-L-/- liver, and that BA was increased in the liver and serum whereas bile was decreased within the gallbladder of Cic-L-/- mice. We also found that levels of proinflammatory cytokine genes were up-regulated in Cic-L-/- liver. Consistent with this finding, levels of hepatic transcriptional regulators, such as hepatic nuclear factor 1 alpha (HNF1α), CCAAT/enhancer-binding protein beta (C/EBPβ), forkhead box protein A2 (FOXA2), and retinoid X receptor alpha (RXRα), were markedly decreased in Cic-L-/- mice. Moreover, induction of tumor necrosis factor alpha (Tnfα) expression and decrease in the levels of FOXA2, C/EBPβ, and RXRα were found in Cic-L-/- liver before BA was accumulated, suggesting that inflammation might be the cause for the cholestasis in Cic-L-/- mice. Our findings indicate that CIC is a critical regulator of BA homeostasis, and that its dysfunction might be associated with chronic liver disease and metabolic disorders.
CIC is an HMG box-containing transcriptional repressor that is evolutionarily conserved from Caenorhabditis elegans to humans1. CIC exists as at least two isoforms, CIC-S and CIC-L, which differ in their amino-terminal regions2. CIC preferentially binds to octameric TGAATGA/GA or TCAATGAA sequences within target promoters and enhancers, and several CIC target genes have been identified in Drosophila and mammals3,4,5. In mammals, CIC has been implicated in cancer progression and pathogenesis of spinocerebellar ataxia type 1 (SCA1). Dozens of mutations in the CIC gene have been identified in the tissues from patients with various types of cancers6,7,8, and the chromosomal translocation generating CIC-DUX4 fusion has been discovered in Ewing-like sarcomas3. Moreover, PEA3 group genes, the best known CIC target genes, are usually overexpressed in various types of cancers, suggesting that CIC deficiency could contribute to tumorigenesis and/or cancer progression through their de-repression9,10. We have previously shown that deficiency of CIC results in alveolarization defects and overexpression of MMP9 in mouse lung tissues11. On the other hand, CIC interacts with ATXN1, the causative protein of SCA1, and its paralog ATXN1-Like (ATXN1L), and this interaction stabilizes these proteins5,11. Haploinsufficiency of CIC alleviates disease severity in SCA1 mice (Atxn1154Q knock-in mice), suggesting that mutant ATXN1 causes SCA1 in part due to enhanced activity of the CIC/ATXN1 complex5.
BA is synthesized from cholesterol in hepatocytes by various enzymes including cholesterol 7α-hydroxylase (CYP7A1), 25-hydroxycholesterol 7α-hydroxylase (CYP7B1), sterol 27-hydroxylase (CYP27A1), and sterol 12α-hydroxylase (CYP8B1), and stored in the gallbladder. After ingestion of food, bile flows into the duodenum, where it contributes to the digestion of lipophilic nutrients. BAs are then absorbed from the terminal ileum and transported back to the liver via the portal vein. This entire process is called enterohepatic circulation of BA. Defects in excretion of hepatic BA cause cholestatic liver damage and chronic liver diseases due to toxicity of BAs and induction of inflammatory response12. Inflammation also can cause cholestasis through dysregulation of hepatobiliary transport system. Proinflammatory signaling cascades lead to repressed expression and activity of several liver-enriched transcription factors and nuclear receptors, which are essential for maintenance of a large number of genes involved in detoxification and hepatobiliary transport of BAs and other toxic compounds13,14, thereby disrupting the enterohepatic circulation of BA.
BA can serve as a ligand for several nuclear receptors. The BA-mediated activation of farnesoid X receptor (FXR, also known as NR1H4) facilitates transcription of small heterodimer partner (Shp, also known as Nr0b2), which in turn suppresses the expression of key genes in BA biosynthesis and Na+-taurocholate cotransporting polypeptide (Ntcp, also known as Slc10a1), a transporter mediating BA uptake from plasma15,16. Therefore, this negative feedback loop maintains BA homeostasis in the liver.
In this study, we show that CIC deficiency impairs BA homeostasis in mice. We also found that levels of several hepatic transcriptional regulators, known to regulate expression of drug metabolism and BA transporter genes, were markedly reduced, whereas proinflammatory cytokine gene expression was increased in liver of Cic-L-/- mice5. Thus, we have identified novel roles of CIC in BA homeostasis and, potentially, inflammatory response.
Results
Serum chemistry for Cic-L-/- mice
To get insight of in vivo physiological roles of CIC, we first checked tissue distribution of CIC and ATXN1L proteins in mice. We found that CIC was expressed in various types of tissues, except for kidney (Fig. 1), and that ATXN1L levels decreased in all tested Cic-L-/- tissues (Fig. 1), suggesting that CIC exists as a complex with ATXN1L in most tissues. Next, we investigated whether Cic-L-/- mice had metabolic abnormalities by measuring the levels of serum metabolites in WT and Cic-L-/- mice at P18, and found that glucose levels were decreased whereas levels of BA and total bilirubin were significantly increased in sera from Cic-L-/- mice compared with WT (Table 1). It is also noteworthy that the serum alanine transaminase (ALT) level, a general parameter indicating liver insult, was increased in Cic-L-/- mice, though not statistically significant (Table 1), suggesting that Cic-L-/- mice might have defects in liver function.
Figure 1. Tissue distribution of CIC and ATXN1L proteins in mice.
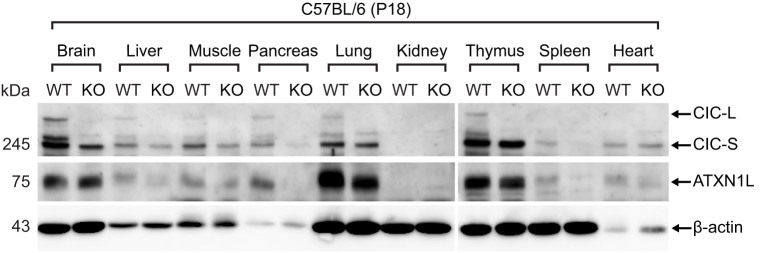
Western blot analysis for tissue distribution of CIC and ATXN1L proteins at P18. Twenty μg of total protein extract was loaded on each lane. KO means Cic-L-/- mouse.
