

Abstract
Intercalated cells are kidney tubule epithelial cells with important roles in the regulation of acid-base homeostasis. However, in recent years the understanding of the function of the intercalated cell has become greatly enhanced and has shaped a new model for how the distal segments of the kidney tubule integrate salt and water reabsorption, potassium homeostasis, and acid-base status. These cells appear in the late distal convoluted tubule or in the connecting segment, depending on the species. They are most abundant in the collecting duct, where they can be detected all the way from the cortex to the initial part of the inner medulla. Intercalated cells are interspersed among the more numerous segment-specific principal cells. There are three types of intercalated cells, each having distinct structures and expressing different ensembles of transport proteins that translate into very different functions in the processing of the urine. This review includes recent findings on how intercalated cells regulate their intracellular milieu and contribute to acid-base regulation and sodium, chloride, and potassium homeostasis, thus highlighting their potential role as targets for the treatment of hypertension. Their novel regulation by paracrine signals in the collecting duct is also discussed. Finally, this article addresses their role as part of the innate immune system of the kidney tubule.
Keywords: renal tubular acidosis, aldosterone, blood pressure, cell and transport, physiology, distal tubule
Introduction
Intercalated cells are epithelial cells traditionally associated with the regulation of acid-base homeostasis in distal segments of the kidney tubule (Figure 1) (1). These cells also participate in potassium and ammonia transport and have a role in the innate immune system. Many early studies emphasized that these tubule segments were not part of the classic nephron because they arise from the mesonephric kidney or Wolffian duct, which also gives origin to the male excurrent duct (2). Most collecting duct cells express the epithelial sodium channel (ENaC) and are grouped together as principal cells (3,4). Until recently, our understanding of the collecting duct and the roles of intercalated cells has lagged behind that of other segments. The collecting duct was initially described as not having a specialized function or as having a role only in water reabsorption (5,6).
Figure 1.

Intercalated cells in rat collecting duct. The cartoon and confocal micrograph illustrate the intercalated cell distribution along the kidney tubule and within the epithelium. Intercalated cells were detected in the cortical and outer medullary collecting duct (green oval) by immunoflorescence labeling using an antibody against one of the H+-ATPase subunits (green). Intercalated cells are also located in the connecting segment (red circle). The apical or luminal labeling of the H+-ATPase indicates that these cells are mostly type A intercalated cells (A-IC). Principal cells were labeled with an antibody against the water channel aquaporin-2 (red). Modified from reference 9, with permission.
Intercalated cells are essential in the response to acid-base status of the organism, and they help dispose of acid that is generated by dietary intake and cannot be eliminated via the lungs, the so-called fixed or nonvolatile acid (Figure 2). The kidney contributes to acid-base homeostasis by recovering filtered bicarbonate in the proximal tubule. Distally, intercalated cells generate new bicarbonate, which is consumed by the titration of nonvolatile acid (7). Dysfunction of the proximal tubule, where approximately 90% of the bicarbonate is reabsorbed, leads to proximal renal tubular acidosis (8). The connecting segment and collecting duct rely mostly on their intercalated cells to reabsorb the normally smaller amount of residual bicarbonate. In addition, intercalated cells participate in the excretion of ammonia/ammonium, a topic reviewed in a separate article in this series (9).
Figure 2.

Transepithelial transport processes and regulatory mechanisms in type A intercalated cells (A-IC) and type B intercalated cells (B-IC). This cartoon illustrates the major transport proteins expressed in the three main epithelial cell types present in the collecting duct: the principal cell, which expresses the epithelial sodium channel; the acid-secreting type A-IC; and type B-IC, which secretes bicarbonate while reabsorbing NaCl. In the cortical and outer medullary collecting duct, type A-ICs express H+-ATPase and the H+/K+-ATPase at the apical/luminal membrane, while they express the Cl−/HCO3− exchanger AE1 at their basolateral membrane. The bicarbonate sensor soluble adenylyl cyclase (sAC) and protein kinase A (PKA) play important roles in the regulation of the H+-ATPase (see Figure 5A). Slc26a11 (A11), an electrogenic Cl− transporter, as well as a Cl−/HCO3− anion exchanger, are also expressed at the apical membrane of the type A-IC. On the other hand, the type B-ICs display an electroneutral NaCl transport/reabsorption pathway at their apical membrane that involves pendrin, a Cl−/HCO3− exchanger, and the Na+-driven Cl−/HCO3− exchanger (NDCBE). The proposed basolateral Na+ extrusion pathway would involve the cotransporter Slc4a9 (AE4). The mechanism of Cl− exit remains to be elucidated. In type B-ICs, reabsorption of NaCl from the lumen is energized by the basolateral H+-ATPase rather than by Na+/K+-ATPase.
The relevance of intercalated cell dysfunction in clinical scenarios is often not as evident as the relevance of principal cell dysfunction, such as in patients who present with diabetes insipidus or the syndrome of inappropriate antidiuretic hormone secretion. In clinical practice, intercalated cell dysfunction is most often associated with metabolic acidosis, although histologic or laboratory confirmation of this dysfunction is seldom performed in the general acute care setting. Moreover, the contribution of intercalated cells in preventing acidemia is often eclipsed by the coordinated compensatory roles of the lung, bone, and more proximal kidney tubule segments. Nonetheless, animals subjected to dietary acid loading have significant increases in the luminal (facing the urine) surface area of intercalated cells, changes that begin within a few hours from the change in diet (reviewed in references 7,10).
Until very recently, intercalated cells were not thought to contribute to extracellular fluid volume regulation, yet now they are firmly established as important contributors to collecting duct NaCl transepithelial transport and the protection of intravascular volume in concert with principal cells (Figure 2) (reviewed by Eladari et al. [4]). An impressive new study has now established that, in vivo, intercalated cells regulate their intracellular volume by a mechanism involving the vacuolar H+-ATPase (H+-ATPase; also referred to as V-ATPase in the literature). This study is important because it establishes that although most animal cells rely on the Na+/K+-ATPase (Na,K-ATPase) to energize other transport processes, intercalated cells instead rely on the H+-ATPase (11). Therefore, now it also must be considered that cell types with high H+-ATPase levels may use this pump for more than just acid-base regulation.
Intercalated Cell Distribution, Nomenclature, Morphology, and Developmental Considerations
The cells lining the proximal tubule, thin limb, and thick ascending limb have a homogeneous ultrastructural appearance. Tubule-specific cells function as a unit within each segment. Distal to the macula densa, however, the epithelia of the distal convoluted tubule, connecting segment, and collecting duct are lined by different types of cells. Most cells in these segments are considered segment-specific principal cells, while the other cells are grouped together and called intercalated cells (5,12). An impediment to the study of these cells has been their pleomorphism and their isolation within the epithelium; they sit surrounded by principal cells. Intercalated cells vary in morphology, localization, and abundance, and these differences are evident not only among species but also among individuals from the same species. In addition, few cell lines replicate their phenotypes in culture. Some of the available immortalized intercalated cell models include the rabbit Clone-C cells, the mouse OMCDis, and the canine MDCKC11 (13–15). The Clone-C cell line is particularly useful because it can switch phenotypes from type B to type A intercalated cells (Figure 2) depending on culture conditions (16,17). Of note, many very relevant studies that have uncovered important regulatory pathways in intercalated cells have been performed in cell lines of inner medullary origin, such as the mIMCD3 cell line (18,19).
In the collecting duct, principal and intercalated cells form a “salt and pepper” epithelium (Figures 1 and 2) (20). This more primitive appearance of the collecting duct is similar to that of frog skin, reptilian bladder, and fish gills (20–22). In the kidney, the transition from the early part of the distal convoluted tubule to an epithelium containing both principal and intercalated cells coincides with the transition to segments that are derived from the ureteric bud outpouching of the Wolffian duct (2). The ratio of principal to intercalated cells varies slightly between tubular segments, with a ratio of 2:1 in outer medullary collecting duct segments and 3:1 in the cortical collecting duct. The ratio also varies between species (10,23).
Intercalated cells have had obscure names such as “dark cell” and “special cell,” a terminology that reflected how little was known about their function from the time of their discovery in 1876 until the late 1960s (24). Their ultrastructural description, however, has remained quite consistent over the years. Intercalated cells are also called “mitochondria-rich cells,” reflecting the high levels of round mitochondria at their apical pole, or in their cytoplasm, a distribution that was quite different from that of other kidney tubule cells, which accumulated mitochondria around the basolateral membrane (Figure 3) (24–27). These cells also have “numerous irregular apical microvilli” compared with the surrounding segment-specific cells (24). Moreover, it became clear that intercalated cells lacked a central cilium, at least in the cortex, which also differentiated them from the adjacent principal cells (Figure 3) (10,28–30). Their peculiar morphology was reminiscent of acid-secreting cells in the turtle bladder (31,32) and frog skin (33), and, like these cells, intercalated cells participate in urinary acid secretion, bicarbonate reabsorption, and bicarbonate secretion (5).
Figure 3.

Morphology of rat cortical collecting duct intercalated cells. (A) The scanning electron micrograph shows the luminal surface of the rat collecting duct. In this image principal cells can be easily identified by their small microprojections into the lumen and by the presence of a single cilium. Two configurations of intercalated cells are present in this tubule. The type A-ICs (arrows) have a large luminal surface covered mostly with microplicae, while a type B-IC (arrowhead) has a more angular cellular outline and smaller apical microvilli (original magnification, ×5500). Reproduced with permission from reference 10. (B) This transmission electron micrograph of rat cortical collecting duct illustrates further the two configurations of intercalated cells. The type A-IC (right) shows a well developed apical tubulovesicular membrane compartment, with prominent microprojections that are part of the microplicae on the luminal membrane. In this image, the type B-IC (left) presents a denser cytoplasm with more abundant mitochondria and many vesicles throughout the cytoplasm. The luminal membrane of the type B cell has a quite smooth luminal membrane (original magnification, ×5000). Reproduced with permission from reference 10.
The question remains of how these two disparate cell types coordinate their function in these epithelia (20,34). The plasma membrane of each epithelial cell organizes into apical and basolateral domains that develop distinct membrane lipid and transport protein compositions. Thus, within a homogeneous epithelium of one cell type, the apical membranes (facing the tubular lumen) of all cells could function as a unit, and similarly for the basolateral domains of the same cells facing the interstitium. The maintenance of the apical and basolateral domains of the kidney tubular epithelium depends on the presence of functional tight junctions, the differential permeabilities of the two membranes, and the generation of gradients between the lumen and the interstitium. Recent findings have uncovered the paracrine signals that integrate the function of intercalated and principal cells in the collecting duct (35).
Three types of intercalated cells are traditionally recognized, based largely on cell morphology and localization (reviewed in reference 36). These cells fall into the categories of type A (also known as “α”), type B (or “β”), and non-A, non-B intercalated cells (Figure 2) (reviewed elsewhere [10,36]). The initial morphologic classification was upheld after both functional and immunolabeling studies once inhibitors and antibodies for individual transport proteins became available. Currently, intercalated cells are classified with respect to the presence of the chloride-bicarbonate exchanger AE1 (Slc4a1) and to the subcellular localization of the multisubunit H+-ATPase (36,37). This classification has been further modified to reflect whether cells express the transport protein pendrin, a subtype of chloride-bicarbonate exchanger (37). Therefore, another definition for type A intercalated cells combines their function in urinary acidification, their lack of pendrin expression, the presence of H+-ATPase at their apical membrane, and the expression of AE1 at their basolateral membrane. On the other hand, the intercalated cells involved in bicarbonate secretion express the chloride/bicarbonate exchanger pendrin at their apical membrane. These cells are further classified as type B and non-A, non-B intercalated cells. While type B intercalated cells express the H+-ATPase at their basolateral pole, the non-A, non-B intercalated cells express both the H+-ATPase and pendrin at their apical membrane. Moreover, non-A, non-B intercalated cells are located in the connecting segment or connecting tubule (10,38). All three types of intercalated cells express carbonic anhydrase in their cytoplasm (36,39). This metalloenzyme reversibly catalyzes the hydration of CO2 (40) and thus leads to the formation of bicarbonate. At least 13 isoforms of carbonic anhydrase have been identified in mammals. Carbonic anhydrase II, acting in tandem with H+-ATPase, facilitates the coordinated proton and bicarbonate secretion from intercalated cells. Table 1 summarizes the current findings on transport or other proteins that can help in the characterization and classification of intercalated cell types.
