

Abstract
This study aimed to induce malignant transformation of endometriosis in Sprague–Dawley rats by hyperestrogenemia and type II diabetes and evaluate its similarity with human disease in biological features. Rats with surgically induced endometriosis were randomized into two groups: those treated with estradiol (5 mg/kg three times/week after surgery), streptozotocin (25 mg/kg, 1 month after surgery), and high carbohydrate-and-fat feed (Es group); and those treated with placebo saline and standard feed (control group). All rats were randomly killed 2, 4, or 8 months after surgery. The endometriosis lesions and the corresponding eutopic endometria were subjected to morphological evaluation, TUNEL, and immunohistochemical analysis for the expressions of proliferating cell nuclear antigen, phosphatase and tensin homolog, phosphorylated protein kinase B, and phosphorylated mammalian target of rapamycin proteins. In the Es group, three cases (6.0%) of endometriosis showed atypical hyperplasia accompanied by simple hyperplastic eutopic endometria, and two cases (4.0%) of endometriosis showed endometrioid carcinoma accompanied by atypical hyperplastic eutopic endometria. In the Es group, the activity of organelles and the expressions of proliferating cell nuclear antigen, phosphorylated protein kinase B, and phosphorylated mammalian target of rapamycin increased, and the level of phosphatase and tensin homolog and TUNEL positivity decreased progressively in the order of endometriosis, atypical endometriosis, and malignant endometriosis. The same tendency was found in the corresponding eutopic endometria. The induced malignant endometriosis showed similarities with human disease in the pathological process and histomorphological and molecular biological features. The method is feasible. The malignant transformations of endometriosis and eutopic endometria may have correlations and similarities, but the former may suffer a higher risk of canceration.
Keywords: Endometrioid adenocarcinoma, endometriosis, hyperestrogenemia, malignant transformation, type II diabetes
Endometriosis (EMs) is a chronic estrogen-dependent gynecological disorder, which is classically defined as the presence of endometrial glands and stroma outside the uterine cavity.1 The rate of malignant transformation of EMs (MTOE) is estimated to be 0.7–1.5%.2 The most common site is the ovary, with endometrioid and clear cell carcinoma most frequently found (endometriosis-associated ovarian carcinoma, EAOC). Compared to the primary ovarian cancer, the patients are younger, diagnosed in earlier stages, have lower grade lesions, and a better survival.3,4 The extra-ovarian MTOE can be widely seen in various locations that are parallel to the frequency and distribution of EMs, with the major pathological type being endometrioid adenocarcinoma.5 Malignant transformation of EMs has always been the research focus due to its intriguing relationship with the leading gynecologic malignancy, ovarian cancer. However, the difficulty in obtaining samples meeting the rigorous diagnostic criteria (demonstration of both cancerous and benign endometrial tissue in the tumor; histology of the neoplasm compatible with an endometrial origin; no other primary tumor sites found) and the absence of in vitro and in vivo models retard the research on this malignancy, leaving many critical problems to be solved, such as the molecular mechanisms and the evidence-based proof for the prevention and intervention timing.
Malignant transformation of EMs has been proposed to be associated with free iron and heme-induced oxidative stress, an aberrant inflammatory milieu, and an estrogen-rich, progesterone-poor hormonal environment.6 Unopposed exposure to endogenous or exogenous estrogen, along with progesterone resistance, is the most widely recognized risk factor of MTOE.7 The estrogens, especially E2, which is accumulated in EMs lesions through excessive synthesis and decreased degradation,8 have been shown to result in direct cell damage with increased mitotic activity, a higher likelihood of DNA errors and somatic mutations,9 and contribute greatly to the overgrowth and oncogenesis of EMs lesions.7,10 In particular, when unopposed estrogens and obesity were considered together, a higher risk of MTOE was found.11 The inactivation of tumor suppressor gene phosphatase and tensin homolog (PTEN) (located on 10q23.3) has been showed to be an early event in MTOE.6 Loss of heterozygosity at locus 10q23.3 and mutation of PTEN could result in its inactivation and the following activation of phosphatidylinositol 3-kinase (PI3K)–protein kinase B (AKT)–mammalian target of rapamycin (mTOR) signaling pathway, which can regulate cell stress response and cell cycle.12 The inactivation of PTEN caused by loss of heterozygosity occurs frequently in EMs, atypical EMs, and also MTOE, which might be a continuum between endometriosis and ovarian cancer.6 Moreover, somatic mutation of the PTEN gene is frequently found in ovarian endometrioid adenocarcinoma but rarely seen in the other pathological types.13 Therefore, PTEN may serve as a characteristic molecular alteration of MTOE into endometrioid carcinoma.