Table 1. Serum chemistry for 18 day-old WT and Cic-L KO mice.
| WT | Cic-L KO | No. of tested animals (WT : KO) | |
|---|---|---|---|
| Glucose (mg/dL)*** | 107.0 ± 8.700 | 53.4 ± 8.235 | 6:5 |
| Insulin (ng/mL) | 0.320 ± 0.026 | 0.285 ± 0.006 | 4:4 |
| Cholesterol (µM) | 5,137.29 ± 701.646 | 5,203.03 ± 436.851 | 4:4 |
| Triglycerides (nmol/µl) | 1.477 ± 0.233 | 0.972 ± 0.017 | 3:3 |
| Bile acid (µM)** | 22.984 ± 3.943 | 49.309 ± 5.058 | 6:6 |
| Alanine transaminase (U/L) | 39.429 ± 61.849 | 61.849 ± 11.205 | 6:6 |
| Total bilirubin (mg/dL)* | 0.312 ± 0.044 | 0.647 ± 0.098 | 6:6 |
| Direct bilirubin (mg/dL) | 0.476 ± 0.128 | 0.745 ± 0.190 | 6:6 |
*p<0.05, **p<0.01, ***p<0.001.
Altered expression of drug metabolism genes in Cic-L-/- liver
Hepatic expression of CIC proteins and significant alterations in levels of several serum metabolites in Cic-L-/- mice led us to investigate roles of CIC in liver given its role in a wide variety of metabolic processes. To examine hepatic gene expression changes caused by deficiency of CIC, we carried out microarray analysis using liver total RNA from WT and Cic-L-/- mice at P18 (four mice per each genotype). A total of 718 differentially expressed genes (DEGs; P < 0.05) between WT and Cic-L-/- mice (405 up-regulated and 313 down-regulated in Cic-L-/- liver) were identified using an integrative statistical method (Fig. 2a and Table S1). To examine cellular pathways represented by the DEGs, we performed KEGG pathway enrichment analysis for the DEGs using DAVID software17. The analysis revealed that the down-regulated genes most significantly represented those involved in drug metabolism (also known as xenobiotic metabolism) (Fig. 2b). Drug metabolism involves a set of enzymatic processes and transporters that modify and remove toxic chemicals from cells in three phases. Phase I is conducted by enzymes mediating oxidation, reduction, or hydroxylation, such as cytochrome P450 enzymes (CYPs), flavin-containing monooxygenases (FMOs), alcohol dehydrogenases (ADHs), and aldehyde dehydrogenases (ALDHs). In phase II, phase I products are conjugated with charged species, such as glutathione, sulfate, and glucuronic acid mediated by glutathione-S-transferases (GSTs), sulfotransferases (SULTs), and UDP-glucuronosyltransferases (UGTs). In phase III, the conjugates can be excreted from cells by a variety of membrane transporters including multidrug resistance protein (MRP) family. Proteins involved in drug metabolism act not only on the pharmaceutical drugs or xenobiotics, but also on endogenous metabolites including BA to maintain their homeostasis. Using qRT-PCR, we confirmed the down-regulation of several phase I and phase II drug metabolism genes in Cic-L-/- liver. Sixteen (84%) out of the 19 tested genes were significantly down-regulated in Cic-L-/- livers obtained from an independent cohort of animals (Fig. 2c). Taken together, these data suggest that drug metabolism is likely the hepatocellular pathway most significantly affected by CIC deficiency.
Figure 2. Down-regulation of drug metabolism genes in Cic-L-/- liver.

(a) Heat map showing up- and down-regulated genes in Cic-L-/- mice (KO) at P18, compared with WT. The numbers in x-axis denote four replicates used in WT and KO. Red and green colors represent increase and decrease in expression levels of the genes, respectively, from the mean expression levels of the eight replicates. Color bar, gradient of log2-difference of the intensities from the median intensities. (b) KEGG pathways significantly represented by the up- and down-regulated genes in Cic-L-/- liver. The significance was represented by -log10(P) where P is the enrichment P-value obtained from the DAVID software. The number of the up- or down-regulated genes involved in each pathway is shown. (c) qRT-PCR analysis for levels of nineteen phase I and phase II drug metabolism genes in livers from 18 day-old WT and Cic-L-/- mice (n = 4~5 per each genotype). *P<0.05, **P<0.01, and ***P<0.001. All error bars show s.e.m.
Impaired BA homeostasis in Cic-L-/- mice
Given that BA levels were significantly increased in sera from Cic-L-/- mice (Table 1), and that some of the drug metabolism genes down-regulated in Cic-L-/- liver, Cyp2b10, Gstm2, and Sult2a1, are known to be involved in detoxification and excretion of BAs18,19, we hypothesized that BA homeostasis might be dysregulated in Cic-L-/- mice. To test this hypothesis, we first checked bile content within gallbladders of 18 day-old WT and Cic-L-/- mice after overnight fasting. Surprisingly, Cic-L-/- mice showed dramatically shrunken gallbladders (Figs. 3a and S1), indicating that they have less bile within the gallbladder in comparison with WT. We next measured the concentration of hepatic and intestinal BAs as well as total BA pool sizes in WT and Cic-L-/- mice. A significant increase in hepatic BA levels was found in Cic-L-/- mice while total BA pool size was comparable between them (Fig. 3b). These data suggest that CIC deficiency impairs the enterohepatic circulation of BA, based on our finding showing accumulation of BAs in serum and liver and dilatory excretion of bile into gallbladder in Cic-L-/- mice.
Figure 3. Impaired BA homeostasis in Cic-L-/- mice.