Table 1.
Relevant proteins expressed in kidney intercalated cells
| Intercalated Cell Type Protein Expressed | Type A | Type B | Non-A, Non-B |
|---|---|---|---|
| Carbonic anhydrase II: catalyzes the reversible hydration of CO2 to bicarbonate and water | Apical | Cytoplasmic | Cytoplasmic |
| H+-ATPase: pumps H+ across the (plasma) membrane, and into the extracellular space | Apicala | Basolaterala | Apical and diffuse vesiculara |
| AE1 (Slc4a1): anion exchanger 1, exchanges a Cl- for a bicarbonate | Basolaterala | ||
| Pendrin (Slc26a4): exchanges a Cl- for a bicarbonate | Apicala | Apicala | |
| H+/K+-ATPase: exchanges an H+ for a K+ at the expense of ATP | Apical | Apical? | |
| AE4 (Slc4a4): anion exchanger 4, exchanges a Cl- for (n) bicarbonate | Basolateral | ||
| RhBG: Rh B glycoprotein, ammonia transporter | Basolateral | Basolateral | |
| RhCG: Rh C Glycoprotein, ammonia transporter | Apical | Apical | |
| NDCBE (Slc4a8): Na+-driven Cl−/bicarbonate exchanger | Apical | ||
| A11 (Slc26a11): electrogenic Cl− transporter and Cl−/HCO3- anion exchanger | Apical | ||
| NKCC1: Na+-K+-2Cl− cotransporter | Basolateral | ||
| BK channel or Maxi-K: K+ secretion | Apical |
Adapted from reference 36. BK, big potassium.
The differential localization patterns pertaining to the transport proteins identify a combination of key markers that aid in the classification of the different intercalated cell subtypes.
Intercalated Cell Development and Plasticity
Interest in intercalated cell development arose, in part, because of the intriguing presence of the non-A, non-B intercalated cell, which some viewed as an intermediate type. Moreover, other intercalated cells did not fit into any of the three available classifications because of the expression of marker proteins in unexpected subcellular localizations. These other types of intercalated cells were thought to represent a series of intermediate forms that could arise if, for example, a type B intercalated cell were transforming into a type A intercalated cell under the pressure of metabolic acidosis. One such “intermediate” cell might express the H+-ATPase diffusely in the cytoplasm or at both the apical and basolateral domains (41,42). There is now evidence that type A and type B intercalated cell types represent different states of differentiation, as proposed by Al-Awqati and confirmed by others (16). In some of these studies carried out in the rabbit model, adaptation to acidosis was not accompanied by changes in the number of intercalated cells but rather by a change from type B to type A intercalated cells (7,43). The transition process from type B to type A intercalated cell depends on the basolateral deposition of the extracellular matrix proteins hensin (DMBT1), galectin 3, and other proteins (7,44). Overall, induction of chronic metabolic acidosis increases the proportion of type A intercalated cells while metabolic alkalosis causes an increase of type B intercalated cells (7,45,46). Moreover, mice with a hensin defect in intercalated cells indeed developed metabolic acidosis. In this mouse model, intercalated cells in the cortex had a type B phenotype (44). Interestingly, the hensin-deficient mice had modified the phenotype of medullary epithelial cells. These modified medullary cells had the ultrastructure of the type B intercalated cells usually found in cortex, while they differed from type B cells in that they did not express pendrin (47).
In addition to the above factors, the Notch signaling pathway also controls the development of primitive epithelia, such as the ones in the connecting segment and the collecting duct (20,48). The Notch signaling cascade is essential for a developmental process called lateral inhibition, where neighboring cells undergo one final step that gives them very different phenotypes. Notch signaling disruption indeed alters collecting duct cellular composition, resulting in more intercalated cells and fewer principal cells; in addition, probably because of the decreased numbers of principal cells, the mice also display a nephrogenic diabetes insipidus phenotype (48). The developmental events would involve ureteric bud cells differentiating into principal cells, with active Notch signaling, while intercalated cells would appear because of inactive Notch signaling (49,50). Intercalated cell development goes one step further to generate cells of type B lineage in the cortex and outer medulla. The expression of grainyhead genes results in the type B intercalated cell phenotype (21,51,52).
In rodent kidney, the appearance of intercalated cells during development has traditionally been tracked by following the expression of several of the intercalated cell–specific H+-ATPase subunits and other markers. This multisubunit proton pump is ubiquitous, but several of its subunits are specific to distal nephron intercalated cells (47,53). These intercalated cell–specific H+-ATPase subunits (such as a4 and B1) appear early in the medulla at embryonic day 15.5 (E15.5), after the detection of the expression of Foxi1, a forkhead family transcription factor that is detected in adult intercalated cells (54). Pendrin-positive intercalated cells have been detected in the mouse kidney at E14. There was also a debate on whether intercalated cells developed from principal cells or vice versa, or whether one cell population could contribute to the repopulation of the collecting duct after injury. Some of this controversy has been clarified by the characterization of a mouse strain lacking a transcription factor of the forkhead family, Foxi1 (55). In adult mice, Foxi1 is detected only in intercalated cells, suggesting that this transcriptional regulator is required for intercalated cell development. Indeed, Foxi1 knockout mice lack intercalated cells, and their collecting duct epithelium instead expresses a more primitive cell type with some principal and intercalated cell characteristics. For example, this undifferentiated cell type expresses the water channel aquaporin-2, which is expressed in principal cells, and the cytosolic enzyme carbonic anhydrase II, which is used as a marker of intercalated cells. Foxi1-null mice did not express H+-ATPase or pendrin in their collecting ducts.
Type A Intercalated Cells, Their Major Transport Proteins, and Their Hormonal Regulation
Type A intercalated cells are present in the late distal convoluted tubule, the connecting segment, cortical and outer medullary collecting duct, and, in some reports, the early part of the inner medullary collecting duct. They are the more abundant type of intercalated cell in the outer stripe of the outer medulla in most mammalian species (10). Morphologically, they lack a cilium, they have numerous apical microplicae, and mitochondria are abundant near the apical pole (10,28). Their most characterized role is as cells that secrete protons into urine, and they can easily be identified for their lack of pendrin expression.
Type A intercalated cells can secrete H+ equivalents into urine via the H+-ATPase or the H+/K+-ATPase (H,K-ATPase) at their apical membrane. The latter pump exchanges one potassium ion for each extruded proton. In addition, these cells express Slc4a1, a splice variant of erythroid band 3, at the basolateral membrane (Figure 1) (42). The secretion of a proton into the tubular lumen, whether it is in exchange for potassium reabsorption or not, results in the generation of intracellular bicarbonate via carbonic anhydrase II, which is reabsorbed into the interstitium in exchange for chloride by AE1. The H+-ATPase is very abundant at the apical membrane of type A intercalated cells and in subapical vesicles or tubulovesicular structures, and they appear as 10-nm spherical structures or “studs” coating these membranes, also described as rod-shaped particles (56,57).
The H+-ATPase facilitates the movement of protons across the apical membrane of type A intercalated cells. Other ion movements, such as Cl− and/or bicarbonate extrusion, compensate H+ transport in proton-secreting cells (32,58). In the case of H+-ATPase, two other main factors affect its function at the plasma membrane: the pH difference across the apical membrane and the transepithelial potential difference (59). For example, this pump mediates H+ transport at a rate that is 0 when the luminal pH is <4.5, while the transport rate of the pump is saturated at a pH of 7.0–8.0. In addition, when the lumen potential is 120 mV relative to the interstitium, the H+ transport rate is negligible. On the other hand, at an applied potential of −30 mV the H+ pumping rate saturates. A hypothesis was then presented that Na+ reabsorption through ENaC would create a more lumen negative transtubular potential difference along the distal nephron. This potential difference would favor H+-ATPase function in type A intercalated cells. Wagner and colleagues elucidated in vivo that ENaC function in the connecting segment was sufficient to induce this upregulation (60).
Both AE1 and H+-ATPase are upregulated in the kidney during metabolic acidosis (45), and the morphology of type A intercalated cells changes (Figure 4) (10). Although the entire collecting duct contributes to urinary acidification, the outer medullary collecting duct is very important in this process, and most intercalated cells in this segment are of type A expressing apical H+-ATPase (5,42). In addition, in collecting duct cells, including intercalated cells, H+/K+-ATPases can be detected by following the expression of their two α subunit isoforms: HKα1 (gastric) or HKα2 (colonic) (reviewed in reference 61). Type A intercalated cells from HKα1,2–null mice had significantly slower acid extrusion compared with A-type intercalated cells from HKα1-null or HKα2-null mice (62), although the lack of either α subunit affected the rate of acid extrusion from both type A and type B cells (63).
Figure 4.

Change in intercalated cell morphology in response to chronic acid-base status changes. This transmission electron micrograph shows the apical membrane of type A cells from the collecting duct from a normal rat (A) and from a rat with acute respiratory acidosis (B). In the control animal prominent studs (arrowheads) are observed on tubulovesicular structures in the control rat (A), while they are more abundant on the apical membrane in the experimental animal (B). In another study, Brown and colleagues showed that these studs are H+-ATPase, both at the membrane and in the tubulovesicular subapical structures (57). The increase in the number of H+-ATPases (studs) at the apical membrane in the animal with acidosis (B) is coupled to an increase in membrane microprojections and a decrease in the number of tubulovesicular structures in the acidotic rat (B). (Original magnification: A, ×48,000; B, ×42,400.) Reproduced with permission from reference 10.
Therefore, type A intercalated cells in the collecting duct participate in active potassium reabsorption via the H+/K+-ATPases (Figure 2), and they undergo hypertrophy when animals are fed a low-potassium diet. However, the contribution of each α isoform in kidney potassium and acid handling remains to be determined, as the null mice for either the α1 or α2 subunits did not have significant changes in their acid-base or potassium excretion (62). In the setting of acidosis, the activity of H+/K+-ATPase increases in the collecting duct (64,65). These findings indicate that this pump represents a key mechanism of proton secretion by the kidney in metabolic acidosis.
In addition to the H+/K+-ATPase, another important regulator of K+ homeostasis, the high-conductance big potassium (BK) or maxi-K channel is also highly expressed at the apical membrane of type A intercalated cells (66), especially in animals fed a high-potassium diet. These channels, which are less abundant in principal cells, are activated by depolarization, stretch/luminal flow, intracellular calcium increases, or hypoosmotic stress (reviewed in reference 67). Moreover, the BK channel is inhibited by the with-no-lysine kinase 4 (WNK4) in a phosphorylation-dependent manner (68,69). In a later study performed in a cell line of intercalated cell characteristics, WNK4 inhibited BK channel activity in part by increasing channel ubiquitination and degradation (70). The researchers investigating flow-induced K secretion from the BK channel in type A intercalated cells found evidence that a basolateral Na-K-Cl cotransporter (NKCC1) could support entry of K+ into the cell, especially in light of the almost negligible levels of Na+/K+-ATPase in this type of intercalated cells (Figure 2) (71).
Acid-Base Sensing in Intercalated Cells
The activity and subcellular localization of acid-base transport proteins can be quickly altered by changes in extracellular or intracellular pH or bicarbonate changes. These findings have led to an intense search for the pH-sensing mechanism within the intercalated cells. For example, the H+-ATPase in type A intercalated cells is acutely activated (within minutes to hours) by kinases such as protein kinase A (PKA), while it is acutely inhibited by the metabolic sensor AMP activated protein kinase (AMPK) (Figure 5) (72–74). This downregulation of the proton pump by AMPK is preliminary evidence that intercalated cell function can be altered when the tubule is under metabolic stress, such as under conditions of ischemia. These two kinases also regulate the H+-ATPase in proximal tubule and epididymis (75,76). We have characterized two major phosphorylation sites in the H+-ATPase A subunit; Ser-175 is required for PKA-mediated pump activation while Ser-384 is required for downregulation of the pump by AMPK (72,73). In particular, Ser-175 is in part responsible for the activation of H+-ATPase in endosomes downstream of G protein–coupled signaling via cAMP/PKA (77).