The similarity and correlation of MTOE and type I endometrial carcinoma is the new viewpoint in the pathogenesis of MTOE.14 Both EAOC (specific to endometrioid adenocarcinoma) and estrogen-dependent (type I) endometrial cancer share the same pathological procedure (from benign to atypical hyperplasia to malignancy) and carcinogenesis. Unopposed exposure to estrogen with progesterone resistance is the risk factor of both. Many of the same genes, such as β-catenin and PTEN, have been shown to be mutated in both diseases, suggesting a shared molecular pathogenesis.13 In addition, an association between type II diabetes (the independent risk factor of endometrial cancer) and MTOE is biologically plausible through perturbations in insulin, insulin-like growth factors, gonadotropin, and steroid hormone metabolism, which could affect cell proliferation.15 Hyperglycemia indirectly stimulates the expression of estrogen receptor so as to promote the stimulatory function of estrogens.16 Also hyperglycemia causes the reduction of tumor suppressor genes like p16 and provides a nutrient-rich microenvironment for rapidly dividing cancer cells.16,17 Moreover, hyperinsulinemia induces proliferative tissue abnormalities because insulin and the cross-activation of the insulin-like growth factor-I receptor family can stimulate DNA synthesis and cell proliferation.18
A rat model of surgically induced EMs involves auto-transplantation of biopsies of uterus in the abdomen,19 which is widely used in the research of EMs. The EMs lesions of rats bear clear similarities to those found in humans: the progress of ectopic growth, the response to steroids, the abnormal levels of cytokines in the site of EMs lesion and peritoneal fluid,20 and clinical presentations.21 In addition, the combination of high carbohydrate-and-fat feed (HCF-feed) and low-dose streptozotocin (STZ)-treated rat serves as an alternative animal model for type II diabetes replicating the natural history and metabolic characteristics of human disease.18 The mutagenic and cytotoxic effects of STZ are selective and confined to liver, kidney, and pancreas, and rare evidence could be found about its association with MTOE.22
Accordingly, we induced MTOE with hyperestrogenemia in a rat EMs and type II diabetes model and evaluated the similarity of this rat MTOE with human disease through detecting the histological appearance and biological behavior and the expressions of PTEN, phosphorylated (p-)AKT, and p-mTOR. This study might be a pioneer of establishing standardized animal models for this malignancy and offering new clues for research into the pathogenesis of MTOE.
Materials and Methods
Animals
Ninety adult female Sprague–Dawley rats (age, 8–12 weeks; weight, 250–300 g), were provided by the Experimental Animal Center of Shengjing Hospital of China Medical University (Shenyang, China). The animals were fed standard feed and housed in a controlled environment (22 ± 2°C) with 12:12 h light:dark cycles. This study was approved by the Ethics Committee of Medical Scientific Research and Technology, Shengjing Hospital of China Medical University (2013PS140K).
Surgical procedures
The rats were randomized into four groups: (i) the Es group (n = 55), autologous endometrium transplantation;19 (ii) the control group (n = 25), autologous endometrium transplantation; (iii) the negative control group (n = 5), autologous epiploon transplantation; and (iv) the blank control group (n = 5), “open–close” surgery. Details are given in Document S1.
Induction of MTOE
After surgery, the rats of the Es group were treated with 17β-estradiol (5 mg/kg, three times/week) i.p. and provided with HCF-feed (10% fat, 20% glucose, and 70% standard feed). One month after surgery, a single dose of STZ (25 mg/kg) was injected i.p. Three days later, fasting blood glucose measurement higher than 250 mg/dL was diagnosed as diabetes. The rats in the control, negative control, and blank control groups were treated with only vehicle saline and provided with standard feed. All the i.p. injections were carried out on the opposite side of the EMs foci to minimize direct simulation.
Sample collection
The rats were killed randomly by cervical dislocation 2 months (Es group, n = 5; control group, n = 6; negative control group, n = 5; blank control group, n = 5), 4 months (Es group, n = 20; control group, n = 7), and 8 months (Es group, n = 25; control group, n = 7) after the surgeries. Details are given in Document S1.
Histomorphological evaluation
For H&E staining, paraffin-embedded samples were sectioned at 5-μm thickness, stained with H&E, and examined under a light microscope. The EMs lesion was determined by the histological confirmation of glandular epithelial and stromal cells. For electron microscopic analysis, Epon 812 epoxy resin-embedded samples were cut into slices of 50-nm thickness, and then the histological ultrastructure of endometrial cells was observed with a transmission electron microscope. Details are given in Document S1.
Proliferation and apoptosis analysis
Cell proliferation was assessed by detection of proliferating cell nuclear antigen (PCNA; Keygen Biotech Company, Nanjing, China). Apoptosis was evaluated with a TUNEL detection kit (Keygen Biotech Company, Nanjing, China). Both detections were carried out following the manufacturer's instructions. The level of apoptosis was established by counting the number of positively stained cells (brown in color).
Immunohistochemistry
For details of the procedure, see Document S1. The reagents and dilution of the primary antibody were as follows: PTEN (1:100, Cell Signaling Technology, Danvers, MA, USA), p-AKT (1:50; Santa Cruz Biotechnology, St Cruz, CA, USA) and p-mTOR (1:50; Cell Signaling Technology). The secondary antibody and 3,3′-diaminobenzidine-tetrachloride kit were purchased from Zhongshan Goldenbridge Biotechnology (Beijing, China).
Results assessment
The assessment criteria for results of the proliferation analysis, apoptosis analysis, and immunohistochemistry were as follows. Five view fields under a high power lens of the microscope (400×) were randomly chosen to observe all the cells in the view fields. The H-score scoring method was used (details provided in Doc. S1).23
Statistical analysis
SPSS version 16.0 software (Armonk, NY, USA) was used. The weight, volume, and H-score value were described by mean ± SEM. The t-test for two independent samples was used to calculate the intergroup difference. The χ2-test and Fisher's exact probability test were adopted for comparative analysis of the count data. P < 0.05 was considered significantly different.
