

Abstract
Breast cancer is a leading cause of cancer-related death in women. Prolonged exposure to the ovarian hormones estrogen and progesterone increases the risk of breast cancer. Although estrogen is known as a primary factor in mammary carcinogenesis, very few studies have investigated the role of progesterone. Receptor activator for nuclear factor-κB (NF-κB) ligand (RANKL) plays an important role in progesterone-induced mammary carcinogenesis. However, the molecular mechanism underlying RANKL-induced mammary carcinogenesis remains unknown. In our current study, we show that RANKL induces glioma-associated oncogene homolog 1 (GLI-1) in estrogen-induced progesterone-mediated mammary carcinogenesis. In vivo experiments were carried out using ACI rats and in vitro experiments were carried out in MCF-7 cells. In ACI rats, mifepristone significantly reduced the incidence of mammary tumors. Likewise, mifepristone also inhibited the proliferation of MCF-7 cells. Hormone treatments induced RANKL, receptor activator of NF-κB (RANK), and NF-κB in a protein kinase B-dependent manner and inhibited apoptosis by activation of anti-apoptotic protein Bcl2 in mammary tumors and MCF-7 cells. Mechanistic studies in MCF-7 cells reveal that RANKL induced upstream stimulatory factor-1 and NF-κB, resulting in subsequent activation of their downstream target GLI-1. We have identified that progesterone mediates estrogen-induced mammary carcinogenesis through activation of GLI-1 in a RANKL-dependent manner.
Keywords: Estradiol, GLI-1, mifepristone, progesterone, RANKL
The ovarian hormones estrogen and progesterone regulate the development of the mammary gland and play a vital role in the normal functioning of breast tissue.1,2 Estrogen and progesterone also regulate various biological processes including reproduction, metabolism, cell proliferation, differentiation, apoptosis, and inflammation.3 Breast neoplastic growth is expected to increase with exposure to long-term hormone replacement therapy (HRT).4,5 Estrogen alone or estrogen combined with medroxyprogesterone acetate increases breast epithelial cell proliferation in postmenopausal women.6 Most epidemiological and experimental studies seem to implicate estrogen as the primary factor in breast cancer etiology.7,8
The Women's Health Initiative studies and the Million Women Study clearly indicated that postmenopausal women who took progestin in combination with estrogen as part of HRT experienced a greater risk of breast cancer than women who took estrogen alone.9,10 Shull et al.11 showed that the long-term exogenous administration of estradiol alone is not capable of inducing mammary cancers in ovariectomized ACI rats. Earlier, we showed that administration of both progesterone and estrogen is required to induce mammary tumors in ovariectomized ACI rats.12 Although the exact underlying mechanism is not known, the evidence suggests that the ovarian hormone progesterone plays an important role in breast cancer.
Primary breast cancer cells and cell lines express receptor activator for nuclear factor-κB (NF-κB) ligand (RANKL) and its receptor, receptor activator for NF-κB (RANK).13 RANKL is essential for the differentiation, proliferation, and migration of cancer cells.13 An earlier study showed that RANKL/RANK is essential for progestin-driven breast cancer in mice.14 Similarly, a RANKL inhibitor effectively inhibited progestin-induced mammary carcinogenesis.15 Despite these advances, the precise molecular circuitry downstream of RANKL/RANK is not completely defined. RANKL/RANK signaling activates the transcription factor NF-κB and upregulates cyclin D1, a target of NF-κB.16 Thus, RANKL appears to mediate its effects through the activation of NF-κB.17 The role of upstream stimulatory factor-1 (USF-1) and USF-2 has been implicated in RANKL-induced osteoclast differentiation;18 USF-1 and-2 were initially identified as regulators of glucose homeostasis.19 Furthermore, USF-1 has also been reported to activate glioma-associated oncogene homolog 1 (GLI-1) by Twist, but this requires binding of USF-1 to E-box homeo domain in rhabdomyosarcomas.20
We carried out in vivo and in vitro experiments to determine whether progesterone promotes mammary carcinogenesis through activation of GLI-1 by RANKL. Here, we report that the progesterone-mediated activation of RANKL enhanced cell proliferation through the increased expression of GLI-1 by NF-κB/USF-1. Further, RANKL activation also inhibited apoptosis by increasing the expression of the anti-apoptotic protein Bcl2.
Materials and Methods
Cell lines and culture conditions
MCF-7 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in phenol red-free DMEM-F12 medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% charcoal-stripped FBS (Equitech-Bio, Kerrville, TX, USA), and penicillin and streptomycin (Life Technologies, Grand Island, NY, USA). We cultured the cells in a humidified incubator at 37°C and 5% CO2.
Hormone treatments
MCF-7 cells were cultured in phenol red-free DMEM-F12 containing 10% FBS and allowed to attach overnight. Next, we maintained the cells in phenol red-free DMEM with 0.2% charcoal-stripped FBS for 24 h. After starving the cells, we treated them with 10 nM estradiol, 10 μM progesterone, 10 nM estradiol + 10 μM progesterone, and 10 nM estradiol + 10 μM mifepristone (Sigma Chemical Co., St. Louis, MO, USA) for 24 h.
Cell proliferation assay
We plated MCF-7 cells (30 000 per well) in 24-well plates, serum starved them for 24 h, and treated as described above. Following the treatment, we assessed cell proliferation using the MTS assay (Promega, Madison, WI, USA) and phenazine methosulfate (Sigma Chemical Co.). We read the plates at 492 nm using the automated VICTOR X Plate Reader (PerkinElmer, Waltham, MA, USA).