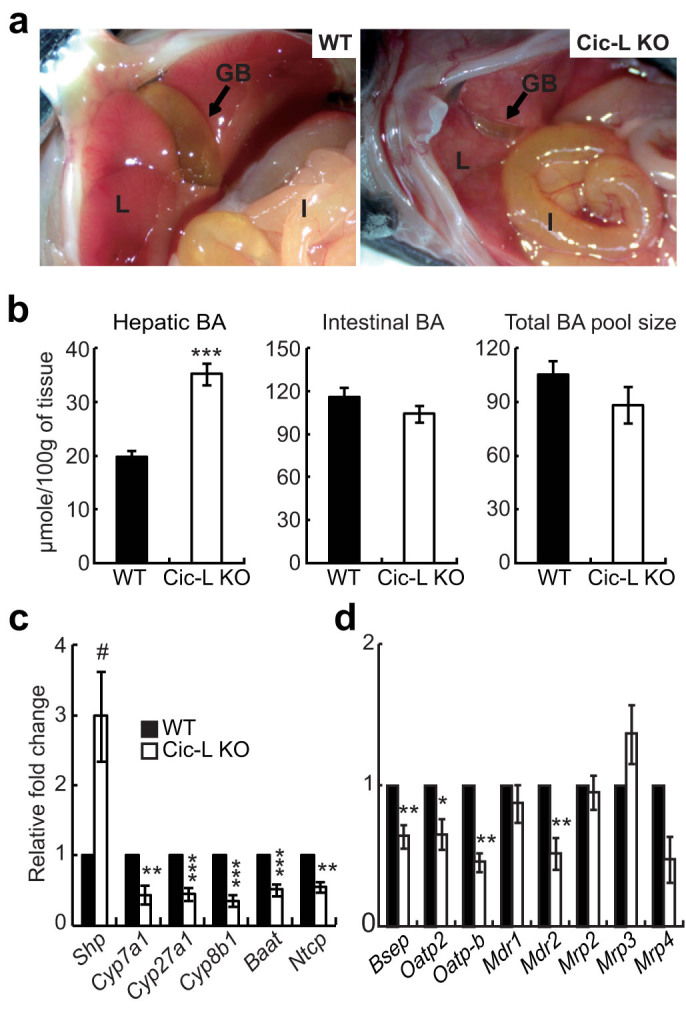
(a) Pictures showing the size of gallbladders in WT and Cic-L-/- mice at P18. GB, L, and I mean gallbladder, liver, and intestine, respectively. Arrows indicate the gallbladder. (b) Hepatic and small intestinal BA levels and total BA pool size in WT and Cic-L-/- mice (n = 4~7 per each genotype) at P18. ***P<0.001. All error bars show s.e.m. (c) qRT-PCR analysis for levels of Shp and key genes involved in BA biosynthesis and transport in livers from 18 day-old WT and Cic-L-/- mice (n = 4~9 per each genotype). #P = 0.054, **P<0.01 and ***P<0.001. All error bars show s.e.m. (d) qRT-PCR analysis for levels of membrane transporter genes in livers from 18 day-old WT and Cic-L-/- mice (n = 3~6 per each genotype). *P<0.05, **P<0.01 and ***P<0.001. All error bars show s.e.m.
Cholestasis can be induced by bile duct malformation; however, histological analysis of the liver showed that hepatic bile duct formation is comparable between WT and Cic-L-/- mice (Fig. S2). We then investigated expression levels of genes implicated in BA biosynthesis and transport. qRT-PCR analysis revealed that the levels of Cyp7a1, Cyp27a1, Cyp8b1, and bile acid CoA:amino acid N-acyltransferase (Baat) were significantly decreased in livers from Cic-L-/- mice compared with WT (Fig. 3c), consistent with the microarray results (Table S1). To test the possibility that down-regulation of those genes in Cic-L-/- mice could be due to elevated levels of hepatic BAs, we checked the level of Shp, a target gene of FXR and a negative regulator of key genes in BA biosynthesis. The level of Shp mRNA was increased approximately three-fold in livers of Cic-L-/- mice compared with WT (Fig. 3c). Consistent with this finding, levels of Ntcp, whose expression can be suppressed by SHP, were also down-regulated in Cic-L-/- mice (Fig. 3c). To determine the cause of impaired enterohepatic circulation of BA in Cic-L-/- mice, we checked hepatic mRNA levels of several basolateral and canalicular membrane transporters mediating bile flow by qRT-PCR. Among eight different membrane transporter genes, levels of bile salt export pump (Bsep, also known as abcb11) and organic anion transporting polypeptide 2 (Oatp2), which are responsible for excretion of bile salts into the canaliculi and hepatic uptake of BAs from plasma, respectively, were significantly decreased in Cic-L-/- mice (Fig. 3d). Moreover, levels of Mdr2 (also known as Abcb4) and Oatp-b (also known as Slco2b1) were significantly decreased in Cic-L-/- mice compared with WT (Fig. 3d). Together with the down-regulation of Ntcp in Cic-L-/- liver (Fig. 3c), these data explain the accumulation of BAs in serum and liver in Cic-L-/- mice at the molecular level.
Histological analysis of liver tissue did not show apparent hepatic damages in Cic-L-/- mice (data not shown). However, it is known that cholestatic liver injury can be induced in the mice with altered BA homeostasis on a diet containing cholic acid16 and that intraperitoneal injection of lithocholic acid can induce necrosis of hepatic cells in the mice with disrupted drug metabolism20,21. To test whether deficiency of CIC contributes to induction of cholestatic liver damages, we placed the nursing females on a diet containing 1% (wt/wt) cholic acid so that the nursing pups will ingest an excess amount of cholic acid through breast milk during postnatal growth. Under this condition, about 30% of Cic-L-/- mice showed hepatic injury at P18, as evidenced by areas of liver necrosis, which was not found in WT littermates (Fig. S3a). To further ascertain hepatic injury in Cic mutants fed with cholic acids, we also determined the serum ALT level and found that it was significantly increased in Cic-L-/- mice compared with WT (Fig. S3b). Together, these data suggest that CIC could play a role in protection of the liver from BA toxicity as well as in BA homeostasis.