Figure 5.
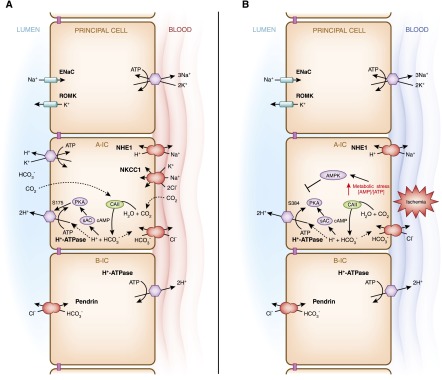
Model of H+-ATPase coregulation at the apical membrane of type A intercalated cells by two kinases, downstream of acid-base status and of cellular metabolic stress. (A) Acute increases in the level of intracellular bicarbonate activates the bicarbonate-sensor sAC, which generates cAMP and then activates PKA. Studies using pharmacological activators have shown that exchange protein directly activated by cAMP (Epac) is not likely to play a role in H+-ATPase regulation (183). Carbonic anhydrase II (CAII) is involved in the generation of intracellular bicarbonate. Downstream of PKA, the A subunit of H+-ATPase is then phosphorylated at Ser-175 (S175) (72). This phosphorylation event is involved in activating H+-ATPase at the apical membrane. (B) This acute stimulatory effect of the sAC/cAMP/PKA signaling cascade on apical H+-ATPase activity is counterbalanced by the inhibitory effect of the metabolic sensor AMP activated protein kinase (AMPK), downstream of ischemia or metabolic stress, for example. Acute metabolic stress leads to an elevation of the levels of AMP compared with ATP, and the increase in cellular [AMP]/[ATP] directly activates AMPK. AMPK mediates the downregulation of H+-ATPase activity by phosphorylating Ser-384 (S384) in the proton pump’s A subunit (73).
In addition, PKA activation of the H+-ATPase has also been linked to the acute activation of the bicarbonate-sensor soluble adenylyl cyclase (sAC) in type A intercalated cells (Figure 5A) (74). This cascade links physiologic changes in plasma and/or intracellular bicarbonate to acute intercalated cell phenotypic changes. For example, the PKA activator cAMP directly stimulates proton secretion in primary cultures of mouse intercalated cells (78) while significantly increasing the size of apical microplicae in these cells (78). The role of bicarbonate-activated sAC in intercalated cell function is also supported in cases of increased chronic luminal bicarbonate delivery (over days) to the collecting duct (79). A recent study found that Slc26a11, recognized as an electrogenic Cl− transporter as well as a Cl−/HCO3− anion exchanger, facilitates H+-ATPase function in type A intercalated cells (80).
Another important sensor of extracellular pH in the kidney is the G protein–coupled receptor 4 (GPR4). Its function was identified and characterized in cell lines in studies that used the mOMCD1 intercalated cell model (18). GPR4-null mice had decreased kidney net acid secretion and a nongap metabolic acidosis, suggesting that GPR4 is a pH sensor with a likely important role in promoting acid secretion in kidney collecting duct intercalated cells in vivo. Moreover, the nonreceptor tyrosine kinase Pyk2 was studied as a potential sensor of pH changes in the distal tubule. Pyk2 is expressed in kidney, and it becomes autophosphorylated by several stimuli, including decreased pH (81). Studies also performed in mOMCD1 cells revealed that Pyk2 and downstream effectors increased H+-ATPase but not H+/K+-ATPase in this intercalated cell model.
Type B Intercalated Cells and Their Transport Processes
The classic type B intercalated cells express a chloride-bicarbonate exchanger, pendrin, at their apical membrane and express H+-ATPase at their basolateral membrane (Figure 6) (41,82). These cells are thought to be responsible for the secretion of OH− equivalents. Pendrin is encoded by the SLC26A4 PDS gene. This transport protein is a member of the multifunctional sulfate transporter solute carrier 26 family (SLC26). Although it does not transport sulfate (83), it can transport anions such as iodide, Cl−, and bicarbonate (84). Pendrin is also expressed in epithelial cells in the inner ear (85) and the thyroid, as well as in other tissues (82,86). This differential tissue expression pattern highlights different transport roles for pendrin. For example, in kidney intercalated cells and in the inner ear, pendrin acts as a chloride-bicarbonate exchanger (85), while in the thyroid gland it mediates iodide transport (86). Therefore, mutations in SLC26A4 involve various organs to differing degrees. Vaughan Pendred described this disorder as the combination of deafness and goiter (87,88). Remarkably, both patients with Pendred syndrome, which is inherited in an autosomal recessive pattern, and pendrin-null mice have normal kidney function under baseline conditions (82,89). However, upon bicarbonate administration, which induces pendrin upregulation in wild-type mice, pendrin-null mice developed a severe metabolic alkalosis. These alkali-loaded animals deficient in pendrin could not secrete bicarbonate, suggesting a critical role for pendrin in the modulation of acid-base status that protects against metabolic alkalosis (90,91).
Figure 6.

Model of major transport processes and regulatory mechanisms in type B intercalated cells. This cartoon is modified from the model proposed by Chambrey and colleagues (35). These cells participate in electroneutral NaCl absorption, which is energized by H+-ATPase. Two cycles of transport via pendrin, when coordinated with one cycle of NDCBE results in net uptake of one Na+ and one Cl− and the extrusion of two bicarbonate (HCO3−) ions. The Cl− ions recycle across the apical membrane. A basolateral channel (not shown) mediates Cl− exit. The basolateral anion exchanger 4 (AE4 or Slc4a9), together with the H+-ATPase also facilitates Na+ and bicarbonate exit.
Initially, the reabsorption of Cl− in the collecting duct was attributed to the paracellular pathway, with some studies indicating that there was transcellular Cl− absorption (92) (reviewed in references 4,37). Indeed, very recent studies have confirmed that the paracellular pathway plays an important role in Cl− reabsorption in the collecting duct (93). On the other hand, the role of type B intercalated cells in Cl− reabsorption became clear when pendrin was identified in these cells (82). Because both sodium and chloride intake can modulate kidney function, pendrin was considered immediately as a potential mediator in the pathogenesis of hypertension (90,94). Several studies have confirmed that pendrin protein expression decreases with maneuvers that increase urinary Cl− excretion, while protocols that caused Cl− depletion, such as furosemide treatment, significantly increased pendrin levels. Later studies showed that metabolic acidosis induced by acetazolamide or ammonium sulfate administration reduced the levels of pendrin irrespective of low Cl− levels in urine (95). Aldosterone and angiotensin II also increase the levels of pendrin (96,97), thus linking this transport protein to the maintenance of adequate extracellular volume and potentially to the development of hypertension. Recently, it has been proposed that pendrin may be activated via a kidney-specific mechanism that does not include circulating aldosterone (98). Furthermore, angiotensin II mediated pendrin upregulation and subcellular localization changes in kidney occur via the angiotensin 1a receptor (99). Pendrin-mediated Cl− uptake leads to vascular volume expansion and pendrin-induced modification of ENaC abundance and function (98). Changes in luminal bicarbonate concentration and/or pH could also contribute to this signaling (100). Thus, pendrin expressed in the kidney might represent a potential target for blood pressure control. This role is illustrated in pendrin-null mice that were resistant to mineralocorticoid-induced hypertension and in turn became hypotensive when fed a low-sodium diet.
Both type B and non-A, non-B intercalated cells (Table 1) express pendrin at their apical membrane and intracellular vesicles (Figure 6) (82,101). Pendrin is more abundant at the apical plasma membrane than intracellularly in non-A, non-B cells under baseline conditions. This finding suggests that pendrin-mediated anion exchange occurs to a greater extent in non-A, non-B cells than in type B cells (102). This difference in pendrin subcellular distribution also suggests that its function is likely regulated by trafficking between the plasma membrane and cytoplasmic vesicles. Nitric oxide also contributes to intercalated cell transport protein regulation as it reduces pendrin protein abundance in the kidney without modulating pendrin subcellular localization in a cAMP-dependent manner (103). The regulation of pendrin also involves glycosylation-dependent processes (104,105). In addition to the regulation of acid-base status, it appears that pendrin expressed in kidney intercalated cells plays a role in controlling iodide homeostasis as well, especially during high water intake (106).
Type B intercalated cells have recently come to prominence with the discovery that the Na+-driven chloride/bicarbonate exchanger (NDCBE) (107) and pendrin colocalize at the apical membrane of these cells (Figure 6). Together these two transport proteins mediate the until recently unexplained thiazide-sensitive electroneutral NaCl reabsorption in the collecting duct (4,11,107). Moreover, pendrin deletion in type B intercalated cells disturbs the function of ENaC in adjacent principal cells (98,100). As in intercalated cells, NaCl absorption is energized by the H+-ATPase and not by the Na+/K+-ATPase (11); Eladari and colleagues hypothesized that Na+ transport in type B intercalated cells was mediated by a bicarbonate-dependent transport protein at the basolateral membrane. This transport protein therefore would be energized by H+-ATPase. Members of the same research group had previously shown that the anion exchanger 4 (AE4/Slc4a9) was expressed basolaterally in type B intercalated cells (Figure 1) (108).
Acidosis induces important changes in the transport proteins expressed in type B intercalated cells. For example, acidosis downregulates pendrin expression at their apical membrane and in apical recycling endosomes, and moreover, AE4 expression is diminished at their basolateral pole (109).
Regulation of Intercalated Cell Transport Proteins by Hormones and Other Mediators: Effects of (Pro)renin, Renin, Angiotensin, and Aldosterone
Steroid hormone receptors, such as the mineralocorticoid receptor (MR), function downstream of the renin-angiotensin-aldosterone system (RAAS). Together with the glucocorticoid receptor these hormones elicit signaling events in the kidney that regulate salt and potassium homeostasis, intravascular volume (110), and cell metabolism (111–113). Aldosterone, via the MR, in turn activates ENaC in principal cells, resulting in an increase in paracellular Cl− transport, H+ secretion from intercalated cells, and activation of basolateral Na+/K+-ATPase (reviewed in references 60,110,114). However, these transcellular transport cascades are mostly relevant to principal cells (11).
During hyperkalemia, aldosterone is also secreted. Its stimulation of ENaC via the MR leads to lumen electronegativity, which promotes K+ secretion via the renal outer medullary small-conductance K+ channel (ROMK) expressed in principal cells and the flow-mediated, large-conductance, calcium-activated potassium channel or BK channel, which is expressed in intercalated cells (Figure 7) (71,115). Thus, potassium secretion is promoted without retaining sodium. It is paradoxical that activation of the same MR in the intercalated cell would result in increases of both NaCl reabsorption and regulation of acid-base transport proteins (110). The RAAS is activated during metabolic acidosis (116), while RAAS blockade inhibits kidney acid excretion in the setting of acidosis (112,117). The regulatory effects of steroid hormones on individual transport proteins in intercalated cells have been extensively studied. For example, decreased dietary potassium increases type A intercalated cell function apical projections and increases the activity of H+/K+-ATPase and the BK channel (66). However, under potassium-replete conditions, which are known to increase aldosterone levels, changes in dietary sodium and serum aldosterone did not affect BK expression in intercalated cells (118).
Figure 7.