Immunofluorescence assay
MCF-7 cells were grown on coverslips and serum starved for 24 h. We treated the cells as described above. Following the hormone treatment, we fixed the cells with 4% paraformaldehyde in PBS for 15 min at room temperature and permeabilized with 0.2% (V/V) Triton X-100 in PBS for 5 min at room temperature. Blocking was done using 3% BSA in PBS with 0.5% Tween-20 for 30 min at 25°C. We incubated the cells with primary RANKL (Epitomics, Burlingame, CA, USA) and RANK antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), then detected the proteins with secondary antibody coupled with fluorophores (Molecular Probes, Eugene, OR, USA). Proteins were visualized by fluorescence microscopy (C2 Si; Nikon, Tokyo, Japan).
Overexpression and silencing of RANKL
MCF-7 cells were transfected with a GFP-tagged ORF clone of TNFSF11 transcript variant 1 cloned into a pCMV6-AC-GFP vector (Fig. S1a) (Origene, Rockville, MD, USA). Plasmid DNA was transfected into MCF-7 cells using Lipofectamine 2000 reagent (Life Technologies). G418 (300 μg/mL; Life Technologies) selection was used. For silencing RANKL, we used four unique 29-mer shRNA constructs in pRFP-C-RS-vectors (Fig. S1b) (Origene). We tested each 29-mer shRNA construct for silencing efficiency and chose the vector with the highest silencing efficiency.
Transcription factor assay
Transcription factor DNA-binding activities were measured using Transcription Factor Activation Profiling Plate Array II (Signosis, Sunnyvale, CA, USA) in nuclear extracts prepared from RANKL overexpressing cells using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific, Rockford, IL, USA). The assay was performed according to the manufacturer's protocol and measured as relative light units on a microplate luminometer (CLARIOstar; BMG, Cary, NC, USA).
Gaussia luciferase assay
The Gaussia luciferase activity for GLI-1 promoter was measured using a Secrete-Pair Gaussia luciferase assay kit (GeneCopoeia, Rockville, MD, USA). The effect of Gluc activity was measured using the media after 72 h of transfection.
Apoptosis detection assay
Apoptosis was detected by evaluating DNA fragmentation by terminal deoxynucleotidyl transferase-mediated TUNEL assay using a DeadEnd Fluorimetric TUNEL assay kit (Promega). Briefly, MCF-7 (1 × 105) cells were plated on coverslip in a 6-well plate and serum starved for overnight. Hormones, estradiol (10 nm), progesterone (10 μM), and mifepristone (10 μM) were treated for 24 h as indicated. The cells were then fixed by incubating with 4% methanol-free paraformaldehyde and permeabilized by using 0.2% Triton X-100. Then TUNEL assay mix containing FITC-labeled dUTP and terminal deoxynucleotidyl transferase was added on top of the cells, incubated for 1 h, and then washed. The reaction was inhibited by incubating the cells with 2× SSC buffer. The coverslips were mounted with mounting media containing DAPI and examined under A Nikon confocal microscope and the images were analyzed using NIS elements software (Nikon Instruments, Melville, NY, USA).
Animals
We obtained female 4-week-old ACI rats from Harlan Sprague-Dawley (San Diego, CA, USA). We housed the rats in a temperature-controlled room with a 12:12 h light:dark schedule. Rats received food (Teklad, Madison, WI, USA) and water ad libitum. All procedures were approved by the Texas Tech University Health Sciences Center Institutional Animal Care and Use Committee guidelines.
Experimental design
Ovary intact ACI rats were divided into five groups (n = 30 per group): control; 30 mg estradiol; 30 mg progesterone; 30 mg estradiol plus 30 mg progesterone; 30 mg estradiol plus 30 mg mifepristone (progesterone antagonist). We have earlier shown that these treatments increase the levels of respective hormone in circulation.21 Hormone treatments were initiated at 6 weeks of age. Ovariectomized rats were not used for this study. All hormones were packed in individual silastic capsules (0.198 cm ID × 0.318 cm OD, 2 cm in length; Dow Corning). The control animals received empty silastic capsules. The silastic capsules were implanted dorsally, s.c. We replaced the capsules every 2 months. We killed a subset of (n = 6) rats at 4 weeks and the remaining 24 rats were killed at 36 weeks after the initiation of treatments. Mammary tumors were surgically excised when they reached a diameter of 1.5 cm. We snap-froze the mammary tumors in liquid nitrogen and stored them at −80°C for molecular analysis.
Wholemount analysis of mammary glands
The mammary glands were fixed in 10% phosphate-buffered formalin for 24 h. Mammary glands were defatted in acetone for 2 days and transferred to 70% ethanol before staining with iron hematoxylin. Subsequently, mammary glands were dehydrated in a series of alcohols and placed in Histo-Clear (National Diagnostics, Atlanta, GA, USA) until microscopic examination.
Western blot analysis
Western blots were done using the mammary tumors and MCF-7 cells treated with hormones. The expression of phosphorylated protein kinase B (pAKT), AKT, Bcl2, P65, P50, cyclin D1 (Cell Signaling Technology, Danvers, MA, USA), RANK (Santa Cruz Biotechnology, Santa Cruz, CA, USA), RANKL, USF-1 (Epitomics), GLI-1 (Abcam, Cambridge, MA, USA), and β-actin (Sigma-Aldrich) were analyzed with an enhanced chemiluminescence kit (Pierce, Rockford, IL, USA) using the Fuji digital imaging system (Fuji Medical Systems USA, Irving, TX, USA).
