

Abstract
Non-muscle invasive bladder cancer is treated with intravesical chemotherapy (IVC) after transurethral resection (TUR) to reduce the probability of recurrence. Despite improvement, the recurrence rate remains high. Intravesical chemotherapeutics at high doses are expected to ablate unresected tumors and floating cancer cells after TUR, although the fate of bladder cancer cells exposed to high-dose chemotherapeutics remains unclear. In this study, we utilized cancer tissue-originated spheroids (CTOS) prepared from bladder cancers or patient-derived xenografts, which may recapitulate human tumors better than 2-D cultures of established cell lines. We exposed CTOS to 1 mg/mL of epirubicin (EPI) or mitomycin C (MMC) for 2 h. EPI was promptly and homogeneously distributed into cancer cells in the CTOS. Two hours after exposure to MMC, the mitochondrial membrane potential decreased and the mitochondria were fragmented, while plasma membrane integrity was maintained. ATP levels rapidly decreased in CTOS after exposure to EPI or MMC. Although activation of the apoptotic pathway was confirmed by the advent of cleaved poly (ADP-ribose) polymerase, fragmentation of DNA (a hallmark of apoptosis) was not observed in CTOS after exposure to EPI and MMC. In the CTOS prepared directly from 19 surgical specimens exposed to EPI and MMC, the decrease of ATP levels varied among patients. Further establishment of the test might help the drug selection and the prediction of recurrence for individual patients.
Keywords: 3-D culture, bladder cancer, cell death, chemosensitivity, intravesical chemotherapy
Bladder cancer is the sixth most common cancer in the USA: the third in men and the eleventh in women.1 Approximately 70% of bladder cancers are non-muscle invasive bladder cancer (NMIBC) at the time of initial diagnosis.2 Although most NMIBC are non-lethal and can be cured by transurethral resection (TUR), approximately half of patients develop intravesical recurrences of NMIBC. The frequent recurrence requires reiterated TUR, which causes additional suffering for patients; in addition, NMIBC has the highest lifetime treatment cost per patient among all cancers.3–5 Therefore, many efforts have been made to reduce the likelihood of intravesical recurrence. According to meta-analyses of seven randomized control trials, intravesical chemotherapy (IVC) immediately after TUR significantly decreased the recurrence rate compared with TUR alone.6,7 Thus, intravesical chemotherapy after TUR has been implemented in clinical practice. Hypothetically, high-dose intravesical chemotherapeutics are expected to ablate unresected tumors and to eliminate floating cancer cells after TUR.8–11 Nonetheless, the fate of cancer cells exposed to high-dose chemotherapeutics remains poorly understood.
Anti-cancer therapies, including chemotherapy and radiotherapy, can trigger the death of cancer cells. Thanks to the understanding of the cell death mechanism induced by such therapies, therapies to target the apoptotic machinery are under development.12–15 Currently, modes of cell death can be classified into three types according to morphologic and biochemical characteristics: apoptosis, necrosis and autophagy.12,13 Each type of cell death involves a variety of molecules and pathways. To improve cancer treatment, it is necessary to know which mode of cell death is associated with a given therapy.
In studies of IVC, apoptotic cell death has been investigated using relatively low chemotherapy doses.14–17 Because the mechanisms of cell death are reportedly different at different drug doses,18,19 understanding the mode of cell death enacted by high-dose chemotherapeutics is a critical issue. Prior preclinical studies with high-dose IVC have not investigated the mode of cell death in detail.20,21
Recently, we developed a novel method for the primary culture of human cancer cells from colorectal, urothelial and lung cancers, which we termed the cancer tissue-originated spheroid (CTOS) method.22–24 Using the CTOS method, we prepare cancer cells as multicellular spheroids from clinical specimens, as well as from patient-derived xenotumors. Because CTOS retain the characteristics of the original cancer cells, one can expect that responses of CTOS to chemotherapeutics reflect those of patient tumors better than established cancer cell lines. In addition, culturing cancer cells as spheroids enables us to 3-D expose them to chemotherapeutics, which is supposed to be more physiologically relevant than conventional 2-D culture. Furthermore, we are able to assess individual responses by preparing CTOS from each patient tumor.22–24
In this study, we exposed CTOS of bladder cancer to high-dose chemotherapeutics to mimic the clinical setting of IVC. We investigated cellular responses in CTOS exposed to the treatment, focusing particularly on the mode of cell death. Finally, by preparing CTOS directly from clinical specimens of bladder cancer, we tested the individual response to high-dose chemotherapeutics.