Results
Macroscopic analyses
Of the 90 rats, 10 (five from the Es group and five from the control group) died and were excluded from the experiment. Of these 10 rats, it was difficult to identify the lesions and the causes of death in four due to severe pelvic adherence; three died of severe intestinal obstruction, and the remaining three died 3 days after the injection of STZ. The EMs lesions were survived in all cases of the Es group and the control group. In most cases, EMs of the Es group grew with time, whereas the ones in the control group showed worsening atrophy with time. The average weights of the EMs lesions in the Es group 2, 4, and 8 months after surgery (424.6 ± 1.9, 453.9 ± 21.6, 523.6 ± 23.2 mg, respectively) were significantly greater than those of the control group in the corresponding months (402.2 ± 2.3, 298.9 ± 39.7, 220.0 ± 27.5 mg, respectively, P < 0.05; Table1). Compared with the volumes of the EMs lesions in the control group 2, 4, and 8 months after surgery (71.5 ± 6.8, 36.2 ± 6.9, 24.4 ± 7.1 mm3, respectively), those in the Es group were significantly greater in the corresponding months (88.7 ± 16.9, 210.8 ± 21.8, 394.8 ± 131.9 mm3, respectively, P < 0.01; Table1).
Table 1.
Mean weight, mean volume, and macroscopic features of lesions in two groups of rats with surgically induced endometriosis (EMs) treated with estradiol and streptozotocin and fed a high carbohydrate-and-fat diet (Es group) or treated with placebo saline and fed a standard diet (control group)
| n | Lesion weight, mg, mean ± SEM | Lesion volume, mm3, mean ± SEM | Lesion atrophy, n (%) | Visible angiogenesis, n (%) | Papilliform hyperplasia of cyst wall, n (%) | |
|---|---|---|---|---|---|---|
| Es group | 50 | 485.8 ± 15.3† | 290.6 ± 67.7‡ | 3 (6.0) | 28 (56.0) | 2 (4.0) |
| 2 months | 5 | 424.6 ± 1.9† | 88.7 ± 16.9‡ | 0 (0.0) | 3 (60.0) | 0 (0.0) |
| 4 months | 20 | 453.9 ± 21.6† | 210.8 ± 21.8‡ | 2 (10.0) | 11 (55.0) | 0 (0.0) |
| 8 months | 25 | 523.6 ± 23.2† | 394.8 ± 131.9‡ | 1 (4.0) | 14 (56.0) | 2 (8.0) |
| Control group | 20 | 326.7 ± 21.0 | 42.7 ± 5.9 | 10 (50.0)§ | 5 (25.0) | 0 (0.0) |
| 2 months | 6 | 402.2 ± 2.3 | 71.5 ± 6.8 | 0 (0.0) | 2 (33.3) | 0 (0.0) |
| 4 months | 7 | 298.9 ± 39.7 | 36.2 ± 6.9 | 4 (57.1)§ | 2 (28.6) | 0 (0.0) |
| 8 months | 7 | 220.0 ± 27.5 | 24.4 ± 7.1 | 6 (85.7)§ | 1 (14.3) | 0 (0.0) |
†Compared with the net weight of the EMs lesions in the control group, P < 0.05.
Compared with the volume of the EMs lesions in the control group, P < 0.01.
Compared with the atrophy rate of the EMs lesions in the Es group, P < 0.05.
Two cases of EMs in the Es group (8 months after surgery) were extraordinarily enlarged (Fig.1a). Grossly, the two were cystic, consisted of thickened capsule wall with papillary hyperplasia and sanguinopurulent fluid (H&E stain revealed malignancy). Endometriosis of the control group (Fig.1b) had the appearance of unilocular cystic structures filled with clear fluid. The uteri of the Es group showed severe edema, with no papillary hyperplasia, whereas uteri of the control group showed no obvious abnormality.
Figure 1.
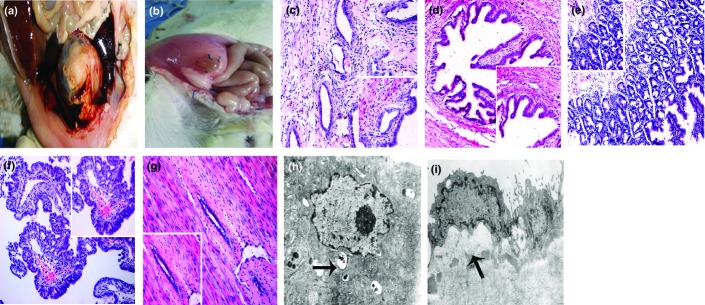
Rat model of malignant transformation in surgically induced endometriosis (EMs). (a) Distinctively enlarged EMs lesion. (b) Viable EMs lesions of the control group (placebo saline and standard feed). (c) EMs of the control group (100×; 400×). (d) EMs with simple hyperplasia showing that the epithelial cells were increased in number, enlarged in size, and distributed in multiple layers, without atypical nuclei (100×; 400×). (e) Atypical EMs showing that the number of endometrial glands was increased, multiple layers were arranged compactly, cytoplasm was reduced, and the cell nuclei were enlarged and moderately irregular (100×; 400×). (f) Malignant transformation of EMs (endometrioid adenocarcinoma): the number of endometrial glands was increased extraordinarily with a disordered arrangement of cells, infiltration of inflammatory cells, reduced cytoplasm, and condensed nuclei irregular in shape and size with the back-to-back phenomenon. There were also bleeding points (100×; 400×). (g) Atrophy EMs showed an apparent decrease of epithelial and stromal cells (100×; 400×). (h, i) Ultrastructure of EMs. (h) Malignant endometriotic cells showing vacuole changes of mitochondria (arrow, 5000×). (i) Simple hyperplastic endometriotic cells of the Es group, treated with estradiol (5 mg/kg three times/week after surgery), streptozotocin (25 mg/kg, 1 month after surgery), and high carbohydrate-and-fat feed, at month 8 showing clearly enlarged and well-defined nuclei with reduced cytoplasm (arrow, 6000×).