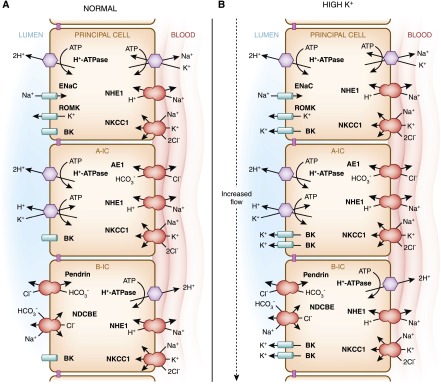
Intercalated cells are involved in K+ secretion in the collecting duct. This cartoon illustrates the location of the voltage-dendent renal outer medullary small-conductance K+ (ROMK) and the flow-dependent big potassium (BK) channels in the intercalated cells (type A-IC and type B-IC) and principal cells. (A) Under normal dietary conditions, ROMK predominantly secretes K+. However, under the influence of a K+-rich diet, because of the increased activity of the basolateral Na+/K+-ATPase, the transport via the epithelial sodium channel (ENaC)– transport increases, generating a driving force for ROMK to secrete more K+ from the principal cells. (B) When high K+ secretion is accompanied by increased flow, it stimulates BK channel synthesis and function in the intercalated cells (type A-IC and type B-IC).
There are several important findings to consider in the regulation of H+-ATPase and other transport proteins in intercalated cells by steroid hormones. For example, Wagner and colleagues showed that angiotensin II increases the expression of the H+-ATPase (119). Moreover, mineralocorticoids, such as aldosterone, which is released in metabolic acidosis, also increase the expression of pendrin and the H+-ATPase (58,97). Recently a novel modulatory pathway that affects the MR in intercalated cells has been uncovered. This pathway, which involves MR phosphorylation, is likely the key to the differential response of these cells when only aldosterone is present (as in hyperkalemia), in contrast to when both aldosterone and angiotensin II are present (as in volume depletion) (see Figure 8) (69,120). The MR can be activated by both aldosterone and cortisol (121–123). While principal cells express the enzyme 11β hydroxysteroid dehydrogenase (11βHSD2), which rapidly converts cortisol to a less active hormone, cortisone, intercalated cells lack this enzyme (69,110,121). Thus, in principal cells the MR is mostly activated by aldosterone and not cortisol, and this activation leads to the coordinated action of ENaC and the Na+/K+-ATPase. A recent study elegantly showed that a single phosphorylation site, Ser-843, in the MR inactivates it, and this inactive receptor is the major species in intercalated cells (69). When there is volume depletion (Figure 8B), both angiotensin II and WNK4 promote dephosphorylation/activation of the MR. Once active, this receptor binds aldosterone and mediates the increase in H+-ATPase and chloride-bicarbonate exchanger activity. This coordinated action increases plasma volume while inhibiting potassium secretion. In the absence of angiotensin II, the receptor remains phosphorylated and thus aldosterone cannot promote H+-ATPase activity. In this scenario, aldosterone promotes K+ secretion from principal cells. Angiotensin II also increases net transcellular chloride absorption across type B cells via a mechanism that involves both pendrin and activation of the H+-ATPase (96). Together these findings show the physiologic relevance of selective mineralocorticoid activation in intercalated cells.
Figure 8.
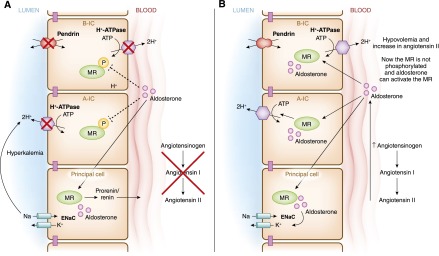
Model of regulatory pathways for the mineralocorticoid receptor (MR) in intercalated cells: hyperkalemia and volume depletion. This figure depicts pathways that involve the differential phosphorylation state of the MR in principal versus intercalated cells (type A-IC; type B-IC), that are likely the key to the distinct responses of these cells in two different scenarios. (A) The first scenario involves conditions when only aldosterone is present (as in hyperkalemia). In this case, hyperkalemia leads to aldosterone secretion while no angiotensin II is present. Here, MR is phosphorylated in intercalated but not in principal cells. These conditions lead to aldosterone-mediated Na+ reabsorption via the epithelial sodium channel in principal cells, which drives K+ secretion also in principal cells. (B) In contrast, when both angiotensin II and aldosterone are present (as in intravascular volume depletion), the MR is dephosphorylated downstream of angiotensin II, and the activity of this receptor is thus restored in intercalated cells. In addition, as a result of aldosterone signaling both pendrin and H+-ATPase are upregulated, and in turn there is a decrease drive for K+ secretion. Modified from reference 69.
The role of renin on intercalated cell function has come to the forefront because of the identification of the (Pro)renin receptor [(P)RR] as an accessory protein of H+-ATPase. However, and just like H+-ATPase, (P)RR is ubiquitously expressed. This receptor protein has a large extracellular domain that binds the enzymes renin and pro-renin (124–126). (P)RR was referred to as the ATP6AP2 (ATPase, H+ transporting, lysosomal accessory protein 2) (127). (P)RR undergoes proteolytic cleavage to generate soluble (P)RR (128,129). (P)RR is easily detected in type A intercalated cells and not in principal cells (130,131). Binding of renin and prorenin to (P)RR increases the catalytic efficiency of these enzymes in the conversion of angiotensinogen to angiotensin by 4-fold. Prorenin secreted from the principal cells binds to the (P)RR located in the apical membrane of intercalated cells or to the soluble (P)RR, and thus enhances local angiotensin I and angiotensin II formation in the collecting duct (Figure 9) (114,132). A (P)RR interaction with the H+-ATPase is required for prorenin-induced activation of Erk1/2 in a cultured cell model of intercalated cells (133). (P)RR has also been shown to play a role in vasopressin-mediated H+-ATPase activity and cAMP accumulation, in a prorenin-independent manner in the intercalated cells. The (P)RR has been implicated in several pathologic conditions, such as diabetic nephropathy, hypertension, albuminuria, and preeclampsia. However, (P)RR blockade would likely have adverse effects through its activation of H+-ATPase (130,134–136).
Figure 9.

Intercalated cells are targets of paracrine angiotensin II signaling. Angiotensin II stimulates secretion of prorenin and renin from principal cells. Prorenin binds to the prorenin receptor at the apical membrane of type A-IC, where in turn it stimulates the synthesis of angiotensin II from angiotensin I via the action of angiotensinogen, which originates in proximal segments of the nephron. Angiotensin II then binds to the angiotensin receptor 1a (AT1aR) in the type A-IC. Moreover, prorenin binds to its receptor in type A-IC to stimulate signaling via p58 or cAMP.
Intercalated Cell Dysfunction and the Pathogenesis of Distal Renal Tubular Acidosis
Mutations in certain intercalated cell transport proteins, or associated enzymes, can lead to distal renal tubular acidosis (dRTA) type 1, a syndrome characterized by a decreased capability of urinary acidification and variable degrees of hyperchloremic, hypokalemic metabolic acidosis (137). When this condition is severe, the patient cannot excrete the daily acid load, which varies from 50 to 100 mEq/d depending on the patient's diet. This continued proton accumulation leads to a normal anion gap metabolic acidosis, with values of plasma bicarbonate that can fall below 10 mEq/L. The treatment involves potassium and alkali repletion. When the disease is not so severe, it is called incomplete, type 1 dRTA. These patients still have an inability to acidify the urine (pH>5.5) without a significant drop in plasma bicarbonate. Some forms of dRTA are due to a back-leak of protons resulting from the exposure of the patient to pharmacologic agents that damage the tubular plasma membrane, such as amphotericin (138). However, these patients will often display signs and symptoms associated with principal as well as intercalated cell dysfunction.
Inherited forms of dRTA most often result from the disruption of normal type A intercalated cell physiology. Patients with dRTA tend to have acidemia because of the lack of acid secretion from type A intercalated cells. This lack of acid secretion, and alkalinization of the urine, can present with nephrocalcinosis and/or nephrolithiasis, as well as hypokalemia (139). The inherited forms of dRTA have been described as having autosomal dominant or recessive modes of transmission. Some disease-causing mutations of H+-ATPase a4 and B1 subunits result in early-onset dRTA (140–142). These findings suggest that the differential distribution of isoforms of H+-ATPase in intercalated versus other epithelial cells may be acquired during early infancy in humans. Another important transport protein in intercalated cells is the chloride-bicarbonate exchanger AE1. A large screen of kindreds with dRTA revealed that some mutations in AE1 cause autosomal dominant dRTA, while other defects identified in AE1 appear to result in autosomal recessive dRTA (143–145). In a mouse animal model, that deletion of carbonic anhydrase II reduces the number of intercalated cells in both type A and type B intercalated cells (146).
In humans, dRTA is often accompanied by some degree of salt, water, and potassium losses, and the causes for these findings were not well understood (147). Recently, a mouse model of dRTA, which was deficient in the intercalated cell–specific B1 subunit of H+-ATPase (ATP6V1B1), also presented with urinary losses, leading to hypovolemia, hypokalemia, and polyuria (Figure 10) (35). Although these mice did not present with acidosis unless challenged with an acid load (148), the molecular defect in H+-ATPase led to dysfunction of both ENaC and the combined pendrin/NDCBE (35). Researchers studying this mouse model discovered that the NaCl urinary loss coincided with increased prostaglandin E2 (PGE2) and ATP levels in urine. PGE2 is a known inhibitor of Na+ transport in the collecting duct (149–151). Indeed, when PGE2 synthesis was blocked in vivo, the animals displayed restored ENaC levels in the cortex (35). This elegant study proposed a mechanism wherein type B intercalated cells would sense increases in NaCl delivery to the distal nephron, leading to release of ATP from intracellular stores. This ATP release would be followed by an increase in PGE2 from the type B intercalated cells, and then to a decrease in Na+ absorption by the adjacent principal cells.
Figure 10.

Intercalated cells are necessary to maintain body fluid and electrolyte balance. This model summarizes the recent findings by Gueutin and colleagues (35), which showed how H+-ATPase dysfunction in type B-IC leads to the urinary water and sodium losses in patients with distal renal tubule acidosis. This group studied mice with significantly decreased H+-ATPase activity in the collecting duct intercalated cells due to knockout of the B1 of the H+-ATPase subunit (ATP6V1B1–/–mice). The specific knockout of the B1 subunit does not disturb H+-ATPase expression in the proximal tubule. These animals presented with a significant urinary loss of NaCl, revealing impaired function of epithelial sodium channel (ENaC) in principal cells, as well as decreased pendrin and NDCBE function in type B-IC in the cortical collecting duct. These animals had an upregulation of ENaC in the medulla. High levels of prostaglandin E2 (PGE2) and ATP were detected in the urine of these animals. When PGE2 was normalized using pharmacologic agents these animals also normalized their ENaC levels in the cortex, and they had improved polyuria and hypokalemia. When the H+-ATPase was inactivated in type B-IC, it resulted in ATP release with the subsequent increases in PGE2 release from these cell as well.
In addition, rare genetic defects, such as carbonic anhydrase II deficiency, can lead to intercalated cell as well as proximal tubular dysfunction. This renal tubular acidosis is also referred to as type 3 or mixed renal tubular acidosis (137,152). The patients with this genetic defect also present with osteopetrosis and cerebral calcification.
Acquired Intercalated Cell Dysfunction
Amphotericin can cause a generalized dysfunction of the kidney tubule, including intercalated cells, because it damages the plasma membrane and causes an H+ back-leak of protons due to the exposure (138,153). On the other hand, some conditions, such as Sjögren syndrome and some cases of systemic lupus erythematosus, that can present with dRTA due to more predominant intercalated cell dysfunction. The estimates of patients with Sjögren syndrome and dRTA vary from 30% to 70%; this condition often presents in the context of tubulointerstitial nephritis (154–156). Autoimmune antibodies isolated from patients interact with the intercalated cells, but the precise antigens have not been determined (157). Kidney biopsies of patients with Sjögren syndrome and dRTA reveal a reduced expression of H+-ATPase and AE1 (158,159). There is also decrease in the total number of intercalated cells in these patients. Taken together, Sjögren syndrome is associated with loss of intercalated cell transport activity associated with defects in acid-base balance, although the precise events in the development of these findings are yet to be explored.