Immunohistochemistry
We carried out anti-rat RANKL, RANK, and cyclin D1 immunohistochemistry on rat mammary tumor sections. We also examined the expression of RANKL, USF-1, GLI-1, and estrogen receptor-α (ER-α) in human breast cancer and adjacent normal tissue that were obtained from our University Breast Care Center. All studies were approved by our Institutional Review Board's guidelines. We visualized RANKL, RANK, and cyclin D1 expression in rat mammary tumors by staining them with DAB chromogen and human tissue with permanent red chromogen.
Statistical analysis
Statistical significance of tumor incidence in rats was analyzed by Fisher's exact test; the influence of hormonal treatment on all other parameters studied was determined by Student's t-test. Results are reported as mean ± SE. Statistical significance was determined at P < 0.05.
Results
Progesterone in combination with estradiol promotes growth of mammary glands and mammary tumors
Remarkable lobulo-alveolar growth of the mammary gland was observed in rats that received estradiol plus progesterone. Estradiol treatment alone was also effective in increasing mammary growth, whereas progesterone induced branching of the mammary gland (Fig.1a). The development of lobulo-alveolar growth of the mammary gland is marked by increased hyperplasia in estradiol, progesterone, and combined estradiol plus progesterone treatment. Treatment with mifepristone drastically inhibited estradiol-induced mammary growth (Fig.1a). Mifepristone treatment reduced mammary gland branching and significantly fewer lobulo-alveolar structures compared with estradiol alone or estradiol plus progesterone treatment.
Figure 1.

Hormonal carcinogenesis. (a) ACI rat mammary gland wholemounts after 4 weeks of hormone treatment (10x magnification). (b) Mammary cancer incidence among rats treated with estradiol (E), progesterone (P), or mifepristone (M) alone or in combination. (c) Mammary cancer latency.
The increase in lobulo-alveolar growth due to hormone treatment is reflected in tumor incidence and tumor burden following hormone treatment. ACI rats treated with estradiol (95%), progesterone (55%), or both estradiol and progesterone (100%) developed a high incidence of mammary tumors (Fig.1b). In contrast, only 3% of ACI rats that received mifepristone along with estradiol developed mammary tumors (Fig.1b). Long-term treatment with ovarian hormones not only increased mammary cancer incidence, but also increased the mammary tumor burden. The ACI rats treated with estradiol, progesterone, or estradiol and progesterone had mammary tumor burdens of 5.9 ± 1, 2.8 ± 0.6, and 6.4 ± 1.1, respectively (Fig.1c). In contrast, the ACI rats treated with estradiol and mifepristone had a tumor burden of 1 ± 0.
Progesterone antagonist inhibits mammary cancer and normal mammary epithelial cell proliferation
We measured cell proliferation using the MTS assay.22 Estradiol alone, progesterone alone, and the combination of estradiol and progesterone increased the proliferation of MCF-7 cells by 23.6%, 21.8%, and 35.2%, respectively (Fig.2a). Similarly, estradiol alone, progesterone alone, and the combination of estradiol and progesterone increased the proliferation of rat primary mammary epithelial cells by 61%, 57%, and 69.7%, respectively (Fig.2b). The combination of estradiol and mifepristone inhibited the proliferation of MCF-7 cells and rat primary mammary-epithelial cells by 17.2% and 16.3%, respectively, when compared with the estradiol alone treated group.
Figure 2.
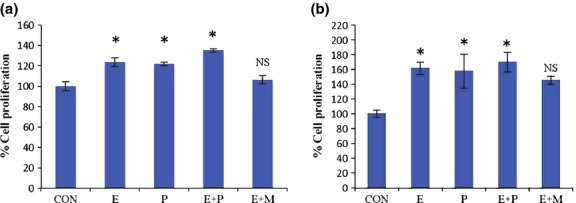
Effects of hormones on cell proliferation. Cell proliferation of (a) MCF-7 cells (b) rat primary epithelial cells treated with estradiol (E), progesterone (P), or mifepristone (M) alone or in combination for 24 h. Data represent the mean ± SEM (n = 6). *P < 0.05 when compared with control (CON). NS, non-significant when compared with control.
Progesterone mediates its effect through activation of RANKL and RANK
We assessed the effects of hormone treatment on RANKL/RANK expression in MCF-7 cells and in ACI rat mammary tumors. Estradiol and progesterone, both separately and in combination, increased RANKL and RANK expression in MCF-7 cells (Figs3a,S2a,b) and mammary tumors (Fig.3b). Mifepristone inhibited RANKL and RANK expression in MCF-7 cells (Fig.3a) and mammary tumors (Figs3b,S2c,d). Furthermore, immunofluorescence in MCF-7 cells (Fig.4a) and immunohistochemical staining in mammary tumors (Fig.4b) confirmed that combined treatment with estradiol and progesterone resulted in a synergistic increase in RANKL/RANK expression compared with either single hormone treatment. Almost 60% of the human breast tumors were estrogen and progesterone receptor positive and of luminal A subtype. We had a small portion of breast tumors that overexpressed human epidermal growth factor receptor-2 (∼25%) and the rest were basal type (∼15%). Immunohistochemical examination of 53 human primary tumors revealed that 86.4% of tumors expressed higher levels of RANKL in cancerous tissue than in normal ducts (Fig.5). RANKL was mainly expressed in the cell surface and cytoplasm.