Histologic analyses
Histologically, the two distinctively enlarged EMs of the Es group (4.0%) were diagnosed as low-grade endometrioid adenocarcinoma, which showed a widespread cribriform arrangement with no invasion of the adjacent peritoneal muscular tissues. The corresponding eutopic endometria showed atypical hyperplasia. Three cases of EMs (6.0%) showed atypical hyperplasia with an increased number of endometrial glands, compactly arranged multiple layers, reduced cytoplasm, and enlarged and moderately irregular cell nuclei. The corresponding eutopic endometria showed simple hyperplasia. The malignant progression of the eutopic endometria was slower than that of the corresponding EMs, which was not synchronized. Endometriosis of the control group showed a similar appearance of the endometrium, consisting of cystically dilated glands with epithelium surrounded by stromal cells. Two cases of eutopic endometria showed diffuse squamous epithelium without columnar glandular epithelium (see Fig. S1). The results are shown in Table2 and Figure1c–g.
Table 2.
Histological features of lesions and eutopic endometria in rats with surgically induced endometriosis (EMs) treated with estradiol and streptozotocin and fed a high carbohydrate-and-fat diet (Es group) or treated with placebo saline and fed a standard diet (control group), n (%)
| n | EMs lesions |
Eutopic endometria† |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EMs | Atrophy | Simple hyperplasia | Atypical hyperplasia | Malignant transformation | Normal | Atrophy | Simple hyperplasia | Atypical hyperplasia | Malignant transformation | ||
| Es group | 50 | 13 (26.0) | 3 (6.0)‡ | 29 (58.0) | 3 (6.0) | 2 (4.0) | 25 (50.0) | 0 (0.0) | 21 (42.0) | 2 (4.0) | 0 (0.0) |
| 2 months | 5 | 3 (60.0) | 0 (0.0) | 2 (40.0) | 0 (0.0) | 0 (0.0) | 5 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 4 months | 20 | 5 (25.0) | 2 (10.0)‡ | 11 (55.0) | 2 (10.0) | 0 (0.0) | 11 (55.0) | 0 (0.0) | 9 (45.0) | 0 (0.0) | 0 (0.0) |
| 8 months | 25 | 5 (20.0) | 1 (4.0)‡ | 16 (64.0) | 1 (4.0) | 2 (8.0) | 9 (36.0) | 0 (0.0) | 12 (48.0) | 2 (8.0) | 0 (0.0) |
| Control group | 20 | 10 (50.0) | 10 (50.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 20 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 2 months | 6 | 6 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 6 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 4 months | 7 | 3 (42.9) | 4 (57.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 7 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| 8 months | 7 | 1 (14.3) | 6 (85.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 7 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
In the Es group, another two cases of eutopic endometria (8%) showed squamous epithelium at 8 months.
Compared with the atrophy rate of EMs of the control group, P < 0.05.
Transmission electron microscopy indicated that the malignant EMs showed remarkable abnormalities in cytomorphology and organelles including swollen mitochondria and vacuole changes (Fig.1h). In addition, compared to the simple hyperplastic EMs of month 4, the ectopic endometrial cells of month 8 with the same pathological change showed more active nuclei, and obviously enlarged and well-defined nuclei with reduced cytoplasm (Fig.1i). The ultrastructure of different pathological changes is shown in Figure S2.
Analysis of proliferation and apoptosis
The atrophy and simple hyperplastic EMs were excluded from the proliferation and apoptosis analysis. In the Es group, the immunostaining of PCNA protein increased (P < 0.01), and the level of TUNEL positivity decreased (P < 0.01) progressively in the order of EMs, atypical EMs, and malignant EMs. The immunostaining of PCNA in EMs of the Es group was stronger than that of the control group (P < 0.05), and the staining intensity of apoptosis in the Es group was lower than that of the control group (P < 0.05) (Fig.2).
Figure 2.