Decrease in protein levels of liver-enriched transcription factors and induction of proinflammatory cytokine gene expression in Cic-L-/- mice
Many previous studies have shown that expression of genes involved in drug metabolism is coordinately regulated by several nuclear receptors and liver-enriched transcription factors22,23,24,25,26,27,28,29,30. It is also known that cholestasis is closely associated with induction of proinflammatory modulators and decreased expression and activity of hepatic transcriptional regulators13,14. In order to explain for the suppressed expression of several drug metabolism genes in liver and impaired BA homeostasis in Cic deficient mice, we examined hepatic levels of several liver-enriched transcription factors and nuclear receptors including FOXA2, HNF1α, C/EBPβ, RXRα, HNF4α, and FOXO1 by western blot analysis using liver total extracts from WT and Cic-L-/- mice at P18. We found that levels of FOXA2, HNF1α, C/EBPβ, and RXRα, but not HNF4α and FOXO1, were significantly down-regulated in livers of Cic-L-/- mice (Fig. 4a), suggesting that down-regulation of drug metabolism genes might result from decreased levels of a subset of liver-enriched transcription factors and nuclear receptors in Cic-L-/- mice. Interestingly, their mRNA levels were not significantly altered in Cic-L-/- mice (Fig. 4b), indicating that levels of these transcription factors are down-regulated at the post-transcriptional level. To validate that the down-regulation of several genes involved in drug metabolism and BA transport in Cic-L-/- liver was, at least in part, due to the decreased levels of hepatic transcriptional regulators, we examined promoter occupancy of FOXA2 and RXRα for their target genes whose levels were significantly down-regulated in Cic-L-/- liver (Figs. 2C and 3D). We carried out chromatin immunoprecipitation (ChIP) using either anti-FOXA2 or anti-RXRα antibody followed by qPCR for promoter regions that contain binding motifs for either FOXA2 or RXRα. ChIP-qPCR analyses revealed that less amount of FOXA2 and RXRα is associated with promoter regions of all the tested genes (Oatp2, Fmo3, and Ugt2b34 as FOXA2 targets16, and Cyp2b10 and Sult2a1 as RXRα targets31,32) in liver of Cic-L-/- mice compared with WT (Fig. S4), suggesting that reduction in the levels of several hepatic transcriptional regulators leads to decrease in their promoter occupancy of target genes, thereby contributing to the down-regulation of a subset of genes mediating drug metabolism and BA transport in Cic-L-/- mice.
Figure 4. Changes in levels of several hepatic transcription factors and proinflammatory cytokine genes in liver of Cic-L-/- mice at P18.
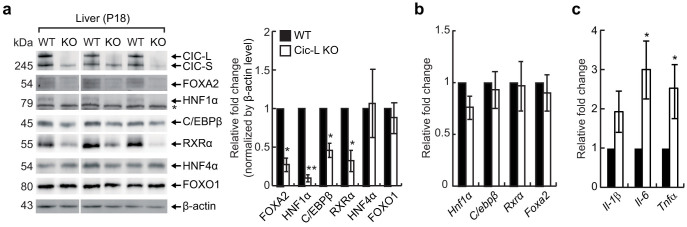
(a) Western blot image showing the decrease in FOXA2, HNF1α, C/EBP1β, and RXRα protein levels in Cic-L-/- liver at P18. The right panel is a bar graph for quantitative analysis on levels of hepatic transcription factors based on Western blot image. The asterisk indicates non-specific bands. *P<0.05 and **P<0.01. All error bars show s.e.m. (b) qRT-PCR analysis of Hnf1α, C/ebpβ, Rxrα, and Foxa2 expression levels using liver total RNAs prepared from 18 day-old WT and Cic-L-/- mice (n = 5 per each genotype). All error bars show s.e.m. (c) qRT-PCR analysis of Il-1β, Il-6, and Tnfα expression levels using liver total RNAs prepared from 18 day-old WT and Cic-L-/- mice (n = 7 per each genotype). *P<0.05. All error bars show s.e.m.
We also checked the levels of proinflammatory cytokine genes to determine induction of inflammatory response in the liver of Cic-L-/- mice. The levels of interleukin-6 (Il-6) and Tnfα were significantly increased in Cic-L-/- mice compared with WT (Fig. 4c), suggesting that the hepatic cholestasis in Cic-L-/- mice might be associated with increased inflammatory response. Altogether, these data indicate that CIC deficiency leads to disruption in BA homeostasis accompanied with dramatic decrease in protein levels of several hepatic transcriptional regulators and induction of proinflammatory cytokine expression.
Decrease in the levels of a subset of hepatic transcription factors and drug metabolism genes and induction of Tnfα expression precede hepatic BA accumulation in Cic-L-/- mice
Because inflammation can cause hepatic cholestasis and vice versa, it was not clear whether the impaired BA homeostasis is the cause or an effect of the reduced protein levels of FOXA2, HNF1α, C/EBPβ, and RXRα, and up-regulation of proinflammatory cytokine genes in Cic-L-/- liver. To address this, we assessed levels of FOXA2, HNF1α, C/EBPβ, and RXRα at P10, when levels of hepatic BA and Shp mRNA are comparable between WT and Cic-L-/- mice (Figs. 5a and 5b). We found that protein levels of FOXA2, C/EBPβ, and RXRα were still decreased in Cic-L-/- liver compared with WT (Fig. 5c), although the fold decrease in FOXA2 and C/EBPβ protein levels was less dramatic at P10 than at P18 (Figs. 4a and 5c). These data suggest that hepatic cholestasis is not the primary cause for the reduction in levels of FOXA2, C/EBPβ, and RXRα in Cic-L-/- mice, but that it could be facilitated as hepatic cholestasis develops. HNF1α proteins were not detectable in livers at P10 under our experimental conditions. Among eighteen drug metabolism genes with decreased levels in Cic-L-/- liver at P18 (Fig. 2c), seven were significantly down-regulated and half of them showed a trend toward down-regulation in livers from 10 day-old Cic-L-/- mice (Fig. 5d), suggesting that decreased levels of a subset of hepatic transcription factors are probably associated with the down-regulation of drug metabolism genes in Cic-L-/- mice. We also checked hepatic levels of the membrane transporter genes in Cic-L-/- mice at P10. Among five genes, only Oatp2 and Mdr2 levels were significantly down-regulated in livers of Cic-L-/- mice at P10 (Fig. 5e), suggesting that decreased levels of Ntcp, Bsep, and Oatp-b might critically contribute to the disruption in BA homeostasis in Cic-L-/- mice at P18. Finally, we examined the expression of proinflammatory cytokine genes in liver of Cic-L-/- mice at P10. We found that Tnfα levels were significantly increased in Cic-L-/- mice compared with WT (Fig. 5f), suggesting that inflammatory response was already induced in Cic-L-/- liver at P10, but amplified during cholestasis, based on more dramatic and significant fold increase in the levels of proinflammatory cytokine genes in Cic-L-/- liver at P18 than at P10 (Figs. 4c and 5f). Taken together, these data suggest that CIC deficiency induces proinflammatory signaling cascades in liver, thereby perturbing BA homeostasis.