Lithium is a proven treatment for bipolar disorder, and approximately 40% of patients receiving this treatment develop urinary concentrating defects and eventually nephrogenic diabetes insipidus, which can contribute to ESRD (reviewed in references 160,161). One of the many renal manifestations of long-term lithium treatment is that patients develop an incomplete form of renal tubular acidosis that is usually not clinically relevant (162). Despite this clinical syndrome, the findings in rats treated with lithium reveal increased numbers of type A intercalated cells that are present even in the inner medulla (163). It has been proposed that lithium decreases the gradient for proton secretion (a voltage-dependent defect) along with a concomitant decrease in H+-ATPase activity. Therefore, the observed increase in the number of intercalated cells may be a potential defense mechanism against the initial mechanism of lithium-induced acidosis. Moreover, lithium inhibits glycogen synthase kinase 3β (reviewed in reference 164). Inhibition of this kinase during kidney development induces nephron differentiation via the Wnt signaling pathway (165). Wnt signaling, a very relevant cascade needed for normal embryonic development, has been reviewed elsewhere (166). Therefore, another explanation for the increased number of intercalated cells seen with lithium treatment could be the disruption of Wnt signaling downstream of lithium-mediated glycogen synthase kinase 3β inhibition.
Findings similar to those observed with long-term lithium treatment were described after dietary K+ depletion, with increases in intercalated cells that were reversed upon dietary K+ repletion (167). These researchers concluded that principal cells and intercalated cells may interconvert in response to changes in dietary K+ and that autophagy was likely involved in the pathways driving this interconversion.
The antirejection medications cyclosporine and tacrolimus affect intercalated cell function and result in the development of dRTA, numerous changes in proximal and distal nephron epithelial transport proteins, and a decrease in the non–type A intercalated cells (168,169). Cyclosporine also inhibits cyclophilin and thus affects hensin polymerization after extracellular deposition, a step that when defective reduces terminal differentiation of intercalated cells (170).
Intercalated Cells and the Innate Immune Response
The epithelial cells lining the urinary tract play a major role in maintaining the sterility of urine, which is achieved by mucus production, urinary flow, and bladder emptying, all of which provide immediate defense against microbial invasion. This innate defense cascade is less specific than the adaptive immune response. Among the better-known components of the innate immune response are the Toll-like receptors (TLRs). TLRs play a key role in containing urinary tract infections and pyelonephritis. More than 13 TLRs have been identified in mammals, and kidney tubular epithelial cells express most of them. Among these TLRs, TLR4 has been implicated in the innate immune defense against uropathogenic Escherichia coli, the most frequent pathogen responsible for urinary tract infection (171). TLR4, which recognizes lipopolysaccharides in the outer membrane of gram-negative bacteria, is expressed in mouse collecting duct intercalated cells (172). TLRs induce a dynamic immune response via several signaling pathways, including the production of antimicrobial peptides (AMPs) (Figure 11) (172,173).
Figure 11.

The intercalated cells as key effectors of the innate immune system. This model presents various signaling cascades triggered in intercalated cells by pathologic conditions that can result in the release of defensins. Collecting duct epithelial cells respond to gram-negative bacteria (uropathogenic Escherichia coli [UPEC]) by activating cascades drownstream from the toll-like receptor 4 (TLR4) and some TLR4-independent pathways (lower). Ischemic injury also induces release of the defensin neutrophil gelatinase-associated lipocalin from intercalated cells (NGAL) (upper), and type A-IC also secrete the defensin RNAase7, which is a key molecule to prevent urinary tract infection. Funtional apical H+-ATPase is also relevant for the type A intercalated cell anti-microbial function.
AMPs are polypeptides of <100 amino acids that at physiologic concentrations are active against bacteria, enveloped viruses, fungi, and protozoa. AMPs are expressed by neutrophils or epithelial cells constitutively or after induction by exposure of the cells to pathogens (174). Urinary tract and kidney epithelial cells synthesize several AMPs, including defensins, cathelicidin, hepcidin, and ribonuclease 7 (RNAse7). Among these AMPs, defensins and RNAse7 are expressed in the kidney collecting duct (175). Defensins, which have broad-spectrum antimicrobial activity, are classified into two main subfamilies: α- and β-defensins. Of the β-defensins, human β-defensin-1 and β-defensin-2 are expressed in the loop of Henle, distal tubule, and collecting duct. In contrast to human β-defensin-1, human β-defensin-2 is not constitutively expressed in the kidney, but instead its upregulation has been implicated in the immune response during chronic kidney infection (176). On the other hand, RNAse7 is a potent AMP constitutively expressed by the uroepithelium of the bladder, ureter, and intercalated cells. Urinary concentrations of RNAse7 are much greater than those of other AMPs, exhibiting a bactericidal activity against both gram-negative and gram-positive bacteria.
Another process that implicates type A intercalated cells in the immune response during urinary tract infection is that these cells secrete the bacteriostatic protein lipocalin 2, also known as neutrophil gelatinase–associated lipocalin (NGAL). An important difference between NGAL and other AMPs is that NGAL expression is intensively upregulated by both septic and aseptic causes of AKI (177,178). Barasch and colleagues showed that type A intercalated cells are key elements in the production of this bacteriostatic protein. They used a mouse model expressing an NGAL reporter gene, which showed NGAL expression after kidney ischemia (179,180). In contrast to most of the AMPs, NGAL is specific in its binding to enterochelin (Ent), a product of gram-negative bacteria that removes Fe3+ from transferrin (181,182). When NGAL is secreted, it binds the Ent:Fe3+ complex and thus it exerts its bacteriostatic effects (181,182). In addition to its effects on Ent, NGAL secretion is required for TLR4 to undergo activation (179). Moreover, mice that lack type A intercalated cells have reduced urinary acidification and lipocalin 2/NGAL secretion into urine, as well as decreased clearance of bacteria from urine (179). The importance of intercalated cells in the innate immune response is highlighted by the recent characterization of yet another scavenger receptor called secretory glycoprotein S5D-SRCRB derived from these cells (183).
Summary and Conclusions
When intercalated cells were first identified, their morphology was distinctive and reminiscent of acid-secreting cells in more primitive epithelia. Since the 1960s, the essential role of the intercalated cells in acid-base homeostasis was solidified, first with the characterization of the transport processes in the distal nephron and later with the identification of individual membrane transport proteins, such as H+-ATPase, AE1, and pendrin, contributing to those processes. The role of intercalated cells in the pathogenesis of distal (type 1) and mixed renal tubule acidosis has been clearly established. In the last few years, the acid-base cellular sensors such as sAC and GPR4, and downstream-activated kinases such as PKA or the metabolic sensor AMPK have been recognized as key regulators of intercalated cell function. Moreover, the role of intercalated cells as regulators of salt reabsorption and intravascular volume preservation was established with the identification of NDCBE and pendrin in type B intercalated cells. Lately, it has come to the forefront that both intercalated cell volume regulation and the functional integrity of the distal nephron depend on H+-ATPase function in these fascinating cells.
Disclosures
Recent funding was received from Sanofi (for a postdoctoral fellowship grant to M.M.A.).
Acknowledgments
We thank Dr. Kenneth R. Hallows for his critical reading of the manuscript and Dr. Arohan Subramanya for helpful suggestions. We thank Mr. Andrew P. Zerby for bibliography research support.
This work was supported in part by the National Institutes of Health grant R01-DK084184 and P30-DK079307, as well as by a Junior Scholar Award of the Department of Medicine at the University of Pittsburgh School of Medicine (N.M.P.-S.) and F32-DK097889 (M.M.A.).
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Crayen ML, Thoenes W: Architecture and cell structures in the distal nephron of the rat kidney. Cytobiologie 17: 197–211, 1978 [PubMed] [Google Scholar]
- 2.Davies JA, Davey MG: Collecting duct morphogenesis. Pediatr Nephrol 13: 535–541, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Meneton P, Loffing J, Warnock DG: Sodium and potassium handling by the aldosterone-sensitive distal nephron: The pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Eladari D, Chambrey R, Peti-Peterdi J: A new look at electrolyte transport in the distal tubule. Annu Rev Physiol 74: 325–349, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madsen KM, Tisher CC: Structural-functional relationship along the distal nephron. Am J Physiol 250: F1–F15, 1986 [DOI] [PubMed] [Google Scholar]
- 6.Smith H: The Kidney: Structure and function in Health and Disease, Oxford, Oxford University Press, 1951 [Google Scholar]
- 7.Al-Awqati Q: Terminal differentiation in epithelia: The role of integrins in hensin polymerization. Annu Rev Physiol 73: 401–412, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez Soriano J: Renal tubular acidosis: The clinical entity. J Am Soc Nephrol 13: 2160–2170, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Weiner ID, Mitch WE, Sands JM: Urea and ammonia metabolism and the control of renal nitrogen excretion [published online ahead of print July 30, 2014). Clin J Am Soc Nephrol 10.2215/CJN.10311013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madsen KM, Verlander JW, Tisher CC: Relationship between structure and function in distal tubule and collecting duct. J Electron Microsc Tech 9: 187–208, 1988 [DOI] [PubMed] [Google Scholar]
- 11.Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hübner CA, Eladari D: Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci U S A 110: 7928–7933, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaissling B, Kriz W: Structural analysis of the rabbit kidney. Adv Anat Embryol Cell Biol 56: 1–123, 1979 [DOI] [PubMed] [Google Scholar]
- 13.Edwards JC, van Adelsberg J, Rater M, Herzlinger D, Lebowitz J, al-Awqati Q: Conditional immortalization of bicarbonate-secreting intercalated cells from rabbit. Am J Physiol 263: C521–C529, 1992 [DOI] [PubMed] [Google Scholar]
- 14.Guntupalli J, Onuigbo M, Wall S, Alpern RJ, DuBose TD, Jr: Adaptation to low-K+ media increases H(+)-K(+)-ATPase but not H(+)-ATPase-mediated pHi recovery in OMCD1 cells. Am J Physiol 273: C558–C571, 1997 [DOI] [PubMed] [Google Scholar]
- 15.Gekle M, Wünsch S, Oberleithner H, Silbernagl S: Characterization of two MDCK-cell subtypes as a model system to study principal cell and intercalated cell properties. Pflugers Arch 428: 157–162, 1994 [DOI] [PubMed] [Google Scholar]
- 16.Al-Awqati Q: Plasticity in epithelial polarity of renal intercalated cells: Targeting of the H(+)-ATPase and band 3. Am J Physiol 270: C1571–C1580, 1996 [DOI] [PubMed] [Google Scholar]
- 17.van Adelsberg J, Edwards JC, Takito J, Kiss B, al-Awqati Q: An induced extracellular matrix protein reverses the polarity of band 3 in intercalated epithelial cells. Cell 76: 1053–1061, 1994 [DOI] [PubMed] [Google Scholar]