Figure 3.
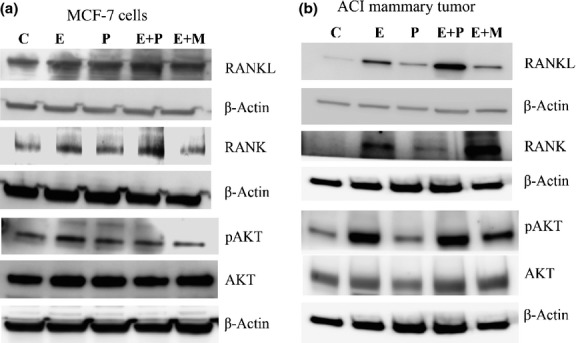
Western blot analysis of receptor activator for nuclear factor-κB ligand (RANKL)/receptor activator for nuclear factor-κB (RANK) signaling in MCF-7 cells and ACI mammary tumors. (a) Expression of RANKL, RANK, phosphorylated protein kinase B (pAKT) and AKT in MCF-7 cells treated with estradiol (E), progesterone (P), or mifepristone (M) alone or in combination for 24 h. (b) Expression of RANKL, RANK, pAKT, and AKT in mammary tumors treated with hormones for 36 weeks.
Figure 4.
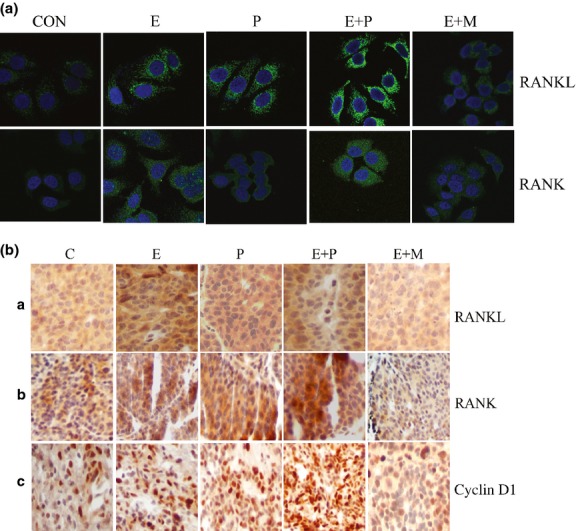
Hormone treatment increases receptor activator for nuclear factor-κB ligand (RANKL) signaling. (a) Activation of RANKL/receptor activator for nuclear factor-κB (RANK) following estradiol (E), progesterone (P), or mifepristone (M) alone or in combination treatment in MCF-7 cells was elucidated using immunofluorescence. CON, control (Magnification 600X). (b) Immunohistochemistry showing the expression of RANKL, RANK, and cyclin D1 in mammary tumors treated with hormones. C, control (Magnification 200X).
Figure 5.
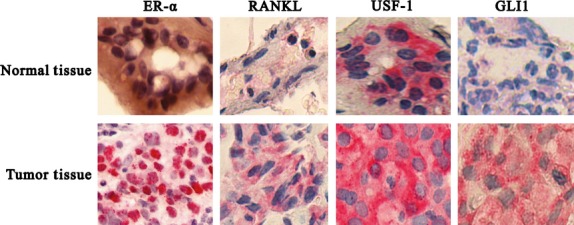
Expression of receptor activator for nuclear factor-κB ligand (RANKL), upstream stimulatory factor-1 (USF-1), glioma-associated oncogene homolog 1 (GLI-1), and estrogen receptor-α (ER-α) in ACI rat normal and tumor tissue. (a) Immunohistochemistry showing the expression of ER-α, RANKL, USF-1, and GLI-1 in human normal and breast cancer tissue (Magnification 1000X).
Receptor activator for nuclear factor-κB ligand activates AKT and NF-κB
Ovarian hormone treatments increased AKT phosphorylation in MCF-7 cells and in ACI rat mammary tumors (Fig.3). Mifepristone completely abrogated the AKT phosphorylation and inhibited cell proliferation both in vitro and in vivo (Figs3,S2e,f). RANKL overexpression in MCF-7 cells also increased phosphorylation of AKT (Figs6a,S3a). Next, we tested the respective abilities of four unique 29-mer shRNAs and a scrambled 29-mer control shRNA to silence RANKL in MCF-7 cells. We selected the shRNA that was the most effective in silencing RANKL (Fig. S4). We found that silencing RANKL with the shRNA attenuated the phosphorylation of AKT (Figs6b,S3b). Because AKT supports cell survival through the activation of NF-κB,23,24 we tested whether the progesterone-mediated activation of RANKL results in the activation of NF-κB. All the hormone treatments resulted in the increased expression levels of NF-κB subunits p50 and p65, both in MCF-7 cells and in rat mammary tumors (Figs7,S4d,e,S5d,e). In contrast, mifepristone significantly downregulated the expression of NF-κB subunits p50 and p65, both in MCF-7 cells and in rat mammary tumors (Figs7,S5a–d). Likewise, overexpression of RANKL increased the expression of NF-κB subunits p50 and p65 (Figs6a,S3c,d); silencing suppressed the activation of the NF-κB subunits (Figs6b,S3e,f).
Figure 6.