Figure 5. Decrease in protein levels of hepatic transcription factors and induction of Tnfα expression in Cic-L-/- liver at P10.

(a) Measurement of hepatic BA concentration in 10 day-old WT and Cic-L-/- mice (n = 5 per each genotype). All error bars show s.e.m. (b) qRT-PCR analysis for levels of Shp mRNA in livers from 10 day-old WT and Cic-L-/- mice (n = 3 per each genotype). Error bar shows s.e.m. (c) Western blot image showing the decrease in FOXA2, C/EBPβ, and RXRα protein levels in Cic-L-/- liver at P10. The right panel is a bar graph for quantitative analysis on levels of FOXA2, C/EBPβ, and RXRα based on Western blot image. *P<0.05 and #P = 0.0587. All error bars show s.e.m. (d) qRT-PCR analysis for levels of eighteen drug metabolism genes in livers from 10 day-old WT and Cic-L-/- mice (n = 3 per each genotype). *P<0.05 and ***P<0.001. All error bars show s.e.m. (e) qRT-PCR analysis for levels of membrane transporter genes in livers from 10 day-old WT and Cic-L-/- mice (n = 3 per each genotype). *P<0.05 and **P<0.01. All error bars show s.e.m. (f) qRT-PCR analysis for levels of Il-1β, Il-6, and Tnfα using liver total RNAs prepared from 10 day-old WT and Cic-L-/- mice (n = 4 per each genotype). *P<0.05. All error bars show s.e.m.
Discussion
In this study, we show that CIC plays a critical role in regulating hepatic inflammatory response and BA homeostasis. Based on our findings, we propose a model to explain how CIC disrupts such processes in mice (Fig. S5). Loss of CIC triggers hepatic proinflammatory signaling cascades that decrease the protein levels of liver-enriched transcription factors and nuclear receptors including FOXA2, C/EBPβ, and RXRα, thereby causing down-regulation of several drug metabolism genes in Cic-L-/- mice before BA accumulates in the liver. Through these processes, prolonged induction of proinflammatory cytokines could eventually perturb BA homeostasis in Cic-L-/- mice. Under cholestatic conditions, proinflammatory signaling is further induced and amplified, and activity of several liver-enriched transcription factors and nuclear receptors including FOXA2, HNF1α, C/EBPβ, and RXRα might become weaker due to a greater reduction in their protein levels and nuclear exclusion14. These changes, in turn, further alter drug metabolism pathways and BA homeostasis in Cic-L-/- mice through more significant down-regulation of larger number of genes that are cooperatively regulated by liver-enriched transcription factors and nuclear receptors.
It is known that inflammation-induced cholestasis is mediated by cholestatic effects of bacterial endotoxins such as lipopolysaccharides (LPS) and/or LPS-induced proinflammatory cytokines33. The liver is a major site of the removal of bacteria and endotoxins from systemic circulation. Kupffer cells, the hepatic resident macrophages, take up bacteria and endotoxins, and are stimulated to release proinflammatory cytokines such as IL-1β, IL-6, and TNFα. The produced cytokines, in turn, trigger intracellular inflammatory signals in hepatocytes, leading to impairment in bile formation, and accumulation of BAs and toxins in liver and serum through down-regulation of hepatic transport systems involved in BA uptake and excretion, as well as down-regulation of phase I and phase II detoxification systems13,34. In this respect, our data suggest that Cic-L-/- mice might have inflammation-induced cholestasis. However, it remains unknown how CIC deficiency induces a subset of proinflammatory cytokines, such as TNFα, in liver. Given that intrahepatic proinflammatory cytokine synthesis is mediated mainly by Kupffer cells33, CIC might be involved in regulation of proinflammatory cytokine gene expression in macrophages. It has been reported that TNFα gene contains ETS transcription factor binding sites in the promoter region35. In addition, Ras-induced proinflammatory cytokine production is known to be dependent on MAPK/ETV4 pathway36. Since CIC represses Etv4 expression in alveolar macrophage cells11, deficiency of CIC might contribute to up-regulation of proinflammatory cytokine genes, such as Tnfα, through de-repression of Etv4 in hepatic macrophages. It is noteworthy that up-regulation of Etv4 levels was found in liver of Cic-L-/- mice at P18 using qRT-PCR (Fig. S6), although it did not appear in microarray results (Table S1). Taken together, investigation on functions of CIC in macrophages and other immune cells involved in proinflammatory cytokine production would be important in uncovering the molecular mechanism for regulation of inflammatory response by CIC.
Our analysis of serum metabolites also revealed hypoglycemia in Cic-L-/- mice. According to the KEGG pathway enrichment analysis, genes involved in metabolism for several different kinds of amino acids were significantly enriched among the down-regulated genes in Cic-L-/- liver (Fig. 2b). Indeed, we confirmed significant down-regulation of nine amino acid metabolism genes (out of fifteen) in Cic-L-/- liver by qRT-PCR using liver total RNA prepared from an independent cohort of animals (Fig. S7a). We also found that levels of glucose 6-phosphatase (G6pc) and phosphoenolpyruvate carboxykinase (Pepck), key genes mediating gluconeogenesis, were not significantly altered in Cic-L-/- mice compared with WT (Fig. S7a), consistent with the microarray results (Table S1). Given that amino acids can be used as carbon precursors in gluconeogenesis37 (Fig. S7b), and that down-regulation of genes encoding enzymes of amino acid catabolism are associated with hypoglycemia in mice38, the decreased levels of amino acid metabolism genes might contribute to hypoglycemia in Cic-L-/- mice. Further studies on the molecular mechanism underlying hypoglycemia in Cic-L-/- mice will demonstrate critical roles of CIC in various metabolic processes.