Materials and Methods
Cancer tissue-originated spheroid preparation and culture
The institutional ethics committees at the Osaka Medical Center for Cancer and Cardiovascular Diseases approved the present study. Human bladder tumor specimens were collected from patients treated at the Department of Urology, Osaka Medical Center for Cancer and Cardiovascular Diseases. CTOS were prepared and cultured according to the previously described protocol.22,24 Briefly, surgical samples or xenograft tumors from NOD/SCID mice were partially digested with Liberase DH (Roche, Mannheim, Germany) and filtered through cell strainers. Fragments were collected on 100 or 40-μm cell strainers (BD Falcon, Franklin Lakes, NJ, USA). CTOS were cultured in StemPro hESC human embryonic stem cell culture medium (Invitrogen, Carlsbad, CA, USA). The growth rate was calculated by dividing the area of CTOS at day 4 by that at day 1.
Animal studies
The institutional animal use committee at Osaka Medical Center for Cancer and Cardiovascular Diseases approved the animal study. A bladder cancer-derived xenograft, BC23, was generated using a urothelial cancer specimen from a patient tumor, as described previously.24 Xenografts were serially passaged by inoculating several hundred CTOS with Matrigel (BD Biosciences, Bedford, MA, USA) subcutaneously at the flank of a 4-week-old male NOD/SCID mouse (CLEA Japan, Shizuoka, Japan).
Reagents
Epirubicin (EPI) and mitomycin C (MMC) were purchased from Wako Pure Chemical Industries (Tokyo, Japan). DPQ was purchased from Abcam (Cambridge, MA, USA). Cyclosporine A (CsA) was kindly provided by Novartis Pharma K.K. (Tokyo, Japan). Z-VAD-FMK was purchased from Promega (Madison, WI, USA).
ATP measurement after exposure to chemotherapeutics
To measure ATP levels in CTOS exposed to chemotherapeutics, one CTOS was put into a well of a 96-well dish. The chemotherapeutics were dissolved in DMEM/F12 medium immediately before use. The CTOS were exposed to EPI or MMC at indicated doses for indicated periods. ATP values were measured using CellTiter Glo (Promega) according to the manufacturer's instructions. Relative ATP values were calculated by dividing the actual ATP value by the area of the CTOS and adjusted with respect to the values of a drug-free control. Images of CTOS were taken using an OLIMPUS IX70 microscope (OLUMPUS, Tokyo, Japan). The CTOS area was measured using ImageJ image analysis software (National Institutes of Health, Bethesda, MD, USA).
Detection of intrinsic fluorescence of epirubicin
Intrinsic EPI fluorescence was detected on formalin-fixed and paraffin-embedded CTOS sections. After sections were dewaxed and rehydrated, nuclei were counterstained with ProLong Gold Antifade Mountant with DAPI (Molecular Probes, Eugene, OR, USA). Fluorescence images were obtained using an OLIMPUS IX70 microscope.
Morphological analysis with confocal microscopy
For whole mount staining, CTOS were incubated with 1 μg/mL propidium iodide (PI) (Molecular Probes) or 500 nM MitoTrackerRed CMXRos (Molecular Probes) with 2 μg/mL Hoechst33342 (Molecular Probes) at 37°C for 15 min. CTOS were washed and mounted with FluorSave Reagent (Calbiochem, San Diego, CA, USA). Fluorescence images were obtained using confocal microscopy (TCS SPE; Leica Microsystems, Wetzlar, Germany). For imaging of mitochondrial morphology, CTOS were dispersed into single cells with 0.25% trypsin/EDTA (Life Technologies, Carlsbad, CA, USA). Dispersed single cells were incubated with 500 nM MitoTrackerRed CMXRos or MitoDeepRed FM (Molecular Probes) with Hoechst33342 at 37°C for 15 min, and images were captured as described above.
Flow cytometry
The treated CTOS were dissociated into single cells with 0.25% trypsin/EDTA. To evaluate drug delivery, EPI fluorescence was analyzed. To evaluate the integrity of the plasma cell membrane and mitochondrial membrane potential (Δψm), single cells were incubated with 1 μg/mL PI or 500 nM MitoTrackerRed CMXRos (Molecular Probes) at 37°C for 15 min without fixation, respectively. Subsequently, flow cytometry was conducted using an Attune Acoustic Focusing Cytometer (Applied Biosystems, Foster City, CA, USA), and the results were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Measurement of reactive oxygen species
Cancer tissue-originated spheroids were labeled with 10 μM dichlorofluorescin diacetate (DCFDA) for 30 min and washed with Hank's balanced salt solution. Then, CTOS were cultured in culture medium, with or without MMC or hydrogen peroxide (Wako Pure Chemical Industries), and analyzed with fluorescence microscopy.