Receptor activator for nuclear factor-κB (NF-κB) ligand (RANKL) signaling activates NF-κB and glioma-associated oncogene homolog 1 (GLI-1). (a) RANKL overexpression in MCF-7 cells activates NF-κB subunits p65, p50, and protein kinase B (AKT) compared to control (C). (b) RANKL silencing in MCF-7 cells results in inactivation of NF-κB subunits and AKT compared to scramble (Scr). (c) RANKL overexpression activates cyclin D1, Bcl2, upstream stimulatory factor-1 (USF-1), and GLI-1 in MCF-7 cells. (d) Expression of cyclin D1, Bcl2, USF-1, and GLI-1 in RANKL-silenced MCF-7 cells (e) RANKL overexpression in MCF-7 cells increases GLI-1 promoter activity. Graphs show luciferase (Luc) activity, mean ± SD of two different experiments. CON, control.
Figure 7.
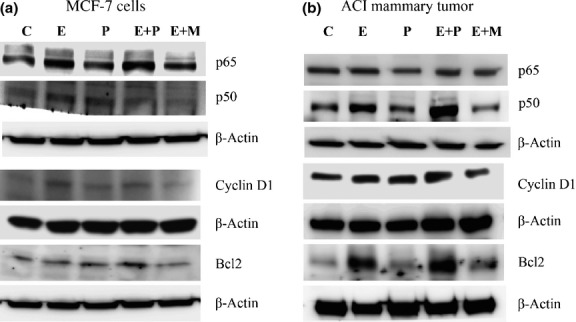
Hormone treatment increases nuclear factor-κB (NF-κB) signaling. (a) Western blots indicate the activation of NF-κB subunits p65, p50, cyclin D1, and Bcl2 in mammary tumors treated with estradiol (E), progesterone (P), or mifepristone (M) alone or in combination. (b) Hormone treatment activates NF-κB subunits p65, p50, cyclin D1, and Bcl2 in MCF-7 cells.
Progesterone induces cyclin D1 and inhibits apoptosis by activation of Bcl2
We found that hormone treatments increased the expression of cyclin D1 in MCF-7 cells and also in mammary tumors (Figs7,S5e,f); mifepristone downregulated the expression of cyclin D1 in MCF-7 cells and mammary tumors, even in the presence of estradiol (Figs7,S5e,f). TUNEL assay also indicated that estradiol and progesterone treatment alone or in combination decreased apoptosis compared to the control MCF-7 cells. In contrast, treatment with mifepristone increased apoptosis (Fig. S7). Immunohistochemistry of mammary tumors revealed that the hormone treatments caused the overexpression of cyclin D1 (Fig.4b). Indeed, RANKL overexpression in MCF-7 cells also increased cyclin D1 expression (Figs6c,S6a). Conversely, silencing RANKL in MCF-7 cells decreased cyclin D1 expression (Figs6d,S6b), revealing that RANKL is essential for optimal cyclin D1 induction, which is in agreement with earlier studies demonstrating a link between RANK signaling and cyclin D1.25
We found that the hormone treatments resulted in the increased expression of Bcl2 in MCF-7 cells and mammary tumors (Fig.7), whereas treatment with mifepristone along with estradiol decreased the expression of Bcl2. Furthermore, the overexpression of RANKL activated Bcl2 (Figs6c,S6e); the silencing of RANKL significantly suppressed Bcl2 (Figs6d,S6f). Taken together, our data suggests that progesterone affects mammary carcinogenesis by altering mammary cell proliferation and apoptosis through RANKL/RANK signaling.
Overexpression of RANKL induces GLI-1
Transcription factor array analysis revealed that RANKL overexpressing cells induces USF-1 and GLI-1 compared to control (Fig. S8). This result was further confirmed by Western blot analysis (Figs6c,S9a,b), suggesting that RANKL induces GLI-1 and USF-1. To illustrate the significance of RANKL in GLI-1 activation, we checked the expression of USF-1 and GLI-1 in RANKL silenced cells. As expected, RANKL silencing diminished USF-1 and its downstream target GLI-1 (Figs6d,S9c,d). We further confirmed the RANKL mediated the effect by using GLI-1 promoter; RANKL overexpressing cells increased GLI-1 promoter activity (Fig.6e). Immunohistochemistry of human primary tumors revealed the increased expression of ER-α, USF-1, and GLI-1 (Fig.5) compared to normal tissue. Analysis of human primary tumors revealed that 59.6% were positive for ER-α, 82.6% were positive for USF-1, and 33.9% were positive for GLI-1 (Table1). The normal breast ductal epithelial cells had a low positivity for ER-α compared to breast cancers. The receptor staining was mainly seen in the nucleus. The majority of staining for USF-1 and GLI-1 was in the cytoplasm.
Table 1.
Percentage positivity of upstream stimulatory factor-1 (USF-1), glioma-associated oncogene homolog 1 (GLI-1), and estrogen receptor-α (ER-α) in human primary breast tumor
| Genes | Positivity in breast cancer tissue, % | Gene co-expression with ERα, % |
|---|---|---|
| ER-α | 59.6 | – |
| USF-1 | 82.6 | 90.3 |
| GLI-1 | 33.9 | 48.3 |
–, not applicable.
Discussion
Hormone replacement therapy eases the symptoms of menopause. Estrogen only is mainly prescribed to women who have undergone hysterectomy; estrogen and progestin together are prescribed to women who have not undergone hysterectomy.26 Multiple studies report that HRT involving estrogen and progesterone increases the risk of breast cancer compared with HRT involving estrogen alone.27–29 An understanding of the mechanism by which progesterone promotes estrogen-induced mammary carcinogenesis will aid in the identification of better HRT supplementation therapy with adjuvants to prevent the development of hormone-induced breast cancer.