Since the long isoform of CIC (CIC-L) harbors a unique amino-terminal region of about 900 amino acids long, the two isoforms of CIC, CIC-L and CIC-S, might have differential roles. It was recently reported that CIC-S, but not CIC-L, predominantly exists in the cytoplasm and regulates cytosolic citrate metabolism through interacting with ATP citrate lyase (ACLY)39, suggesting differential functions of the CIC isoforms in mammals. Given that CIC-L expression is completely abolished whereas a small amount of CIC-S is still expressed in Cic-L-/- mice5 (Fig. 1), it is unclear whether the defects observed in Cic-L-/- mice reflect a CIC-L specific requirement, a partial loss of CIC-S, or a reduction of total CIC activity. To better understand molecular pathways for the CIC-mediated biological processes including regulation of BA homeostasis and glucose metabolism, comparative studies on molecular characteristics and in vivo functions of CIC isoforms will be required.
Although the functions of CIC have mostly been investigated in neurodegeneration and cancer, this study provides new insight into the critical role of CIC in normal liver function and metabolism. Our findings not only suggest that CIC is important in regulation of BA homeostasis and hepatic inflammatory response, but also raise the possibility that CIC dysfunction might be associated with pathogenesis of chronic liver disease and metabolic disorders in humans.
Methods
Ethics Statement
All experimental procedures and animal care were performed in accordance with the guidelines approved by the ethics committee in Pohang University of Science and Technology (POSTECH).
Mice
Generation of Cic-L-/- mice was described previously5. Mice were backcrossed for more than 10 generations onto C57BL/6J background. Animals were maintained in a pathogen-free animal facility under standard 12 hr light/12 hr dark cycle. Mice were fed standard rodent chow and water ad libitum. For the experiments to study the effect of cholic acid diet on liver functions in Cic-L-/- mice, the normal chow was replaced with 1% cholic acid-supplemented chow (Purina, Bethlehem, PA) a day before delivery. In all the experiments except for microarray analyses, mice were fasted overnight, then anesthetized with isoflurane before collecting tissue and serum samples. All procedures were approved by the POSTECH Institutional Animal Care and Use Committee (IACUC).
Serum Chemistry
Blood samples were collected from 18 day-old mice by heart puncture using 26G 1/2 needles. After blood clot formation, samples were centrifuged for 30 min at 3,000 rpm at 4°C. Supernatants were collected to new tubes and used for subsequent experiments. Serum glucose levels were measured by a glucometer. Insulin levels were determined by mouse insulin EIA (ALPCO, Salem, NH). Levels of serum triglyceride, cholesterol, alanine transaminase (ALT), and bilirubin were measured using Triglyceride Quantifcation Colormetic/Fluorometric Kit (Biovision, Milpitas, CA), Total Cholesterol Assay Kit (Cell biolabs, San Diego, CA), EnzyChrom™ Alanine Transaminase Assay Kit (Bioassay system, Hayward, CA), and QuantiChrom™ Bilirubin Assay Kit (Bioassay systems, Hayward, CA), respectively, according to the manufacturers' instructions.
BA Measurement
The concentration of BAs was measured using Diazyme Total Bile Acids Assay kit (Diazyme Laboratories, Poway, CA) according to the manufacturer's instructions. For measurement of hepatic BA levels, median lobes of liver were used. For determination of total BA pool size, the whole liver tissue, intact gallbladder, and small intestine were collected in one tube and subjected to BA measurement.
RNA Extraction and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Liver tissues were dissected, flash-frozen in liquid nitrogen, and stored at −80°C until RNA preparation. Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA), then two to three μg of total RNA was subjected to cDNA synthesis using GoScript™ Reverse Transcription System (Promega, Madison, WI) according to the manufacturer's instructions. Primers used for qRT-PCR are listed in Table S2. SYBR Green real-time PCR master mix (TOYOBO, Osaka, Japan) was used for PCR reactions.
Microarray Analysis
Gene expression profiles for four replicates of liver tissues obtained from independent 18 day-old WT and Cic-L-/- mice were generated using Illumina MouseRef-8 v2.0 Expression BeadChip (Illumina, San Diego, CA) including 26,821 probes corresponding to 18,016 annotated genes. The probes were annotated using Manifest file from Illumina. Total RNA (500 ng) was isolated from liver tissues using RNeasy Mini Kit (Qiagen, GmbH, Germany). RNA integrity number (RIN) was measured using an Agilent 2100 Bioanalyzer (RIN > 9 in all samples). According to the Illumina protocols, RNA was reverse transcribed and amplified, in vitro transcription was carried out to prepare cRNA, and the cRNAs were hybridized to the array and then labeled with Cy3-streptavidin. The fluorescent signal on the array was measured with a BeadStation 500 System.
Identification of Differentially Expressed Genes (DEGs)
The log2-intensities were first normalized using the quantile normalization method40. The probes were annotated using Lumi 1.8.3. The expressed genes were identified as previously described41. To identify DEGs, an integrative statistical hypothesis testing previously reported was applied to the normalized log2-intensities41. Briefly, both t-test and log2-median-ratio test were performed to calculate T values and log2-median-ratios for all genes. Empirical distributions of the null hypothesis (a gene is not differentially expressed) were estimated by performing random permutations of the samples and then applying the Gaussian kernel density estimation method42 to T values and log2-median-ratios resulted from the random permutations. The adjusted P-values of each gene for the individual tests were then computed by the two-tailed test using their empirical null distributions and combined using Stouffer's method to calculate the overall adjusted P-value43. Finally, the DEGs were selected as the ones with the overall P<0.05 and absolute log2-fold-changes > 0.43 (1.35-fold), the mean of 2.5th and 97.5th percentiles of the null distribution of log2-fold-changes.
Generation of Rabbit Polyclonal Antibodies for CIC and ATXN1L
Bacterially expressed C-terminal region (214 amino acids) of CIC44 was used to generate rabbit polyclonal anti-CIC serum. For generation of polyclonal anti-ATXN1L antibody, rabbits were immunized with the peptide of 39 amino acids corresponding to the C-terminus of mouse ATXN1L protein (SFQRFSMQGEEARAAMLRPSFIPQEVKLSIEGRSNAGK).