- 18.Sun X, Yang LV, Tiegs BC, Arend LJ, McGraw DW, Penn RB, Petrovic S: Deletion of the pH sensor GPR4 decreases renal acid excretion. J Am Soc Nephrol 21: 1745–1755, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz JH, Li G, Yang Q, Suri V, Ross JJ, Alexander EA: Role of SNAREs and H+-ATPase in the targeting of proton pump-coated vesicles to collecting duct cell apical membrane. Kidney Int 72: 1310–1315, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Sampogna RV, Al-Awqati Q: Salt and pepper distribution of cell types in the collecting duct. J Am Soc Nephrol 24: 163–165, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Al-Awqati Q, Gao XB: Differentiation of intercalated cells in the kidney. Physiology (Bethesda) 26: 266–272, 2011 [DOI] [PubMed] [Google Scholar]
- 22.Wieczorek H, Brown D, Grinstein S, Ehrenfeld J, Harvey WR: Animal plasma membrane energization by proton-motive V-ATPases. BioEssays 21: 637–648, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM: Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol 285: F998–F1012, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Griffith LD, Bulger RE, Trump BF: Fine structure and staining of mucosubstances on “intercalated cells” from the rat distal convoluted tubule and collecting duct. Anat Rec 160: 643–662, 1968 [DOI] [PubMed] [Google Scholar]
- 25.Hancox NM, Komender J: Quantitative and qualitative changes in the “dark” cells of the renal collecting tubules in rats deprived of water. Q J Exp Physiol Cogn Med Sci 48: 346–354, 1963 [DOI] [PubMed] [Google Scholar]
- 26.Clark SL, Jr: Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol 3: 349–362, 1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flume JB, Ashworth CT, James JA: An electron microscopic study of tubular lesions in human kidney biopsy specimens. Am J Pathol 43: 1067–1087, 1963 [PMC free article] [PubMed] [Google Scholar]
- 28.Latta H, Maunsbach AB, Madden SC: Cilia in different segments of the rat nephron. J Biophys Biochem Cytol 11: 248–252, 1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfaller W, Klima J: A critical reevaluation of the structure of the rat uriniferous tubule as revealed by scanning electron microscopy. Cell Tissue Res 166: 91–100, 1976 [DOI] [PubMed] [Google Scholar]
- 30.Schwartz EA, Leonard ML, Bizios R, Bowser SS: Analysis and modeling of the primary cilium bending response to fluid shear. Am J Physiol 272: F132–F138, 1997 [DOI] [PubMed] [Google Scholar]
- 31.Stetson DL, Steinmetz PR: Alpha and beta types of carbonic anhydrase-rich cells in turtle bladder. Am J Physiol 249: F553–F565, 1985 [DOI] [PubMed] [Google Scholar]
- 32.Schwartz JH, Steinmetz PR: CO2 requirements for H+ secretion by the isolated turtle bladder. Am J Physiol 220: 2051–2057, 1971 [DOI] [PubMed] [Google Scholar]
- 33.Harvey BJ: Energization of sodium absorption by the H(+)-ATPase pump in mitochondria-rich cells of frog skin. J Exp Biol 172: 289–309, 1992 [DOI] [PubMed] [Google Scholar]
- 34.Kleyman TR, Satlin LM, Hallows KR: Opening lines of communication in the distal nephron. J Clin Invest 123: 4139–4141, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gueutin V, Vallet M, Jayat M, Peti-Peterdi J, Cornière N, Leviel F, Sohet F, Wagner CA, Eladari D, Chambrey R: Renal β-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest 123: 4219–4231, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kriz W, Kaissling B: Structural Organization of the Mammalian Kidney. In: Seldin and Giebisch’s The Kidney: Physiology and Pathophysiology, edited by Alpern RJ, Caplan MJ, Moe OW, London, Academic Press, 2013, pp 595–691 [Google Scholar]
- 37.Wall SM, Weinstein AM: Cortical distal nephron Cl(-) transport in volume homeostasis and blood pressure regulation. Am J Physiol Renal Physiol 305: F427–F438, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Kim YH, Cha JH, Tisher CC, Madsen KM: Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J Am Soc Nephrol 10: 1–12, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Spicer SS, Stoward PJ, Tashian RE: The immunohistolocalization of carbonic anhydrase in rodent tissues. J Histochem Cytochem 27: 820–831, 1979 [DOI] [PubMed] [Google Scholar]
- 40.Schwartz GJ: Physiology and molecular biology of renal carbonic anhydrase. J Nephrol 15[Suppl 5]: S61–S74, 2002 [PubMed] [Google Scholar]
- 41.Brown D, Hirsch S, Gluck S: An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature 331: 622–624, 1988 [DOI] [PubMed] [Google Scholar]
- 42.Alper SL, Natale J, Gluck S, Lodish HF, Brown D: Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. Proc Natl Acad Sci U S A 86: 5429–5433, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purkerson JM, Tsuruoka S, Suter DZ, Nakamori A, Schwartz GJ: Adaptation to metabolic acidosis and its recovery are associated with changes in anion exchanger distribution and expression in the cortical collecting duct. Kidney Int 78: 993–1005, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao X, Eladari D, Leviel F, Tew BY, Miró-Julià C, Cheema FH, Miller L, Nelson R, Paunescu TG, McKee M, Brown D, Al-Awqati Q: Deletion of hensin/DMBT1 blocks conversion of beta- to alpha-intercalated cells and induces distal renal tubular acidosis. Proc Natl Acad Sci U S A 107: 21872–21877, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bastani B, Purcell H, Hemken P, Trigg D, Gluck S: Expression and distribution of renal vacuolar proton-translocating adenosine triphosphatase in response to chronic acid and alkali loads in the rat. J Clin Invest 88: 126–136, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabolić I, Brown D, Gluck SL, Alper SL: Regulation of AE1 anion exchanger and H(+)-ATPase in rat cortex by acute metabolic acidosis and alkalosis. Kidney Int 51: 125–137, 1997 [DOI] [PubMed] [Google Scholar]
- 47.Forgac M: Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8: 917–929, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Jeong HW, Jeon US, Koo BK, Kim WY, Im SK, Shin J, Cho Y, Kim J, Kong YY: Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J Clin Invest 119: 3290–3300, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Al-Awqati Q: Segmental expression of Notch and Hairy genes in nephrogenesis. Am J Physiol Renal Physiol 288: F939–F952, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Liu Y, Pathak N, Kramer-Zucker A, Drummond IA: Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development 134: 1111–1122, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Yamaguchi Y, Yonemura S, Takada S: Grainyhead-related transcription factor is required for duct maturation in the salivary gland and the kidney of the mouse. Development 133: 4737–4748, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Quigley IK, Stubbs JL, Kintner C: Specification of ion transport cells in the Xenopus larval skin. Development 138: 705–714, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Breton S, Brown D: Regulation of luminal acidification by the V-ATPase. Physiology (Bethesda) 28: 318–329, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vidarsson H, Westergren R, Heglind M, Blomqvist SR, Breton S, Enerbäck S: The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLoS ONE 4: e4471, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, Persson AE, Bergström GG, Enerbäck S: Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest 113: 1560–1570, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown D, Weyer P, Orci L: Nonclathrin-coated vesicles are involved in endocytosis in kidney collecting duct intercalated cells. Anat Rec 218: 237–242, 1987 [DOI] [PubMed] [Google Scholar]
- 57.Brown D, Gluck S, Hartwig J: Structure of the novel membrane-coating material in proton-secreting epithelial cells and identification as an H+ATPase. J Cell Biol 105: 1637–1648, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP: Renal vacuolar H+-ATPase. Physiol Rev 84: 1263–1314, 2004 [DOI] [PubMed] [Google Scholar]
- 59.Andersen OS, Silveira JE, Steinmetz PR: Intrinsic characteristics of the proton pump in the luminal membrane of a tight urinary epithelium. The relation between transport rate and delta mu H. J Gen Physiol 86: 215–234, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kovacikova J, Winter C, Loffing-Cueni D, Loffing J, Finberg KE, Lifton RP, Hummler E, Rossier B, Wagner CA: The connecting tubule is the main site of the furosemide-induced urinary acidification by the vacuolar H+-ATPase. Kidney Int 70: 1706–1716, 2006 [DOI] [PubMed] [Google Scholar]
- 61.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS: The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol 298: F12–F21, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lynch IJ, Rudin A, Xia SL, Stow LR, Shull GE, Weiner ID, Cain BD, Wingo CS: Impaired acid secretion in cortical collecting duct intercalated cells from H-K-ATPase-deficient mice: role of HKalpha isoforms. Am J Physiol Renal Physiol 294: F621–F627, 2008 [DOI] [PubMed] [Google Scholar]
- 63.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS: The renal H,K-ATPases. Curr Opin Nephrol Hypertens 19: 478–482, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Silver RB, Frindt G, Mennitt P, Satlin LM: Characterization and regulation of H-K-ATPase in intercalated cells of rabbit cortical collecting duct. J Exp Zool 279: 443–455, 1997 [PubMed] [Google Scholar]
- 65.Silver RB, Mennitt PA, Satlin LM: Stimulation of apical H-K-ATPase in intercalated cells of cortical collecting duct with chronic metabolic acidosis. Am J Physiol 270: F539–F547, 1996 [DOI] [PubMed] [Google Scholar]
- 66.Najjar F, Zhou H, Morimoto T, Bruns JB, Li HS, Liu W, Kleyman TR, Satlin LM: Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am J Physiol Renal Physiol 289: F922–F932, 2005 [DOI] [PubMed] [Google Scholar]
- 67.Woda CB, Miyawaki N, Ramalakshmi S, Ramkumar M, Rojas R, Zavilowitz B, Kleyman TR, Satlin LM: Ontogeny of flow-stimulated potassium secretion in rabbit cortical collecting duct: functional and molecular aspects. Am J Physiol Renal Physiol 285: F629–F639, 2003 [DOI] [PubMed] [Google Scholar]
- 68.Zhuang J, Zhang X, Wang D, Li J, Zhou B, Shi Z, Gu D, Denson DD, Eaton DC, Cai H: WNK4 kinase inhibits Maxi K channel activity by a kinase-dependent mechanism. Am J Physiol Renal Physiol 301: F410–F419, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shibata S, Rinehart J, Zhang J, Moeckel G, Castañeda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, Lifton RP: Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Z, Subramanya AR, Satlin LM, Pastor-Soler NM, Carattino MD, Kleyman TR: Regulation of large-conductance Ca2+-activated K+ channels by WNK4 kinase. Am J Physiol Cell Physiol 305: C846–C853, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palmer LG, Frindt G: High-conductance K channels in intercalated cells of the rat distal nephron. Am J Physiol Renal Physiol 292: F966–F973, 2007 [DOI] [PubMed] [Google Scholar]