Our results support the hypothesis that estrogen and progesterone work together to influence mammary development and carcinogenesis. Our observation that mifepristone, a progesterone antagonist, abrogated the increased ductal branching and lobulo-alveologenesis caused by exogenous estrogen is in agreement with previous findings,11,12 and suggests that progesterone is involved in estrogen-induced mammary growth. The results of the present study support our previous findings regarding the role of progesterone in driving estrogen-induced mammary carcinogenesis in the ACI rat model.12 This is in agreement with the work of others who showed that estrogen alone is not capable of inducing mammary carcinogenesis in ovariectomized rats.11 Thus, in vivo and in vitro evidence indicates that progesterone may be the principal mediator of the protumorigenic effects of estrogen in the mammary gland.
We show that combined estradiol/progesterone treatment clearly has a synergistic effect on RANKL/RANK expression. RANKL is implicated in the survival and migration of multiple cell types, including breast cancer cells.30 Others have shown that the inhibition of RANKL actively inhibits the proliferation of mammary epithelial cells and reduces mammary tumor multiplicity.15,16 Our results illustrate that progesterone is a key regulator of mammary RANKL/RANK expression. The present study also indicates that RANKL mediates its effect in an AKT-dependent manner. Earlier studies showed that RANKL increases survival through the AKT pathway,23 illustrating that RANKL is crucial for the induction of cell survival. Using MCF-7 cells that overexpressed RANKL and MCF-7 cells in which RANKL was silenced, we confirmed the finding that AKT phosphorylation is regulated by RANKL. Compared with control cells, the cells overexpressing RANKL displayed a significant increase in the phosphorylation of AKT, which coincided with the increased expression of cyclin D1, Bcl2, and active NF-κB. The converse was true in the RANKL silenced cells. Together, these findings clearly indicate that progesterone mediates estrogen-induced effects through the activation of RANKL in an AKT-dependent manner.
The RelA/p65 transactivation subunit of NF-κB is activated by RANKL/AKT.24,31,32 The hormone-induced increase in the expression of the p50 and p65 subunits of NF-κB was observed both in vitro and in vivo. The reduction in p50 and p65 expression caused by mifepristone suggests that NF-κB is activated downstream of RANKL/AKT signaling in response to progesterone. Our results show that progesterone and RANKL/AKT signaling have a positive impact on the expression of the NF-κB target cyclin D1. Our results agree with previous reports that RANKL mediates its effects, in part, through NF-κB activation and the subsequent upregulation of cyclin D1.16,33 Taken together with previous findings, our results suggest that progesterone mediates its effect through the activation of cyclin D1. In addition to the upregulation of proteins like cyclin D1 that positively regulate cell-cycle entry, another method by which progesterone may enhance cell proliferation is through the upregulation of proteins like Bcl2, which supports cell survival through the inhibition of apoptosis.34,35 The activation of AKT supports the survival of MCF-7 cells by inhibiting apoptosis,36 partly through the increased expression of Bcl2.37 We hypothesize that the in vitro and in vivo increases in the expression of Bcl2 in response to exogenous hormones in our study were caused by increases in AKT phosphorylation. RANKL signaling leads to the increased activation of AKT and NF-κB, resulting in the increased expression of cyclin D1 and Bcl2. The activation of these pathways favors tumorigenesis by causing the overexpression of proteins that favor progression and inhibit apoptosis.
Although NF-κB signaling is the well characterized downstream target for RANKL, our present work revealed that RANKL may regulate GLI-1 through USF-1. In our current study, transcription factor profiling shows increased activity of USF-1 and GLI-1 transcription factors in cells overexpressing RANKL. It has been reported that USF-1 and-2 are involved in RANKL-induced osteoblast differentiation.18 Furthermore, USF-1 has been shown to be involved in activation of GLI-1, by binding to the E-box homeo domain,20 in agreement with our finding that RANKL-overexpressing cells increased expression of USF-1 and GLI-1. Silencing of RANKL abolished USF-1 and thereby significantly reduced GLI-1 expression. Promoter analysis revealed the activation of GLI-1 promoter activity in RANKL-overexpressing cells, suggesting that activation of GLI-1 is due to RANKL. Based on earlier reports involving the role of USF-1 in induction of GLI-1 activity,20 the observed increase in promoter activity of GLI-1 in RANKL overexpressing cells might have occurred through USF-1. As RANKL has been shown to activate NF-κB,17 and NF-κB has also been reported to activate the hedgehog signaling pathway,38 the observed activation of RANKL might have induced GLI-1 through increased activity of USF-1 or in part through the increased activity of NF-κB. Human primary breast tumor analysis revealed that all RANKL-expressing tumors are positive for USF-1 and GLI-1, suggesting the activation of USF-1 in GLI-1 is inevitable. Even though the significance of GLI-1 regulation by RANKL has been established by silencing studies, further studies are required to complete our knowledge on RANKL-induced activation of GLI-1.
Based on the findings presented here, we propose that progesterone mediates mammary carcinogenesis by enhancing GLI-1 through USF-1/NF-κB in a RANKL-dependent manner and it also inhibits apoptosis in an AKT-dependent manner through RANKL activation. Determining the exact mechanism by which progesterone mediates its effects on GLI-1 through RANKL signaling remains a challenge and will require further experiments specifically designed to study the increased activity of GLI-1 promoter.