Western Blot Analysis
Liver tissues were homogenized and sonicated in RIPA buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% Sodium deoxycholate, 0.1% SDS, 1% Triton X-100) containing protease and phosphatase inhibitors and incubated on ice for 15 min. After centrifugation at 13,300 rpm for 15 min, the supernatant was taken and 20~25 μg of that was used for sample preparation. Protein samples were loaded on tris-glycine SDS-PAGE gel and transferred to Trans-Blot nitrocellulose membrane (Bio-Rad, Hercules, CA). Antibodies used for western blot analyses are as follows: anti-CIC (1:1000 dilution, rabbit), anti-ATXN1L (1:500 dilution, rabbit), anti-RXRα (sc-553, 1:2000 dilution, rabbit), anti-HNF1α (sc-10791, 1:500 dilution, rabbit), anti-FOXA2 (sc-6554, 1:500 dilution, goat), anti-C/EBPβ (sc-150, 1:1000 dilution, rabbit), anti-HNF4α (sc-8987, 1:500 dilution, rabbit), anti-FOXO1 (sc-11350, 1:500 dilution, rabbit), and anti-β-actin (sc-47778, 1:5000 dilution, mouse). All antibodies, except for anti-CIC and anti-ATXN1L antibodies, were purchased from Santa Cruz Biotechnology (Dallas, TX). The band intensities were quantified using ImageJ.
Liver Histology
Liver tissues from 18 day-old WT and Cic-L-/- mice were fixed in 10% formalin and paraffin-embedded before sectioning. The tissues were cut into 6 μm sections, and sections were deparaffinized and dehydrated by using xylene, 100% ethanol, and 95% ethanol sequentially. They were washed by distilled water and stained with hematoxylin and eosin (Sigma, St. Louis, MO).
Chromatin Immunoprecipitation (ChIP)
The liver tissue was chopped with a razor more than 20 times and crosslinked in 1% formaldehyde for 12 min with constant shaking then rinsed with cold PBS twice. After centrifugation for 5 min, the pellet was resuspended in Buffer A (100 mM Tris pH 9.4, 1X protease inhibitor cocktail (Roche, Basel, Switzerland), Buffer 1 (10 mM HEPES, 10 mM EDTA, 0.5 mM EGTA, 0.25% Triton-X, 1X protease inhibitor cocktail), and Buffer 2 (10 mM HEPES, 0.2 M NaCl, 1 mM EDTA, 0.5 mM EGTA, 1X protease inhibitor cocktail), successively. Ten mg of liver lysate was resuspended in 100 μl of nuclei lysis buffer (5 mM Tris pH 8.1, 10 mM EDTA, 1% SDS, 1X protease inhibitor cocktail) and sonicated. After microcentrifugation, the supernatant was pre-cleared with protein G agarose (Millipore, Billerica, MA), diluted 1:3 with dilution buffer (16.7 mM Tris pH 8.1, 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS, 1.1% Triton-X, protease inhibitor), and divided into aliquots. One and a half μg of anti-FOXA2 antibody (sc-6554, Santa Cruz Biotechnology, Dallas, TX) or anti-RXRα antibody (sc-553, Santa Cruz Biotechnology, Dallas, TX) was added to each aliquot of chromatin and incubated on a rotating platform overnight at 4°C. The chromatin and antibody mixtures were further incubated with protein G agarose for 2–4 h at 4°C. After extensive washing, bound chromatins were eluted twice by elution buffer (0.5% SDS and 0.1 M NaHCO3) and reverse-crosslinked with 200 mM NaCl for at least 4 h at 65°C. Proteins were digested by proteinase K and DNA was purified by AccuPrep Gel Purification Kit (Bioneer, Daejeon, Korea). For quantification of the relative enrichment of FOXA2 and RXRα targets in the immunoprecipitated DNA fragments, quantitative real-time PCR was performed. The FOXA2 and RXRα binding sites in promoter regions of the tested genes were predicted using PROMO website45, and the primers were designed to amplify promoter regions harboring one of the putative binding motifs for either FOXA2 or RXRα. The primer sequences are listed in Table S2.
Statistical Analysis
For statistical analysis, all experiments were performed more than three times independently. Statistical analyses were carried out using the Student t-test (two-tailed, two-sample unequal variance). All data were expressed as the mean ± standard error. A p-value less than 0.05 was considered as significant.
Author Contributions
E.K. and Y.L. designed the experiments. E.K., S.P., N.C., J.L., S.K. and H.-Y.J. performed the experiments. Data analyses and interpretation were conducted by E.K., S.P., K.-T.K., H.K., J.D.F., H.Y.Z., D.H. and Y.L. E.K. and Y.L. wrote the paper. J.Y., J.D.F., H.Y.Z. and Y.L. edited the paper.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korean Ministry of Science, ICT and Future Planning (NRF-2012R1A1A1005631 to Y.L. and NRF-2013R1A1A2063089 to N.C.), the POSTECH Basic Science Research Institute (4.0010906.01), and T.J. Park Science Fellowship of POSCO TJ Park Foundation. E.K., S.P., J.Y. and N.C. were supported by the BK21 Plus Program (Program of Bio-Molecular Function, POSTECH). HHMI and NINDS (NS27699) support H.Y.Z. D.H. was supported by the grant from the Institute for Basic Science (CA1308).
Accession number The GEO accession number of microarray data is GSE60375.