- 72.Alzamora R, Thali RF, Gong F, Smolak C, Li H, Baty CJ, Bertrand CA, Auchli Y, Brunisholz RA, Neumann D, Hallows KR, Pastor-Soler NM: PKA regulates vacuolar H+-ATPase localization and activity via direct phosphorylation of the a subunit in kidney cells. J Biol Chem 285: 24676–24685, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alzamora R, Al-Bataineh MM, Liu W, Gong F, Li H, Thali RF, Joho-Auchli Y, Brunisholz RA, Satlin LM, Neumann D, Hallows KR, Pastor-Soler NM: AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am J Physiol Renal Physiol 305: F943–F956, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gong F, Alzamora R, Smolak C, Li H, Naveed S, Neumann D, Hallows KR, Pastor-Soler NM: Vacuolar H+-ATPase apical accumulation in kidney intercalated cells is regulated by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 298: F1162–F1169, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Al-bataineh MM, Gong F, Marciszyn AL, Myerburg MM, Pastor-Soler NM: Regulation of proximal tubule vacuolar H(+)-ATPase by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 306: F981–F995, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hallows KR, Alzamora R, Li H, Gong F, Smolak C, Neumann D, Pastor-Soler NM: AMP-activated protein kinase inhibits alkaline pH- and PKA-induced apical vacuolar H+-ATPase accumulation in epididymal clear cells. Am J Physiol Cell Physiol 296: C672–C681, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gidon A, Al-Bataineh MM, Jean-Alphonse FG, Stevenson HP, Watanabe T, Louet C, Khatri A, Calero G, Pastor-Soler NM, Gardella TJ, Vilardaga JP: Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat Chem Biol 10: 707–709, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Păunescu TG, Ljubojevic M, Russo LM, Winter C, McLaughlin MM, Wagner CA, Breton S, Brown D: cAMP stimulates apical V-ATPase accumulation, microvillar elongation, and proton extrusion in kidney collecting duct A-intercalated cells. Am J Physiol Renal Physiol 298: F643–F654, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paunescu TG, Da Silva N, Russo LM, McKee M, Lu HA, Breton S, Brown D: Association of soluble adenylyl cyclase with the V-ATPase in renal epithelial cells. Am J Physiol Renal Physiol 294: F130–F138, 2008 [DOI] [PubMed] [Google Scholar]
- 80.Xu J, Barone S, Li H, Holiday S, Zahedi K, Soleimani M: Slc26a11, a chloride transporter, localizes with the vacuolar H(+)-ATPase of A-intercalated cells of the kidney. Kidney Int 80: 926–937, 2011 [DOI] [PubMed] [Google Scholar]
- 81.Fisher KD, Codina J, Petrovic S, DuBose TD, Jr: Pyk2 regulates H+-ATPase-mediated proton secretion in the outer medullary collecting duct via an ERK1/2 signaling pathway. Am J Physiol Renal Physiol 303: F1353–F1362, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED: Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A 98: 4221–4226, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED: Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17: 411–422, 1997 [DOI] [PubMed] [Google Scholar]
- 84.Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP: The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 21: 440–443, 1999 [DOI] [PubMed] [Google Scholar]
- 85.Everett LA, Morsli H, Wu DK, Green ED: Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci U S A 96: 9727–9732, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED: Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 141: 839–845, 2000 [DOI] [PubMed] [Google Scholar]
- 87.Pendred V: Deaf-mutism and goitre. Lancet 148: 532, 1896 [Google Scholar]
- 88.Morgans ME, Trotter WR: Association of congenital deafness with goitre; the nature of the thyroid defect. Lancet 1: 607–609, 1958 [DOI] [PubMed] [Google Scholar]
- 89.Kim YH, Verlander JW, Matthews SW, Kurtz I, Shin W, Weiner ID, Everett LA, Green ED, Nielsen S, Wall SM: Intercalated cell H+/OH- transporter expression is reduced in Slc26a4 null mice. Am J Physiol Renal Physiol 289: F1262–F1272, 2005 [DOI] [PubMed] [Google Scholar]
- 90.Verlander JW, Kim YH, Shin W, Pham TD, Hassell KA, Beierwaltes WH, Green ED, Everett L, Matthews SW, Wall SM: Dietary Cl(-) restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am J Physiol Renal Physiol 291: F833–F839, 2006 [DOI] [PubMed] [Google Scholar]
- 91.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW: NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: Role in Cl- conservation. Hypertension 44: 982–987, 2004 [DOI] [PubMed] [Google Scholar]
- 92.Sansom SC, Weinman EJ, O’Neil RG: Microelectrode assessment of chloride-conductive properties of cortical collecting duct. Am J Physiol 247: F291–F302, 1984 [DOI] [PubMed] [Google Scholar]
- 93.Gong Y, Yu M, Yang J, Gonzales E, Perez R, Hou M, Tripathi P, Hering-Smith KS, Hamm LL, Hou J: The Cap1-claudin-4 regulatory pathway is important for renal chloride reabsorption and blood pressure regulation. Proc Natl Acad Sci U S A 111: E3766–E3774, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kurtz TW, Al-Bander HA, Morris RC, Jr: “Salt-sensitive” essential hypertension in men. Is the sodium ion alone important? N Engl J Med 317: 1043–1048, 1987 [DOI] [PubMed] [Google Scholar]
- 95.Hafner P, Grimaldi R, Capuano P, Capasso G, Wagner CA: Pendrin in the mouse kidney is primarily regulated by Cl- excretion but also by systemic metabolic acidosis. Am J Physiol Cell Physiol 295: C1658–C1667, 2008 [DOI] [PubMed] [Google Scholar]
- 96.Pech V, Zheng W, Pham TD, Verlander JW, Wall SM: Angiotensin II activates H+-ATPase in type A intercalated cells. J Am Soc Nephrol 19: 84–91, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mohebbi N, Perna A, van der Wijst J, Becker HM, Capasso G, Wagner CA: Regulation of two renal chloride transporters, AE1 and pendrin, by electrolytes and aldosterone. PLoS ONE 8: e55286, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim YH, Pech V, Spencer KB, Beierwaltes WH, Everett LA, Green ED, Shin W, Verlander JW, Sutliff RL, Wall SM: Reduced ENaC protein abundance contributes to the lower blood pressure observed in pendrin-null mice. Am J Physiol Renal Physiol 293: F1314–F1324, 2007 [DOI] [PubMed] [Google Scholar]
- 99.Verlander JW, Hong S, Pech V, Bailey JL, Agazatian D, Matthews SW, Coffman TM, Le T, Inagami T, Whitehill FM, Weiner ID, Farley DB, Kim YH, Wall SM: Angiotensin II acts through the angiotensin 1a receptor to upregulate pendrin. Am J Physiol Renal Physiol 301: F1314–F1325, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, Walp E, Kim YH, Sutliff RL, Bao HF, Eaton DC, Wall SM: Pendrin modulates ENaC function by changing luminal HCO3-. J Am Soc Nephrol 21: 1928–1941, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW: Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol 284: F229–F241, 2003 [DOI] [PubMed] [Google Scholar]
- 102.Kim YH, Kwon TH, Frische S, Kim J, Tisher CC, Madsen KM, Nielsen S: Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am J Physiol Renal Physiol 283: F744–F754, 2002 [DOI] [PubMed] [Google Scholar]
- 103.Scott DA, Karniski LP: Human pendrin expressed in Xenopus laevis oocytes mediates chloride/formate exchange. Am J Physiol Cell Physiol 278: C207–C211, 2000 [DOI] [PubMed] [Google Scholar]
- 104.Azroyan A, Laghmani K, Crambert G, Mordasini D, Doucet A, Edwards A: Regulation of pendrin by pH: dependence on glycosylation. Biochem J 434: 61–72, 2011 [DOI] [PubMed] [Google Scholar]
- 105.Alesutan I, Daryadel A, Mohebbi N, Pelzl L, Leibrock C, Voelkl J, Bourgeois S, Dossena S, Nofziger C, Paulmichl M, Wagner CA, Lang F: Impact of bicarbonate, ammonium chloride, and acetazolamide on hepatic and renal SLC26A4 expression. Cell Physiol Biochem 28: 553–558, 2011 [DOI] [PubMed] [Google Scholar]
- 106.Kim YH, Pham TD, Zheng W, Hong S, Baylis C, Pech V, Beierwaltes WH, Farley DB, Braverman LE, Verlander JW, Wall SM: Role of pendrin in iodide balance: going with the flow. Am J Physiol Renal Physiol 297: F1069–F1079, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D: The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hentschke M, Hentschke S, Borgmeyer U, Hübner CA, Kurth I: The murine AE4 promoter predominantly drives type B intercalated cell specific transcription. Histochem Cell Biol 132: 405–412, 2009 [DOI] [PubMed] [Google Scholar]
- 109.Purkerson JM, Heintz EV, Nakamori A, Schwartz GJ: Insights into acidosis-induced regulation of SLC26A4 (pendrin) and SLC4A9 (AE4) transporters using three-dimensional morphometric analysis of β-intercalated cells. Am J Physiol Renal Physiol 307: F601–F611, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Verrey F, Hummler E, Schild L, Rossier B: Control of Na+ transport by aldosterone. In: The Kidney: Physiology and Pathophysiology, edited by Seldin DW, Giebisch G, Philadelphia, Williams & Wilkins, 2000, pp 1441–1471 [Google Scholar]
- 111.Karim Z, Szutkowska M, Vernimmen C, Bichara M: Renal handling of NH3/NH4+: Recent concepts. Nephron, Physiol 101: 77–81, 2005 [DOI] [PubMed] [Google Scholar]
- 112.Sebastian A, Sutton JM, Hulter HN, Schambelan M, Poler SM: Effect of mineralocorticoid replacement therapy on renal acid-base homeostasis in adrenalectomized patients. Kidney Int 18: 762–773, 1980 [DOI] [PubMed] [Google Scholar]
- 113.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE: Collecting duct principal cell transport processes and their regulation [published online ahead of print May 29, 2014]. Clin J Am Soc Nephrol 10.2215/CJN.05760513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Prieto-Carrasquero MC, Botros FT, Kobori H, Navar LG: Collecting duct renin: a major player in angiotensin II-dependent hypertension. J Am Soc Hypertens 3: 96–104, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Frindt G, Palmer LG: Low-conductance K channels in apical membrane of rat cortical collecting tubule. Am J Physiol 256: F143–F151, 1989 [DOI] [PubMed] [Google Scholar]
- 116.Schambelan M, Sebastian A, Katuna BA, Arteaga E: Adrenocortical hormone secretory response to chronic NH4Cl-induced metabolic acidosis. Am J Physiol 252: E454–E460, 1987 [DOI] [PubMed] [Google Scholar]
- 117.Wagner CA, Devuyst O, Bourgeois S, Mohebbi N: Regulated acid-base transport in the collecting duct. Pflugers Arch 458: 137–156, 2009 [DOI] [PubMed] [Google Scholar]
- 118.Estilo G, Liu W, Pastor-Soler N, Mitchell P, Carattino MD, Kleyman TR, Satlin LM: Effect of aldosterone on BK channel expression in mammalian cortical collecting duct. Am J Physiol Renal Physiol 295: F780–F788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rothenberger F, Velic A, Stehberger PA, Kovacikova J, Wagner CA: Angiotensin II stimulates vacuolar H+ -ATPase activity in renal acid-secretory intercalated cells from the outer medullary collecting duct. J Am Soc Nephrol 18: 2085–2093, 2007 [DOI] [PubMed] [Google Scholar]
- 120.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, Loffing J: In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: Differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 299: F1473–F1485, 2010 [DOI] [PubMed] [Google Scholar]
- 121.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH: 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998 [DOI] [PubMed] [Google Scholar]
- 122.Farman N, Rafestin-Oblin ME: Multiple aspects of mineralocorticoid selectivity. Am J Physiol Renal Physiol 280: F181–F192, 2001 [DOI] [PubMed] [Google Scholar]
- 123.Stanton BA: Regulation by adrenal corticosteroids of sodium and potassium transport in loop of Henle and distal tubule of rat kidney. J Clin Invest 78: 1612–1620, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD: Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417–1427, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Batenburg WW, Krop M, Garrelds IM, de Vries R, de Bruin RJ, Burcklé CA, Müller DN, Bader M, Nguyen G, Danser AH: Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor. J Hypertens 25: 2441–2453, 2007 [DOI] [PubMed] [Google Scholar]
- 126.Jan Danser AH, Batenburg WW, van Esch JH: Prorenin and the (pro)renin receptor—an update. Nephrol Dial Transplant 22: 1288–1292, 2007 [DOI] [PubMed] [Google Scholar]
- 127.Ludwig J, Kerscher S, Brandt U, Pfeiffer K, Getlawi F, Apps DK, Schägger H: Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem 273: 10939–10947, 1998 [DOI] [PubMed] [Google Scholar]
- 128.Yoshikawa A, Aizaki Y, Kusano K, Kishi F, Susumu T, Iida S, Ishiura S, Nishimura S, Shichiri M, Senbonmatsu T: The (pro)renin receptor is cleaved by ADAM19 in the Golgi leading to its secretion into extracellular space. Hypertens Res 34: 599–605, 2011 [DOI] [PubMed] [Google Scholar]
- 129.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, Nguyen G: Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension 53: 1077–1082, 2009 [DOI] [PubMed] [Google Scholar]
- 130.Ichihara A, Kaneshiro Y, Suzuki F: Prorenin receptor blockers: effects on cardiovascular complications of diabetes and hypertension. Expert Opin Investig Drugs 15: 1137–1139, 2006 [DOI] [PubMed] [Google Scholar]
- 131.Gonzalez AA, Lara LS, Luffman C, Seth DM, Prieto MC: Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension 57: 859–864, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, Zhang S, Cheng T, Inagami T, Ward K, Terreros DA, Lalouel JM: Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension 34: 1265–1274, 1999 [DOI] [PubMed] [Google Scholar]