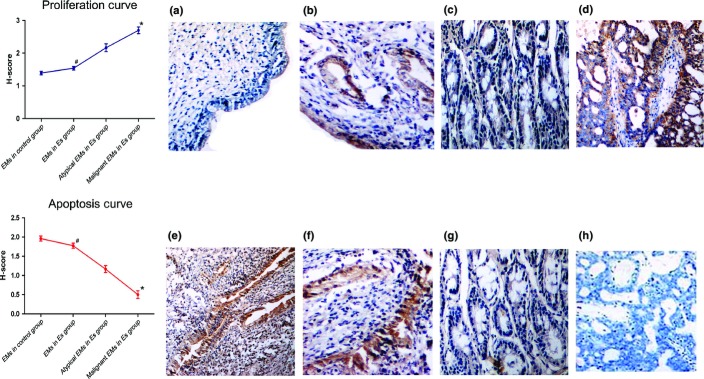
H-score value of apoptosis and proliferation of endometriosis (EMs) lesions in a rat model of malignant transformation, expressed as mean ± SEM. *Compared with EMs and atypical EMs of the Es group (treated with estradiol [5 mg/kg three times/week after surgery], streptozotocin [25 mg/kg, 1 month after surgery], and high carbohydrate-and-fat feed), P < 0.01. #Compared to EMs of the control group (placebo saline and standard feed), P < 0.05. (a, e) EMs in the control group (400×); (b, f) EMs in the Es group (400×); (c, g) atypical EMs in the Es group (400×); (d, h) malignant EMs in the Es group (400×).
Immunohistochemical analysis
The samples with simple hyperplasia and atrophy were excluded. The staining of PTEN was weakened, and stainings of p-AKT and p-mTOR were intensified progressively in the order of EMs, atypical EMs, and malignant EMs (P < 0.05). The samples with the same pathological changes showed no significant difference in the abovementioned stainings, regardless of the treatment group or whether the samples were eutopic or ectopic (P > 0.05). The differences in the H-score for PTEN, p-AKT, and p-mTOR among groups are shown in Figure3 and Table3.
Figure 3.

Immunostaining of phosphatase and tensin homolog (PTEN) in benign (a), atypical (b), and malignant endometriosis (EMs) (c) (400×) in a rat model. Immunostaining of phosphorylated protein kinase B (p-AKT) in benign (e), atypical (f) and malignant EMs (g) (400×). Immunostaining of phosphorylated mammalian target of rapamycin (p-mTOR) in benign (i), atypical (j), and malignant EMs (k) (400×). H-score values of PTEN (d), p-AKT (h), and p-mTOR (l) immunostaining in EMs, atypical EMs, and malignant EMs.
Table 3.
Statistical comparison of the expressions of phosphatase and tensin homolog (PTEN), phosphorylated protein kinase B (p-AKT), and phosphorylated mammalian target of rapamycin (p-mTOR) protein by an immunohistochemical method, P-values
| PTEN | p-AKT | p-mTOR | |
|---|---|---|---|
| Es group versus control group | |||
| EMs | 0.1030 | 0.7900 | 0.8730 |
| Normal eutopic endometria | 0.4800 | 0.7600 | 0.8320 |
| Comparison within the Es group | |||
| EMs versus eutopic endometria | |||
| EMs versus normal eutopic endometria | 0.7540 | 0.7730 | 0.5920 |
| Atypical EMs versus atypical endometria | 0.9840 | 0.8970 | 0.7910 |
| Ectopic endometria | |||
| EMs versus atypical EMs | 0.0210* | 0.0260* | 0.0001** |
| Atypical EMs versus malignant EMs | 0.0230* | 0.0280* | 0.0320* |
| EMs versus malignant EMs | 0.0001** | 0.0001** | 0.0001** |
| Eutopic endometria | |||
| Normal endometria versus atypical hyperplasia | 0.0050** | 0.0310* | 0.0220* |
| Comparison within the control group | |||
| EMs versus eutopic endometria | 0.6410 | 0.8630 | 0.8960 |
P < 0.05, statistical difference;
P < 0.01, significant statistical difference. Control group, rats with surgically induced endometriosis (EMs) treated with placebo saline and fed a standard diet; Es group, rats with surgically induced EMs treated with estradiol and streptozotocin and fed a high carbohydrate-and-fat diet.
Discussion
The present study explored a feasible way of inducing malignant transformation of rat EMs in combination with hyperestrogenemia and type II diabetes. The results indicated that three cases of ectopic endometria had atypical hyperplasia (two cases in 4 months and one case in 8 months), and two cases had endometrioid adenocarcinoma (both in 8 months), which were similar to the pathological appearances of human disease. The process of pathological changes, from benign to atypical and then to malignant change, is consistent with that of the human disease, especially the occurrence of atypical EMs (3/50). Ogawa et al.24 found atypical EMs in 78% EAOC, and it can act as a premalignant lesion. Atypical EMs was also reported to have spatial and chronological association with MTOE.25,26 It may represent the transition from benign EMs to malignant carcinoma. In the Es group, most EMs lesions grew along with time, the ectopic endometrial cells had rigorous hyperplasia, and the organelles were active. In the control group without intervention, atrophy worsened along with time, indicating estrogen dependency and the significant role of hyperestrogenism in this study. In our previous study, we injected once only with estradiol, without STZ and HCF-feed. After 8 months, only 9 of 14 cases of EMs presented with simple hyperplasia, with no malignant or atypical cases (unpublished results, 2014). This illustrated the indispensible role of type II diabetes in this study, and the combination of both treatments was more effective and feasible. Although STZ and estradiol were both give i.p., special attention was paid during the surgery and post-surgery treatment to assure that both the eutopic and ectopic endometrial side was not directly exposed to the peritoneal cavity and carcinogenetic stimulations. So the route of administration was not supposed to take an influential part in the hyperplastic progression of disease. Rare reports have been published regarding the correlation between STZ with MTOE, but indeed the possibility cannot be ruled out that STZ might be an unknown interference, which may be worthy of further investigation.