References
- Jiménez G., Guichet A., Ephrussi A. & Casanova J. Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev. 14, 224–231 (2000). [PMC free article] [PubMed] [Google Scholar]
- Jiménez G., Shvartsman S. Y. & Paroush Z. The Capicua repressor--a general sensor of RTK signaling in development and disease. J. Cell. Sci. 125, 1383–1391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura-Saito M. et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 15, 2125–2137 (2006). [DOI] [PubMed] [Google Scholar]
- Ajuria L. et al. Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development 138, 915–924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer J. D. et al. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science 334, 690–693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöblom T. et al. The consensus coding sequences of human breast and colorectal cancers. Science 314, 268–274 (2006). [DOI] [PubMed] [Google Scholar]
- Kan Z. et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466, 869–873 (2010). [DOI] [PubMed] [Google Scholar]
- Alentorn A., Sanson M. & Idbaih A. Oligodendrogliomas: new insights from the genetics and perspectives. Curr Opin Oncol 24, 687–693 (2012). [DOI] [PubMed] [Google Scholar]
- Kurpios N. A., Sabolic N. A., Shepherd T. G., Fidalgo G. M. & Hassell J. A. Function of PEA3 Ets transcription factors in mammary gland development and oncogenesis. J Mammary Gland Biol Neoplasia 8, 177–190 (2003). [DOI] [PubMed] [Google Scholar]
- Dissanayake K. et al. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicúa. Biochem. J. 433, 515–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. et al. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev. Cell 21, 746–757 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K., Jaeschke H. & Copple B. L. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 178, 175–186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosters A. & Karpen S. J. The role of inflammation in cholestasis: clinical and basic aspects. Semin. Liver Dis. 30, 186–194 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollner G. et al. Role of nuclear receptors and hepatocyte-enriched transcription factors for Ntcp repression in biliary obstruction in mouse liver. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G798–805 (2005). [DOI] [PubMed] [Google Scholar]
- Sinal C. J. et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744 (2000). [DOI] [PubMed] [Google Scholar]
- Bochkis I. M. et al. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat. Med. 14, 828–836 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G. Jr et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4, P3 (2003). [PubMed] [Google Scholar]
- Moschetta A. Welcoming Foxa2 in the bile acid entourage. Cell Metab. 8, 276–278 (2008). [DOI] [PubMed] [Google Scholar]
- Wagner M. et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 42, 420–430 (2005). [DOI] [PubMed] [Google Scholar]
- Zhang J., Huang W., Qatanani M., Evans R. M. & Moore D. D. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J. Biol. Chem. 279, 49517–49522 (2004). [DOI] [PubMed] [Google Scholar]
- Uppal H. et al. Combined loss of orphan receptors PXR and CAR heightens sensitivity to toxic bile acids in mice. Hepatology 41, 168–176 (2005). [DOI] [PubMed] [Google Scholar]
- Song C. S. et al. Tissue-specific and androgen-repressible regulation of the rat dehydroepiandrosterone sulfotransferase gene promoter. J. Biol. Chem. 273, 21856–21866 (1998). [DOI] [PubMed] [Google Scholar]
- Elizondo G., Corchero J., Sterneck E. & Gonzalez F. J. Feedback inhibition of the retinaldehyde dehydrogenase gene ALDH1 by retinoic acid through retinoic acid receptor alpha and CCAAT/enhancer-binding protein beta. J. Biol. Chem. 275, 39747–39753 (2000). [DOI] [PubMed] [Google Scholar]
- Gardner-Stephen D. A. & Mackenzie P. I. Liver-enriched transcription factors and their role in regulating UDP glucuronosyltransferase gene expression. Curr. Drug Metab. 9, 439–452 (2008). [DOI] [PubMed] [Google Scholar]
- Hashita T. et al. Forkhead box A2-mediated regulation of female-predominant expression of the mouse Cyp2b9 gene. Drug Metab. Dispos. 36, 1080–1087 (2008). [DOI] [PubMed] [Google Scholar]
- Klick D. E., Shadley J. D. & Hines R. N. Differential regulation of human hepatic flavin containing monooxygenase 3 (FMO3) by CCAAT/enhancer-binding protein beta (C/EBPbeta) liver inhibitory and liver activating proteins. Biochem. Pharmacol. 76, 268–278 (2008). [DOI] [PubMed] [Google Scholar]
- Hwang-Verslues W. W. & Sladek F. M. HNF4α--role in drug metabolism and potential drug target? Curr Opin Pharmacol 10, 698–705 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. B. & Klaassen C. D. Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor alpha, and nuclear factor erythroid 2-related factor 2. Drug Metab. Dispos. 37, 847–856 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksunes L. M. & Klaassen C. D. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARα-, and Nrf2-null mice. Drug Metab. Dispos. 40, 1366–1379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Cheng X., Aleksunes L. & Klaassen C. D. Transcription factor-mediated regulation of carboxylesterase enzymes in livers of mice. Drug Metab. Dispos. 40, 1191–1197 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y. J. et al. Hepatocyte-specific mutation establishes retinoid X receptor alpha as a heterodimeric integrator of multiple physiological processes in the liver. Mol. Cell. Biol. 20, 4436–4444 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo Y.-K. et al. Xenobiotic- and vitamin D-responsive induction of the steroid/bile acid-sulfotransferase Sult2A1 in young and old mice: the role of a gene enhancer in the liver chromatin. Gene 386, 218–223 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M., Fickert P. & Stauber R. E. Inflammation-induced cholestasis. J. Gastroenterol. Hepatol. 14, 946–959 (1999). [DOI] [PubMed] [Google Scholar]
- Chand N. & Sanyal A. J. Sepsis-induced cholestasis. Hepatology 45, 230–241 (2007). [DOI] [PubMed] [Google Scholar]
- Krämer B., Wiegmann K. & Krönke M. Regulation of the human TNF promoter by the transcription factor Ets. J. Biol. Chem. 270, 6577–6583 (1995). [DOI] [PubMed] [Google Scholar]
- Catanzaro J. M. et al. Oncogenic Ras induces inflammatory cytokine production by upregulating the squamous cell carcinoma antigens SerpinB3/B4. Nat Commun 5, 3729 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio A., D'Antona G. & Nisoli E. Branched-chain amino acids, mitochondrial biogenesis, and healthspan: an evolutionary perspective. Aging (Albany NY) 3, 464–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray S. et al. Regulation of gluconeogenesis by Krüppel-like factor 15. Cell Metab. 5, 305–312 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittaranjan S. et al. Mutations in CIC and IDH1 cooperatively regulate 2-hydroxyglutarate levels and cell clonogenicity. Oncotarget 5, 7960–7979 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad B. M., Irizarry R. A., Astrand M. & Speed T. P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 (2003). [DOI] [PubMed] [Google Scholar]
- Lee H.-J. et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman A. W. & Azzalini A. Applied Smoothing Techniques for Data Analysis: The Kernel Approach with S-Plus Illustrations: The Kernel Approach with S-Plus Illustrations. (OUP Oxford, 1997). [Google Scholar]
- Hwang D. et al. A data integration methodology for systems biology. Proc. Natl. Acad. Sci. U.S.A. 102, 17296–17301 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam Y. C. et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335–1347 (2006). [DOI] [PubMed] [Google Scholar]
- Messeguer X. et al. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information