Acknowledgments
We would like to thank Texas Tech University Health Sciences Center – Paul L. Foster School of Medicine for funding this study.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Vector map and insert sequences.
Fig. S2. Densitometric analysis of receptor activator for nuclear factor-κB (RANK), receptor activator for nuclear factor-κB ligand (RANKL), and phosphorylated protein kinase B pAKT immunoblots in ACI tumor and MCF-7 breast cancer cells.
Fig. S3. Densitometric analysis of immunoblots of MCF-7 cells overexpressing receptor activator for nuclear factor-κB ligand (RANKL) and depleted RANKL.
Fig. S4. Knockdown efficiency of receptor activator for nuclear factor-κB ligand (RANKL) shRNA.
Fig. S5. Densitometric analysis of immunoblots of nuclear factor-κB (NF-κB) signaling in MCF-7 breast cancer cells.
Fig. S6. Densitometric analysis of immunoblots of cyclin D1 and Bcl2 in MCF-7 cells overexpressing receptor activator for nuclear factor-κB ligand (RANKL) and depleted RANKL and ACI tumor.
Fig. S7. Apoptosis analysis by TUNEL assay.
Fig. S8. Upstream stimulatory factor-1 (USF-1) and glioma-associated oncogene homolog 1 (GLI-1) expression in MCF-7 cells overexpressing receptor activator for nuclear factor-κB ligand (RANKL).
Fig. S9. Densitometric analysis of immunoblots of upstream stimulatory factor-1 (USF-1) and glioma-associated oncogene homolog 1 (GLI-1).
References
- Nandi S. Endocrine control of mammarygland development and function in the C3H/he crgl mouse. J Natl Cancer Inst. 1958;21:1039–63. [PubMed] [Google Scholar]
- Haslam SZ, Shyamala G. Effect of oestradiol on progesterone receptors in normal mammary glands and its relationship with lactation. Biochem J. 1979;182(1):127–31. doi: 10.1042/bj1820127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–76. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- Magnusson C, Baron JA, Correia N, Bergstrom R, Adami HO, Persson I. Breast-cancer risk following long-term oestrogen-and oestrogen-progestin-replacement therapy. Int J Cancer. 1999;81:339–44. doi: 10.1002/(sici)1097-0215(19990505)81:3<339::aid-ijc5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Kenemans P, Bosman A. Breast cancer and post-menopausal hormone therapy. Best Pract Res Clin Endocrinol Metab. 2003;17(1):123–37. doi: 10.1016/s1521-690x(02)00084-2. [DOI] [PubMed] [Google Scholar]
- Hofseth LJ, Raafat AM, Osuch JR, Pathak DR, Slomski CA, Haslam SZ. Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J Clin Endocrinol Metab. 1999;84:4559–65. doi: 10.1210/jcem.84.12.6194. [DOI] [PubMed] [Google Scholar]
- Harris JR, Lippman ME, Veronesi U, Willett W. Breast cancer (1) N Engl J Med. 1992;327(5):319–28. doi: 10.1056/NEJM199207303270505. [DOI] [PubMed] [Google Scholar]
- Travis RC, Key TJ. Oestrogen exposure and breast cancer risk. Breast Cancer Res. 2003;5(5):239–47. doi: 10.1186/bcr628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women's health initiative randomized controlled trial. JAMA. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Beral V. Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the million women study. Lancet. 2003;362(9382):419–27. doi: 10.1016/s0140-6736(03)14065-2. [DOI] [PubMed] [Google Scholar]
- Shull JD, Spady TJ, Snyder MC, Johansson SL, Pennington KL. Ovary-intact, but not ovariectomized female ACI rats treated with 17beta-estradiol rapidly develop mammary carcinoma. Carcinogenesis. 1997;18(8):1595–601. doi: 10.1093/carcin/18.8.1595. [DOI] [PubMed] [Google Scholar]
- Blank EW, Wong PY, Lakshmanaswamy R, Guzman R, Nandi S. Both ovarian hormones estrogen and progesterone are necessary for hormonal mammary carcinogenesis in ovariectomized ACI rats. Proc Natl Acad Sci U S A. 2008;105(9):3527–32. doi: 10.1073/pnas.0710535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DH, Nakashima T, Sanchez OH, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440(7084):692–6. doi: 10.1038/nature04524. [DOI] [PubMed] [Google Scholar]
- Schramek D, Leibbrandt A, Sigl V, et al. Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. 2010;468(7320):98–102. doi: 10.1038/nature09387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez E, Jacob AP, Jones J, et al. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature. 2010;468(7320):103–7. doi: 10.1038/nature09495. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Suarez E. RANKL inhibition: a promising novel strategy for breast cancer treatment. Clin Transl Oncol. 2011;13:222–8. doi: 10.1007/s12094-011-0646-5. [DOI] [PubMed] [Google Scholar]
- Hauer J, Puschner S, Ramakrishnan P, et al. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci U S A. 2005;102(8):2874–9. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Shi Z, Silveira A, et al. Involvement of upstream stimulatory factors 1 and 2 in RANKL-induced transcription of tartrate-resistant acid phosphatase gene during osteoclast differentiation. J Biol Chem. 2003;278:20603–11. doi: 10.1074/jbc.M212093200. [DOI] [PubMed] [Google Scholar]
- Vallet VS, Casado M, Henrion AA, et al. Differential roles of upstream stimulatory factors 1 and 2 in the transcriptional response of liver genes to glucose. J Biol Chem. 1998;273:20175–9. doi: 10.1074/jbc.273.32.20175. [DOI] [PubMed] [Google Scholar]