Western blot
Cancer tissue-originated spheroid were lysed with cell lysis buffer (10 mM Tris [pH 7.4], 0.15 M NaCl, 1% NP40, 0.25% sodium deoxycholate, 0.05 M NaF, 2 mM EDTA, 0.1% SDS, 2 mM NaVO4, 10 μg/mL aprotinin, 10 μg/mL leupeptin and 1 mM PMSF). Western blotting was performed as previously described.24 Primary antibodies raised against poly (ADP-ribose) polymerase (PARP) were obtained from BD Biosciences, cleaved caspase-3, caspase-8 and caspase-9 from Cell Signaling Technologies (Danvers, MA, USA), and β-actin from Sigma (Saint Louis, MO, USA), respectively.
Detection of DNA laddering
Four hundred CTOS were treated with 1 mg/mL or 0.01 mg/mL EPI or MMC. The CTOS exposed to 1 mg/mL EPI or MMC were washed after 2 h exposure, and incubated in StemPro hESC. Three days after treatment, CTOS were collected and analyzed to detect DNA laddering using the Apoptotic DNA Ladder Kit (Roche, Mannheim, Germany) according to the manufacturer's protocol. Briefly, the collected CTOS were lysed and the DNA was extracted. The extracted DNA was electrophoresed on a 1% agarose gel. The gel was immersed in 10 mg/mL ethidium bromide solution for 15 min. The electrophoresed DNA was visualized under UV light.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). The R2 value was calculated to assess the correlation of the results. Data are expressed as mean, SD.
Results
We previously reported the preparation and culture of bladder cancer cells from surgically resected samples based on the CTOS method.24 Here, we focused on CTOS from TUR samples. CTOS were prepared from 176 TUR samples, including 135 from the previous study. More than 20 CTOSs were obtained from each sample; the success rate of CTOS preparation was better in NMIBC (76.2%) than in MIBC (42.1%) (Fig.1a). Low-grade (in pathological findings) and papillary (in cystoscopic appearance) types had better success rates than others. The CTOS from high-grade tumors were irregularly shaped, with a high nucleus-to-cytoplasm ratio (Fig.1b). In contrast, CTOS from low-grade tumors were round, with a low nucleus-to-cytoplasm ratio. These findings were comparable to the original tumors. There was no correlation between clinicopathological factors and CTOS growth in 14 patient samples examined (Suppl. Table S1).
Figure 1.
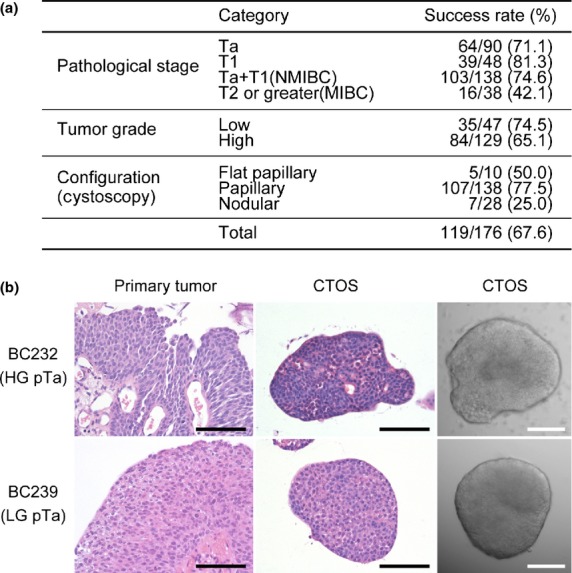
Successful preparation of cancer-tissue origi-nated spheroids (CTOS) in non-muscle invasive bladder cancer. (a) Success rate of CTOS preparation from transurethrally resected specimens of bladder cancers according to clinicopathological factors. Success of preparation was defined as ≥20 CTOS in the culture dish 1 day after preparation. Pathological T stage (pT) was classified according to the 2009 TNM classification. (b) H&E staining of the original tumor (left column) and CTOS (middle column) and phase contrast image of CTOS (right column) for BC232 (upper row); high-grade (HG) and pTa tumor, and BC239 (lower row); low-grade (LG) and pTa tumor. Scale bar, 100 μm.