The inactivation of the PTEN gene and the following activation of the PI3K–AKT–mTOR pathway were reported to be associated with MTOE.27 Silencing PTEN could lead to the abnormal activation of PI3K and the phosphorylation of AKT, a serine/threonine kinase involved in cell growth and survival. Activated AKT (or p-AKT) could further activate mTOR by phosphorylation, which could regulate cell stress response and cell cycle.12 As PTEN mutations are seen early in endometrioid ovarian carcinoma and endometrioid ovarian cancer is thought to arise from EMs, it has been proposed that somatic genetic alterations in the PTEN gene may be an early event in MTOE.6 The ovarian endometrioid adenocarcinoma with an activated PI3K–AKT–mTOR signaling pathway is more likely to be of a low grade instead of a high grade.28 In our present research, PTEN expression was progressively reduced in the order of EMs, atypical EMs, and malignant EMs. The most remarkable abnormality was seen in malignant cases, with conspicuously decreased PTEN and increased p-AKT and p-mTOR. These results implied a possible role of the loss of PTEN and the following activation of the PI3K–AKT–mTOR pathway in the development of the present rat model of MTOE and a shared molecular alternation of the present rat model of MTOE with human low-grade endometrioid adenocarcinoma.
Recently, there is growing evidence pointing to hereditary predisposition and biological characteristics of the eutopic endometrium as the determinative factors in the pathogenesis of MTOE.29 Vigano et al.30 proposed that the malignant transformation of eutopic and ectopic endometria shared similar incidence and pathological type, and that abnormal gene expressions in the eutopic endometrium could be related to MTOE. Clinically, it was reported that patients with ovarian tumor arising from EMs suffered higher risks of endometrial pathology (endometrial adenocarcinoma, hyperplasia, and polyps).14 Studies have also shown that women with EAOC will also have a simultaneous endometrial adenocarcinoma, and almost 90% of synchronous tumors identified in the ovary and in the endometrium were of the endometrioid cell type.31,32 In the present study, we found that the pathological grade of the eutopic endometria corresponding with malignant EMs was higher than that with atypical EMs. The former was atypical hyperplasia, whereas the latter was simple hyperplasia. It is assumed that rats with malignant EMs might have abnormal gene expressions in their eutopic endometria. Such endometrium and its EMs cysts may be more sensitive to carcinogenic stimulations and thus more susceptible to malignant transformation. Moreover, the EMs lesions and the corresponding eutopic endometria in the Es group showed the same progressively reduced tendency with regard to PTEN, p-AKT, and p-mTOR expressions (no malignant case in the eutopic ones). But in a certain pathological grade of either eutopic or ectopic endometria, the expressions of the above proteins showed no statistical differences. These results contributed to imply the similarity and correlation between malignant transformation of EMs and endometrium.
However, as two independent disorders, MTOE and endometrial carcinoma must have many distinctions. Malignant transformation of EMs commonly occurs at reproductive age and seems to be associated with oxidative stress induced by free iron and heme accumulated during periodic and repeated bleeding, and inflammation.6 In contrast, type I endometrial carcinoma often occurs in premenopausal and younger postmenopausal women, is strongly associated with the estrogen-related pathway, and arises in association with unopposed estrogen stimulation.33 In the present study, we found that the progression of hyperplasia of EMs was more advanced than the corresponding eutopic endometria, indicating a higher risk of malignization of EMs. This phenomenon may be attributed to the specific local microenvironment and enhanced sensitivity to carcinogenic risk factors of EMs. Studies on humans and rats both proposed that there were more activated inflammatory cells and cytokines in the site of EMs than the corresponding eutopic endometria,34,35 indicating an inflammatory microenvironment of EMs. Inflammatory cells and their secreted cytokines can stimulate angiogenesis and cell proliferation, inhibit cell apoptosis, facilitate invasion and metastasis, and produce active oxygen capable of inducing DNA damage and mutation.36 So inflammation can prompt growth and invasion of EMs, which constitutes a link between EMs and ovarian cancer.2 In addition, the electron microscopy of the malignant endometriotic cells in our study revealed the presence of mitochondrial swelling and vacuole changes, which reflected that the EMs lesions might grow in a hypoxic microenvironment and implied a possible role of the hypoxic damage to mitochondria in the relatively higher malignant transformation risk of the endometriotic lesions. Apart from these, ectopic survival and growth might induce a higher sensitivity to various simulations.37,38 Wu et al.38 found that ectopic endometrium was at least 100 times more sensitive to interleukin-1β, compared with eutopic endometrium. Therefore, the hypersensitive EMs lesions might exaggerate the carcinogenic simulations and trigger the cascade chain reaction of oncogenesis.
In summary, the present study found that the combination of hyperestrogenemia with type II diabetes in a rat EMs model can induce MTOE, which showed similar morphological changes and molecular abnormalities to human disease. The method of induction is easy and feasible, and provides a good foundation for establishing an animal model of MTOE. Although eutopic endometrium and EMs showed similar responses and pathological changes to the carcinogenic simulations, EMs had higher susceptibility and risk for oncogenesis. Therefore, malignant transformation of EMs and eutopic endometrium may share hereditary correlations, which are worthy of further investigation. Future studies might also focus on raising the malignant transformation rate by improving the induction method and exploring the genetic molecular mechanism of MTOE in more depth.