- 133.Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, Connelly KA, Yuen D, Trogadis J, Herzenberg AM, Kuliszewski MA, Leong-Poi H, Gilbert RE: The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension 54: 261–269, 2009 [DOI] [PubMed] [Google Scholar]
- 134.Ichihara A, Sakoda M, Kurauchi-Mito A, Kaneshiro Y, Itoh H: Renin, prorenin and the kidney: a new chapter in an old saga. J Nephrol 22: 306–311, 2009 [PubMed] [Google Scholar]
- 135.Kaneshiro Y, Ichihara A, Takemitsu T, Sakoda M, Suzuki F, Nakagawa T, Hayashi M, Inagami T: Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int 70: 641–646, 2006 [DOI] [PubMed] [Google Scholar]
- 136.Deinum J, Tarnow L, van Gool JM, de Bruin RA, Derkx FH, Schalekamp MA, Parving HH: Plasma renin and prorenin and renin gene variation in patients with insulin-dependent diabetes mellitus and nephropathy. Nephrol Dial Transplant 14: 1904–1911, 1999 [DOI] [PubMed] [Google Scholar]
- 137.DuBose TD, Jr, Goode AK: Isolated renal tubular disorder: Mechanisms and clinical expression. In: Schrier's Diseases of the Kidney, edited by Coffman TM, Falk RJ, Molitoris B, Neilson EG, Schrier RW, Philadelphia, Wolters Kluwer Lippincott, Williams & Wilkins, 2007, pp 587-614. [Google Scholar]
- 138.Douglas JB, Healy JK: Nephrotoxic effects of amphotericin B, including renal tubular acidosis. Am J Med 46: 154–162, 1969 [DOI] [PubMed] [Google Scholar]
- 139.Sebastian A, McSherry E, Morris RC, Jr: Renal potassium wasting in renal tubular acidosis (RTA): Its occurrence in types 1 and 2 RTA despite sustained correction of systemic acidosis. J Clin Invest 50: 667–678, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Karet FE, Finberg KE, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Medina JF, Lifton RP: Localization of a gene for autosomal recessive distal renal tubular acidosis with normal hearing (rdRTA2) to 7q33-34. Am J Hum Genet 65: 1656–1665, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP: Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90, 1999 [DOI] [PubMed] [Google Scholar]
- 142.Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch AB, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE: Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 39: 796–803, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Karet FE, Gainza FJ, Györy AZ, Unwin RJ, Wrong O, Tanner MJ, Nayir A, Alpay H, Santos F, Hulton SA, Bakkaloglu A, Ozen S, Cunningham MJ, di Pietro A, Walker WG, Lifton RP: Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci U S A 95: 6337–6342, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, Unwin RJ, Wrong O, Tanner MJ: Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Invest 100: 1693–1707, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ribeiro ML, Alloisio N, Almeida H, Gomes C, Texier P, Lemos C, Mimoso G, Morlé L, Bey-Cabet F, Rudigoz RC, Delaunay J, Tamagnini G: Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood 96: 1602–1604, 2000 [PubMed] [Google Scholar]
- 146.Breton S, Alper SL, Gluck SL, Sly WS, Barker JE, Brown D: Depletion of intercalated cells from collecting ducts of carbonic anhydrase II-deficient (CAR2 null) mice. Am J Physiol 269: F761–F774, 1995 [DOI] [PubMed] [Google Scholar]
- 147.Sebastian A, McSherry E, Morris RC, Jr: Impaired renal conservation of sodium and chloride during sustained correction of systemic acidosis in patients with type 1, classic renal tubular acidosis. J Clin Invest 58: 454–469, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Finberg KE, Wagner CA, Bailey MA, Paunescu TG, Breton S, Brown D, Giebisch G, Geibel JP, Lifton RP: The B1-subunit of the H(+) ATPase is required for maximal urinary acidification. Proc Natl Acad Sci U S A 102: 13616–13621, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Guan Y, Zhang Y, Breyer RM, Fowler B, Davis L, Hébert RL, Breyer MD: Prostaglandin E2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J Clin Invest 102: 194–201, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hébert RL, Jacobson HR, Fredin D, Breyer MD: Evidence that separate PGE2 receptors modulate water and sodium transport in rabbit cortical collecting duct. Am J Physiol 265: F643–F650, 1993 [DOI] [PubMed] [Google Scholar]
- 151.Ando Y, Asano Y: Luminal prostaglandin E2 modulates sodium and water transport in rabbit cortical collecting ducts. Am J Physiol 268: F1093–F1101, 1995 [DOI] [PubMed] [Google Scholar]
- 152.Krupin T, Sly WS, Whyte MP, Dodgson SJ: Failure of acetazolamide to decrease intraocular pressure in patients with carbonic anhydrase II deficiency. Am J Ophthalmol 99: 396–399, 1985 [DOI] [PubMed] [Google Scholar]
- 153.Zietse R, Zoutendijk R, Hoorn EJ: Fluid, electrolyte and acid-base disorders associated with antibiotic therapy. Nat Rev Nephrol 5: 193–202, 2009 [DOI] [PubMed] [Google Scholar]
- 154.Pertovaara M, Korpela M, Kouri T, Pasternack A: The occurrence of renal involvement in primary Sjögren’s syndrome: A study of 78 patients. Rheumatology (Oxford) 38: 1113–1120, 1999 [DOI] [PubMed] [Google Scholar]
- 155.Ren H, Wang WM, Chen XN, Zhang W, Pan XX, Wang XL, Lin Y, Zhang S, Chen N: Renal involvement and followup of 130 patients with primary Sjögren’s syndrome. J Rheumatol 35: 278–284, 2008 [PubMed] [Google Scholar]
- 156.Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL: Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol 3: 264–271, 1992 [DOI] [PubMed] [Google Scholar]
- 157.Devuyst O, Lemaire M, Mohebbi N, Wagner CA: Autoantibodies against intercalated cells in Sjögren’s syndrome. Kidney Int 76: 229, 2009 [DOI] [PubMed] [Google Scholar]
- 158.DeFranco PE, Haragsim L, Schmitz PG, Bastani B: Absence of vacuolar H(+)-ATPase pump in the collecting duct of a patient with hypokalemic distal renal tubular acidosis and Sjögren’s syndrome. J Am Soc Nephrol 6: 295–301, 1995 [DOI] [PubMed] [Google Scholar]
- 159.Walsh S, Turner CM, Toye A, Wagner C, Jaeger P, Laing C, Unwin R: Immunohistochemical comparison of a case of inherited distal renal tubular acidosis (with a unique AE1 mutation) with an acquired case secondary to autoimmune disease. Nephrol Dial Transplant 22: 807–812, 2007 [DOI] [PubMed] [Google Scholar]
- 160.Trepiccione F, Christensen BM: Lithium-induced nephrogenic diabetes insipidus: new clinical and experimental findings. J Nephrol 23[Suppl 16]: S43–S48, 2010 [PubMed] [Google Scholar]
- 161.Grünfeld JP, Rossier BC: Lithium nephrotoxicity revisited. Nat Rev Nephrol 5: 270–276, 2009 [DOI] [PubMed] [Google Scholar]
- 162.Batlle D, Gaviria M, Grupp M, Arruda JA, Wynn J, Kurtzman NA: Distal nephron function in patients receiving chronic lithium therapy. Kidney Int 21: 477–485, 1982 [DOI] [PubMed] [Google Scholar]
- 163.Christensen BM, Marples D, Kim YH, Wang W, Frøkiaer J, Nielsen S: Changes in cellular composition of kidney collecting duct cells in rats with lithium-induced NDI. Am J Physiol Cell Physiol 286: C952–C964, 2004 [DOI] [PubMed] [Google Scholar]
- 164.Kishore BK, Ecelbarger CM: Lithium: A versatile tool for understanding renal physiology. Am J Physiol Renal Physiol 304: F1139–F1149, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kuure S, Popsueva A, Jakobson M, Sainio K, Sariola H: Glycogen synthase kinase-3 inactivation and stabilization of beta-catenin induce nephron differentiation in isolated mouse and rat kidney mesenchymes. J Am Soc Nephrol 18: 1130–1139, 2007 [DOI] [PubMed] [Google Scholar]
- 166.Pulkkinen K, Murugan S, Vainio S: Wnt signaling in kidney development and disease. Organogenesis 4: 55–59, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Park EY, Kim WY, Kim YM, Lee JH, Han KH, Weiner ID, Kim J: Proposed mechanism in the change of cellular composition in the outer medullary collecting duct during potassium homeostasis. Histol Histopathol 27: 1559–1577, 2012 [DOI] [PubMed] [Google Scholar]
- 168.Heering P, Ivens K, Aker S, Grabensee B: Distal tubular acidosis induced by FK506. Clin Transplant 12: 465–471, 1998 [PubMed] [Google Scholar]
- 169.Mohebbi N, Mihailova M, Wagner CA: The calcineurin inhibitor FK506 (tacrolimus) is associated with transient metabolic acidosis and altered expression of renal acid-base transport proteins. Am J Physiol Renal Physiol 297: F499–F509, 2009 [DOI] [PubMed] [Google Scholar]
- 170.Peng H, Vijayakumar S, Schiene-Fischer C, Li H, Purkerson JM, Malesevic M, Liebscher J, Al-Awqati Q, Schwartz GJ: Secreted cyclophilin A, a peptidylprolyl cis-trans isomerase, mediates matrix assembly of hensin, a protein implicated in epithelial differentiation. J Biol Chem 284: 6465–6475, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Korhonen TK, Virkola R, Holthöfer H: Localization of binding sites for purified Escherichia coli P fimbriae in the human kidney. Infect Immun 54: 328–332, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chassin C, Goujon JM, Darche S, du Merle L, Bens M, Cluzeaud F, Werts C, Ogier-Denis E, Le Bouguénec C, Buzoni-Gatel D, Vandewalle A: Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J Immunol 177: 4773–4784, 2006 [DOI] [PubMed] [Google Scholar]
- 173.Gluba A, Banach M, Hannam S, Mikhailidis DP, Sakowicz A, Rysz J: The role of Toll-like receptors in renal diseases. Nat Rev Nephrol 6: 224–235, 2010 [DOI] [PubMed] [Google Scholar]
- 174.Ali AS, Townes CL, Hall J, Pickard RS: Maintaining a sterile urinary tract: The role of antimicrobial peptides. J Urol 182: 21–28, 2009 [DOI] [PubMed] [Google Scholar]
- 175.Spencer JD, Schwaderer AL, Dirosario JD, McHugh KM, McGillivary G, Justice SS, Carpenter AR, Baker PB, Harder J, Hains DS: Ribonuclease 7 is a potent antimicrobial peptide within the human urinary tract. Kidney Int 80: 174–80, 2011. [DOI] [PubMed] [Google Scholar]
- 176.Lehmann J, Retz M, Harder J, Krams M, Kellner U, Hartmann J, Hohgräwe K, Raffenberg U, Gerber M, Loch T, Weichert-Jacobsen K, Stöckle M: Expression of human beta-defensins 1 and 2 in kidneys with chronic bacterial infection. BMC Infect Dis 2: 20, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P: Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 14: 2534–2543, 2003 [DOI] [PubMed] [Google Scholar]
- 178.Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J: Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest 115: 610–621, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Paragas N, Kulkarni R, Werth M, Schmidt-Ott KM, Forster C, Deng R, Zhang Q, Singer E, Klose AD, Shen TH, Francis KP, Ray S, Vijayakumar S, Seward S, Bovino ME, Xu K, Takabe Y, Amaral FE, Mohan S, Wax R, Corbin K, Sanna-Cherchi S, Mori K, Johnson L, Nickolas T, D’Agati V, Lin CS, Qiu A, Al-Awqati Q, Ratner AJ, Barasch J: α-Intercalated cells defend the urinary system from bacterial infection. J Clin Invest 124: 2963–2976, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Paragas N, Qiu A, Zhang Q, Samstein B, Deng SX, Schmidt-Ott KM, Viltard M, Yu W, Forster CS, Gong G, Liu Y, Kulkarni R, Mori K, Kalandadze A, Ratner AJ, Devarajan P, Landry DW, D’Agati V, Lin CS, Barasch J: The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 17: 216–222, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK: The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10: 1033–1043, 2002 [DOI] [PubMed] [Google Scholar]
- 182.Barasch J, Mori K: Cell biology: Iron thievery. Nature 432: 811–813, 2004 [DOI] [PubMed] [Google Scholar]
- 183.Miró-Julià C, Escoda-Ferran C, Carrasco E, Moeller JB, Vadekaer DF, Gao X, Paragas N, Oliver J, Holmskov U, Al-Awqati Q, Lozano F: Expression of the innate defense receptor S5D-SRCRB in the urogenital tract. Tissue Antigens 83: 273–285, 2014 [DOI] [PubMed] [Google Scholar]
- 184.Pastor-Soler NM, Hallows KR, Smolak C, Gong F, Brown D, Breton S: Alkaline pH- and cAMP-induced V-ATPase membrane accumulation is mediated by protein kinase A in epididymal clear cells. Am J Physiol Cell Physiol 294: C488–C494, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]