We studied the responses of CTOS to high-dose chemotherapeutics, which are used in IVC for patients with NMIBC after TUR. To provide a reproducible setting, we first analyzed CTOS from a patient-derived xenograft of bladder cancer, BC23.24 We investigated delivery of EPI into CTOS after exposure to high-dose EPI (1 mg/mL), which is used in clinical settings. It is clinically recommended that high-dose chemotherapeutics be retained for 1–2 h after they are instilled into the patient's bladder. Thus, we conducted a 2-h time-course experiment. We took advantage of the intrinsic fluorescence of EPI to monitor drug delivery. Fluorescence microscopy analysis (Fig.2a) and flow cytometric analysis (Fig.2b) revealed that EPI was promptly and homogeneously distributed into the cancer cells comprising CTOS. According to flow cytometric analysis, EPI levels in cancer cells reached a plateau within 1 h. Notably, spheroidal shape was maintained intact even after 2 h exposure to high-dose EPI.
Figure 2.
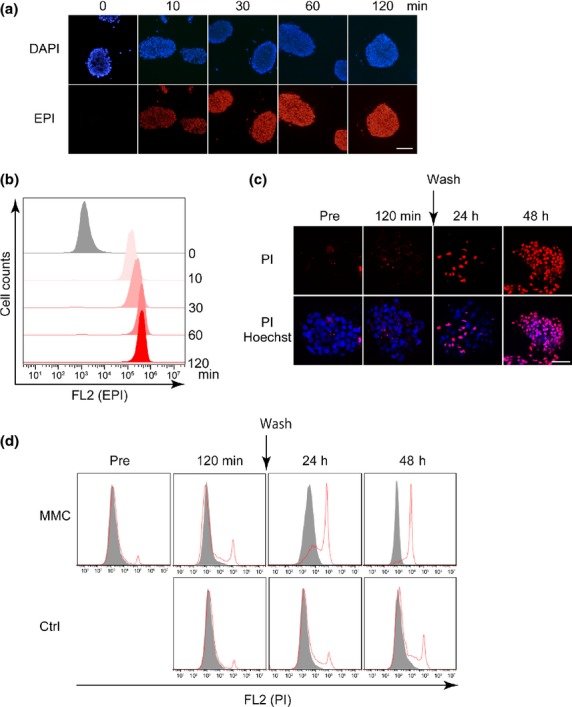
Delivery of chemotherapeutic agents into cancer-tissue originated spheroids (CTOS), and plasma membrane integrity of CTOS after exposure to high-dose chemotherapeutics. (a) Intrinsic fluorescence of epirubicin (EPI) (red) was captured on formalin-fixed, paraffin-embedded CTOS sections. CTOS were exposed to EPI (1 mg/mL) for the indicated time. Nuclei were counterstained with DAPI (blue). Scale bar, 100 μm. (b) Flow cytometric analysis of the intrinsic fluorescence of EPI in CTOS. CTOS were exposed to EPI (1 mg/mL) for the indicated time. (c, d) Loss of plasma membrane integrity was evaluated by propidium iodide (PI) (red) using fluorescence microscopy (c [scale bar, 100 μm]) and flow cytometry (d). CTOS were exposed to control medium or mitomycin C (MMC) (1 mg/mL) for 2 h, and were washed and incubated for an additional 48 h.
To assess cell fate after exposure to high-dose chemotherapeutics, we examined the integrity of the plasma cell membrane in CTOS after exposure to MMC. CTOS were exposed to high-dose MMC for 2 h, washed and incubated in fresh medium. At 2 h after exposure to MMC, almost no cells exhibited intake of PI (Fig.2c,d). Some cells exhibited intake of PI at 24 h after the 2-h exposure to MMC. In contrast, at 48 h all cells had incorporated PI (Fig.2c,d). Thus, plasma cell membrane integrity was lost in CTOS late after exposure to high-dose MMC.
Mitochondrial dysfunction plays a pivotal role in mechanisms of cell death, including both apoptosis and necrosis.25,26 Therefore, we monitored mitochondrial membrane potential, Δψm, in CTOS before and after exposure to MMC using fluorescence microscopy and flow cytometric analysis. After 2 h exposure to MMC, Δψm in CTOS decreased remarkably (Fig.3a,b). Next, we investigated the morphology of mitochondria with a fluorescent probe that stains mitochondria independent of Δψm. Fluorescence microscopy revealed notable mitochondrial fragmentation after 2 h exposure to MMC (Fig.3c). Thus, mitochondrial function and morphology were severely impaired after 2 h exposure to high-dose MMC.