Acknowledgments
We thank Ji Lian from the Experimental Animal Center and Chang Xiao-ying from the Department of Pathology (both Shengjing Hospital, China Medical University, Shenyang, China) for their technical assistance.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Hematoxylin–eosin staining of eutopic endometria with squamous epithelium in a rat model of surgically induced endometriosis.
Fig. S2. Ultrastructure of different pathological changes in a rat model of surgically induced endometriosis.
Doc S1. Methods of surgery, electron microscopic analysis, immunohistochemistry, and the H-score.
References
- Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364:1789–99. doi: 10.1016/S0140-6736(04)17403-5. [DOI] [PubMed] [Google Scholar]
- Ness RB. Endometriosis and ovarian cancer: thoughts on shared pathophysiology. Am J Obstet Gynecol. 2003;189:280–94. doi: 10.1067/mob.2003.408. [DOI] [PubMed] [Google Scholar]
- Erzen M, Rakar S, Klancnik B, Syrjanen K. Endometriosis-associated ovarian carcinoma (EAOC): an entity distinct from other ovarian carcinomas as suggested by a nested case–control study. Gynecol Oncol. 2001;83:100–8. doi: 10.1006/gyno.2001.6382. [DOI] [PubMed] [Google Scholar]
- Modesitt SC, Tortolero-Luna G, Robinson JB, Gershenson DM, Wolf JK. Ovarian and extraovarian endometriosis-associated cancer. Obstet Gynecol. 2002;100:788–95. doi: 10.1016/s0029-7844(02)02149-x. [DOI] [PubMed] [Google Scholar]
- Benoit L, Arnould L, Cheynel N, et al. Malignant extraovarian endometriosis: a review. Eur J Surg Oncol. 2006;32:6–11. doi: 10.1016/j.ejso.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Munksgaard PS, Blaakaer J. The association between endometriosis and ovarian cancer: a review of histological, genetic and molecular alterations. Gynecol Oncol. 2002;124:164–9. doi: 10.1016/j.ygyno.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Oxholm D, Knudsen UB, Kryger-Baggesen N, Ravn P. Postmenopausal endometriosis. Acta Obstet Gynecol Scand. 2007;4:1–7. doi: 10.1080/00016340701619407. [DOI] [PubMed] [Google Scholar]
- Zeitoun K, Takayama K, Sasano H, et al. Deficient 17beta-hydroxysteroid dehydrogenase type 2 expression in endometriosis: failure to metabolize 17beta-estradiol. J Clin Endocrinol Metab. 1998;83:4474–80. doi: 10.1210/jcem.83.12.5301. [DOI] [PubMed] [Google Scholar]
- Turbiner J, Moreno-Bueno G, Dahiya S, et al. Clinicopathological and molecular analysis of endometrial carcinoma associated with tamoxifen. Mod Pathol. 2008;21:925–36. doi: 10.1038/modpathol.2008.49. [DOI] [PubMed] [Google Scholar]
- Heaps JM, Nieberg RK, Berek JS. Malignant neoplasms arising in endometriosis. Obstet Gynecol. 1990;75:1023–8. [PubMed] [Google Scholar]
- Zanetta GM, Webb MJ, Li H, Keeney GL. Hyperestrogenism: a relevant risk factor for the development of cancer from endometriosis. Gynecol Oncol. 2000;79:18–22. doi: 10.1006/gyno.2000.5905. [DOI] [PubMed] [Google Scholar]
- Masaki M, Yamaguchi K, Matsumura N, Tsukasa B, Ikuo K. Ovarian cancer in endometriosis: molecular biology, pathology, and clinical management. Int J Clin Oncol. 2009;14:383–91. doi: 10.1007/s10147-009-0935-y. [DOI] [PubMed] [Google Scholar]
- Kurman RJ, Shih I-M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer. Shifting the Paradigm. Hum Pathol. 2011;42:918–31. doi: 10.1016/j.humpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gorp GT, Amant F, Neven P, Vergote I, Moerman P. Endometriosis and the development of malignant tumours of the pelvis. A review of literature. Best Pract Res Clin Obstet Gynaecol. 2004;18:349–71. doi: 10.1016/j.bpobgyn.2003.03.001. [DOI] [PubMed] [Google Scholar]
- Soliman PT, et al. Association between adiponectin, insulin resistance, and endometrial cancer. Cancer. 2006;106:2376–81. doi: 10.1002/cncr.21866. [DOI] [PubMed] [Google Scholar]
- AI-Jarrah M, Matalka I, Aseri HA, et al. Exercise training prevents endometrial hyperplasia and biomarkers for endometrial cancer in rat model of type I diabetes. J Clin Med Res. 2010;2:207–14. doi: 10.4021/jocmr444e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaafar DK, Zaitone SA, Moustafa YM. Role of metformin in suppressing 1,2-dimethylhydrazine-induced colon cancer in diabetic and non-diabetic mice: effect on tumor angiogenesis and cell proliferation. PLoS ONE. 2014;9:e100562. doi: 10.1371/journal.pone.0100562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon MW, Wilson EA. Studies on the surgical induction of endometriosis in the rat. Fertil Steril. 1985;44:684–94. [PubMed] [Google Scholar]