- Villavicencio EH, Yoon JW, Frank DJ, Fuchtbauer EM, Walterhouse DO, Iannaccone PM. Cooperative E-box regulation of human GLI1 by TWIST and USF. Genesis. 2002;32(4):247–58. doi: 10.1002/gene.10078. [DOI] [PubMed] [Google Scholar]
- Rajkumar L, Guzman RC, Yang J, et al. Short-term exposure to pregnancy levels of estrogens prevents mammary carcinogenesis. Proc Natl Acad Sci USA. 2001;98:11755–9. doi: 10.1073/pnas.201393798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg K, Zhai L, Chen M, Kharazmi A, Owen TC. The use of a water-soluble formazan complex to quantitate the cell number and mitochondrial function of leishmania major promastigotes. Parasitol Res. 1994;80(3):235–9. doi: 10.1007/BF00932680. [DOI] [PubMed] [Google Scholar]
- Kim HH, Shin HS, Kwak HJ, et al. RANKL regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. FASEB J. 2003;17:2163–5. doi: 10.1096/fj.03-0215fje. [DOI] [PubMed] [Google Scholar]
- Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr Akt stimulates the transactivation potential of the RelA/p65 subunit of NF-kappa B through utilization of the ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276:18934–40. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves TN, et al. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107:763–75. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- Weiderpass E, Adami HO, Baron JA, et al. Risk of endometrial cancer following estrogen replacement with and without progestins. J Natl Cancer Inst. 1999;91:1131–7. doi: 10.1093/jnci/91.13.1131. [DOI] [PubMed] [Google Scholar]
- Ross RK, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328–32. doi: 10.1093/jnci/92.4.328. [DOI] [PubMed] [Google Scholar]
- Moorman PG, Kuwabara H, Millikan RC, Newman B. Menopausal hormones and breast cancer in a biracial population. Am J Public Health. 2000;90:966–71. doi: 10.2105/ajph.90.6.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergkvist L, Adami HO, Persson I, Hoover R, Schairer C. The risk of breast cancer after estrogen and estrogen-progestin replacement. N Engl J Med. 1989;321(5):293–7. doi: 10.1056/NEJM198908033210505. [DOI] [PubMed] [Google Scholar]
- Papanastasiou AD, Sirinian C, Kalofonos HP. Identification of novel human receptor activator of nuclear factor-kB isoforms generated through alternative splicing: implications in breast cancer cell survival and migration. Breast Cancer Res. 2012;14(4):R112. doi: 10.1186/bcr3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390(6656):175–9. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Galibert L, Tometsko ME, Anderson DM, Cosman D, Dougall WC. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-kappaB, a member of the TNFR superfamily. J Biol Chem. 1998;273:34120–7. doi: 10.1074/jbc.273.51.34120. [DOI] [PubMed] [Google Scholar]
- Fernandez-Valdivia R, Mukherjee A, Ying Y, et al. The RANKL signaling axis is sufficient to elicit ductal side-branching and alveologenesis in the mammary gland of the virgin mouse. Dev Biol. 2009;328(1):127–39. doi: 10.1016/j.ydbio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM. Bcl-2, a novel regulator of cell death. BioEssays. 1995;17(7):631–8. doi: 10.1002/bies.950170709. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM. Bcl-2 in cancer, development and apoptosis. J Cell Sci Suppl. 1994;18:51–5. doi: 10.1242/jcs.1994.supplement_18.7. [DOI] [PubMed] [Google Scholar]
- Bratton MR, Duong BN, Elliott S, et al. Regulation of ERalpha-mediated transcription of bcl-2 by PI3K-AKT crosstalk: implications for breast cancer cell survival. Int J Oncol. 2010;37:541–50. doi: 10.3892/ijo_00000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, et al. Akt/protein kinase B up-regulates bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–6. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- Nakashima H, Nakamura M, Yamaguchi H, et al. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006;66:7041–9. doi: 10.1158/0008-5472.CAN-05-4588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Vector map and insert sequences.
Fig. S2. Densitometric analysis of receptor activator for nuclear factor-κB (RANK), receptor activator for nuclear factor-κB ligand (RANKL), and phosphorylated protein kinase B pAKT immunoblots in ACI tumor and MCF-7 breast cancer cells.
Fig. S3. Densitometric analysis of immunoblots of MCF-7 cells overexpressing receptor activator for nuclear factor-κB ligand (RANKL) and depleted RANKL.
Fig. S4. Knockdown efficiency of receptor activator for nuclear factor-κB ligand (RANKL) shRNA.
Fig. S5. Densitometric analysis of immunoblots of nuclear factor-κB (NF-κB) signaling in MCF-7 breast cancer cells.
Fig. S6. Densitometric analysis of immunoblots of cyclin D1 and Bcl2 in MCF-7 cells overexpressing receptor activator for nuclear factor-κB ligand (RANKL) and depleted RANKL and ACI tumor.
Fig. S7. Apoptosis analysis by TUNEL assay.
Fig. S8. Upstream stimulatory factor-1 (USF-1) and glioma-associated oncogene homolog 1 (GLI-1) expression in MCF-7 cells overexpressing receptor activator for nuclear factor-κB ligand (RANKL).
Fig. S9. Densitometric analysis of immunoblots of upstream stimulatory factor-1 (USF-1) and glioma-associated oncogene homolog 1 (GLI-1).