Figure 3.
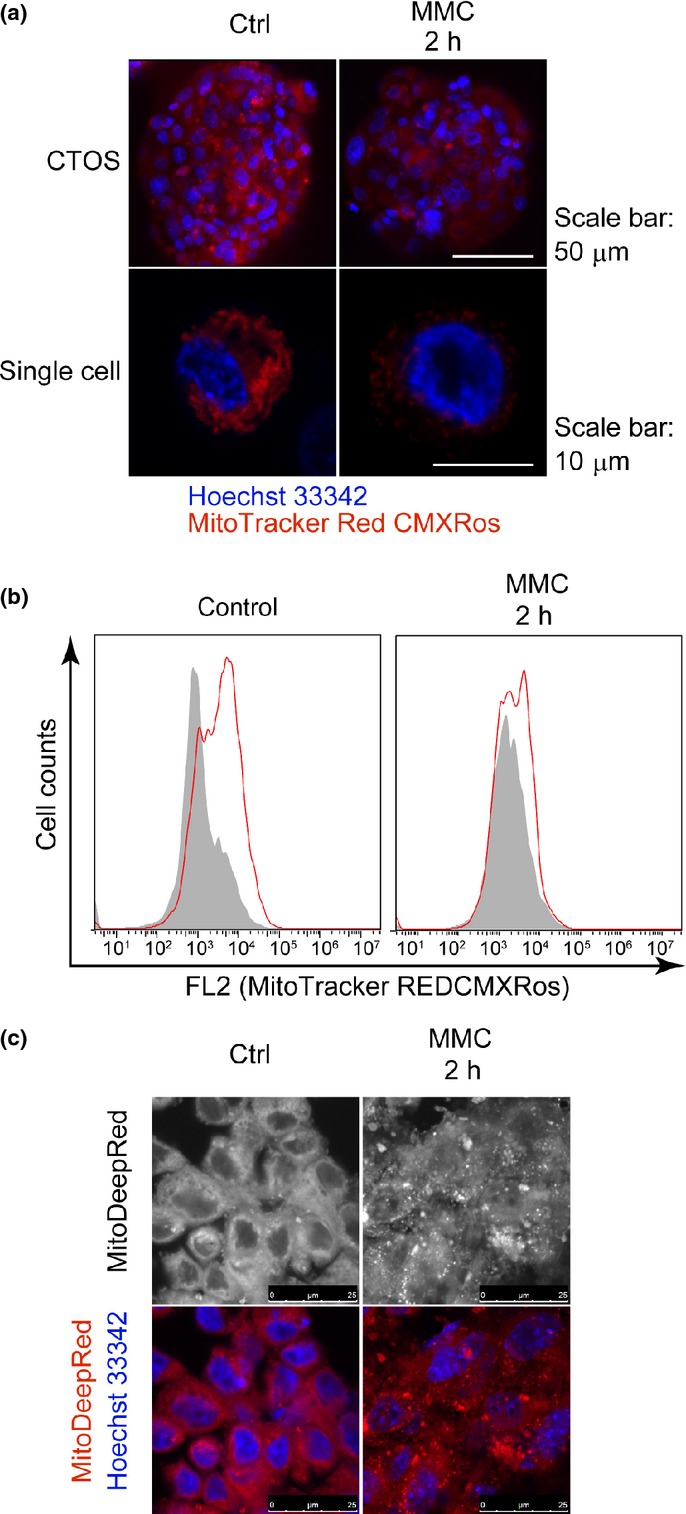
Decrease of mitochondrial membrane potential (Δψm) in cancer-tissue originated spheroids (CTOS) after exposure to high-dose mitomycin C (MMC). (a, b) MitoTracker Red CMXRos (red), Δψm-dependent dye, was evaluated by fluorescence microscopy (a) and flow cytometry (b) in CTOS after 2 h exposure to control medium or MMC. (c) MitoDeepRed (red), Δψm-independent dye, depicts morphology of mitochondria in CTOS after 2 h exposure to control medium or MMC (1 mg/mL).
Because damaged mitochondria can be a source of reactive oxygen species (ROS), which initiate cell death pathways,25,28 we assessed ROS levels using the ROS-sensitive dye DCFDA. Basal DCFDA fluorescence was detected in CTOS and enhanced by treatment with hydrogen peroxide (Suppl. Fig. S1). Because basal DCFDA fluorescence was reduced within 5 min after high-dose MMC, it is unlikely that ROS are a cause of mitochondrial damage.