- Sharpe-Timms KL. Using rats as a research model for the study of endometriosis. Ann N Y Acad Sci. 2002;955:318–27. doi: 10.1111/j.1749-6632.2002.tb02792.x. [DOI] [PubMed] [Google Scholar]
- Cason A, Samuelsen C, Berkley K. Estrous changes in vaginal nociception in a rat model of endometriosis. Horm Behav. 2003;44:123–31. doi: 10.1016/s0018-506x(03)00121-1. [DOI] [PubMed] [Google Scholar]
- Vinerean HV, Gazda LS, Hall RD, Smith BH. Streptozotocin is responsible for the induction and progression of renal tumorigenesis in diabetic wistar-furth rats treated with insulin or transplanted with agarose encapsulated porcine islets. Islets. 2011;3:196–203. doi: 10.4161/isl.3.4.16129. [DOI] [PubMed] [Google Scholar]
- Peng Y, Biliang C, Yanhong H, Xiaoyan X. Long-term regression of experimental endometriosis in a rat model treated with local application of levonorgestrel-loaded biodegradable microspheres. Hum Reprod. 2012;27:2089–95. doi: 10.1093/humrep/des142. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Kaku T, Amada S, et al. Ovarian endometriosis associated with ovarian carcinoma: a clinicopathological and immunohistochemical study. Gynecol Oncol. 2000;77:298–304. doi: 10.1006/gyno.2000.5765. [DOI] [PubMed] [Google Scholar]
- LaGrenade A, Silverberg SG. Ovarian tumors associated with atypical endometriosis. Hum Pathol. 1988;19:1080–4. doi: 10.1016/s0046-8177(88)80090-x. [DOI] [PubMed] [Google Scholar]
- Moll UM, Chumas JC, Chalas E, Mann WJ. Ovarian carcinoma arising in atypical endometriosis. Obstet Gynecol. 1990;75:537–9. [PubMed] [Google Scholar]
- Obata K, Morland SJ, Watson RH, et al. Frequent PTEN/MMAC1 mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998;58:2095–7. [PubMed] [Google Scholar]
- Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/b-Catenin and PI3K/PTEN signaling pathways. Cancer Cell. 2007;11:321–33. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Bulun SE. Endometriosis. N Engl J Med. 2009;360:268–79. doi: 10.1056/NEJMra0804690. [DOI] [PubMed] [Google Scholar]
- Vigano P, Somigliana E, Parazzini F, Vercellini P. Bias versus causality: interpreting recent evidence of association between endometriosis and ovarian cancer. Fertil Steril. 2007;88:588–93. doi: 10.1016/j.fertnstert.2006.11.180. [DOI] [PubMed] [Google Scholar]
- Falkenberry SS, Steinhoff MM, Gordinier M, Rappoport S, Gajewski W, Granai CO. Synchronous endometrioid tumors of the ovary and endometrium. A clinicopathologic study of 22 cases. J Reprod Med. 1996;41:713–8. [PubMed] [Google Scholar]
- Zaino R, Whitney C, Brady MF, De Geest K, Burger RA, Buller RE. Simultaneously detected endometrial and ovarian carcinomas-a prospective clinicopathologic study of 74 cases: a gynecologic oncology group study. Gynecol Oncol. 2001;83:355–62. doi: 10.1006/gyno.2001.6400. [DOI] [PubMed] [Google Scholar]
- Takahashi-Shiga N, Utsunomiya H, Miki Y, et al. Local biosynthesis of estrogen in human endometrial carcinoma through tumor-stromal cell interactions. Clin Cancer Res. 2009;15:6028–34. doi: 10.1158/1078-0432.CCR-09-1013. [DOI] [PubMed] [Google Scholar]
- Machado DE, Berardo PT, Palmero CY, Nasciutti LE. Higher expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 (Flk-1) and metalloproteinase-9 (MMP-9) in a rat model of peritoneal endometriosis is similar to cancer diseases. J Exp Clin Cancer Res. 2010;29:4. doi: 10.1186/1756-9966-29-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater M, Cooper M, Murphy CR. Human growth hormone and interleukin-6 are upregulated in endometriosis and endometrioid adenocarcinoma. Acta Histochem. 2006;108:13–8. doi: 10.1016/j.acthis.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Matsuzaki S, Murakami T, Uehara S, Canis M, Sasano H, Okamura K. Expression of estrogen receptor alpha and beta in peritoneal and ovarian endometriosis. Fertil Steril. 2001;75:1198–205. doi: 10.1016/s0015-0282(01)01783-6. [DOI] [PubMed] [Google Scholar]
- Wu MH, Wang CA, Lin CC, Chen LC, Chang WC, Tsai SJ. Distinct regulation of cyclooxygenase-2 by interleukin-1beta in normal and endometriotic stromal cells. J Clin Endocrinol Metab. 2005;90:286–95. doi: 10.1210/jc.2004-1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Hematoxylin–eosin staining of eutopic endometria with squamous epithelium in a rat model of surgically induced endometriosis.
Fig. S2. Ultrastructure of different pathological changes in a rat model of surgically induced endometriosis.
Doc S1. Methods of surgery, electron microscopic analysis, immunohistochemistry, and the H-score.