Because a collapse of Δψm can lead to ATP depletion,27,29 we measured ATP levels in CTOS after exposure to high-dose chemotherapeutics. The ATP levels in CTOS decreased rapidly after exposure to high-dose EPI and MMC, reaching a nadir within 1 h. At 2 h, ATP levels in EPI-treated and MMC-treated CTOS were approximately 20% and 30% of the levels of non-treated CTOS, respectively, and were reduced in a dose-dependent manner (Fig.4a,b). An inhibitor of cyclophilin D, CsA, and an inhibitor of PARP, DPQ, reportedly rescue necrosis by suppressing the ATP decrease caused by oxidative stress30 and alkylating DNA damage,31 respectively. Neither CsA nor DPQ rescued the reduction of ATP levels in CTOS caused by exposure to high-dose MMC (Fig.4c).
Figure 4.
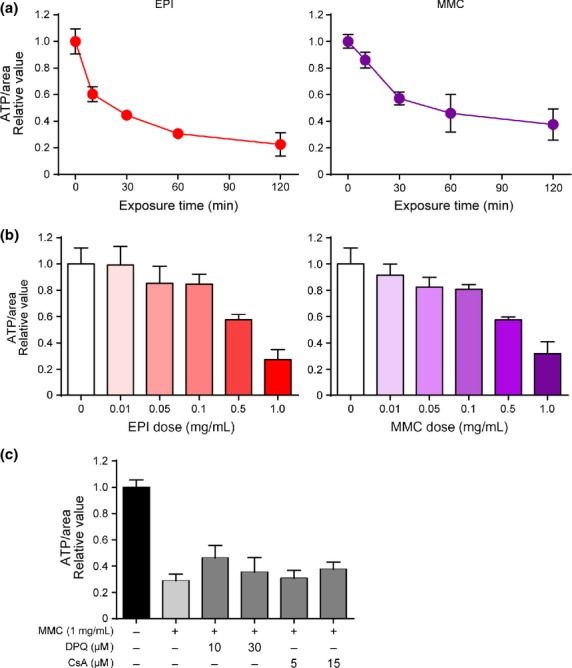
Rapid decrease of ATP levels in cancer-tissue originated spheroids (CTOS) after exposure to high-dose epirubicin (EPI) and mitomycin C (MMC). (a) Time course of ATP levels in CTOS after exposure to EPI or MMC (1 mg/mL). (b) ATP levels in CTOS after 2-h exposure to indicated concentrations of EPI or MMC. (c) ATP levels were measured after 2-h exposure to MMC (1 mg/mL) with the indicated concentration of DPQ or cyclosporine A (CsA). DMSO was used as a vehicle control. All values are presented relative to control.
Mitochondrial dysfunction can trigger the apoptotic pathway. Thus, we tested whether the apoptotic pathway was activated after exposure to high-dose chemotherapeutics. Cleaved PARP, a substrate of caspase-3, appeared after 1 h of exposure to high-dose MMC and after 2 h of exposure to EPI; PARP cleavage was inhibited by the caspase inhibitor Z-VAD-FMK (Fig.5a,d). In addition to the PARP cleavage, we observed cleaved caspase-3, caspase-8 and caspase-9 after 2 h treatment of MMC, and increase of cleaved caspase-8 by EPI treatment (Suppl. Fig. S2). However, 48 h after MMC exposure, the nuclei of cells in the CTOS remained round, without any sign of fragmentation (Fig.5b,e). In contrast, when the CTOS were exposed to low-dose MMC (0.01 mg/mL), cell viability was also completely lost at 72 h after the exposure (Suppl. Fig. S3). In contrast to high-dose MMC, obvious fragmentation of the nuclei was observed in CTOS after exposure to low-dose MMC (0.01 mg/mL) (Fig.5b). DNA laddering was also observed in CTOS after continuous exposure to low-dose MMC or EPI, but not in CTOS at 48 h after a 2-h exposure to high-dose EPI or MMC (Fig.5c). Thus, DNA fragmentation, a hallmark of apoptosis, was not observed in CTOS after exposure to high-dose drugs, although the apoptotic pathway was initiated. Because the cell death resulting from high-dose chemotherapeutics was not rescued by Z-VAD-FMK (Fig.5e), it was independent of caspase activation.
Figure 5.
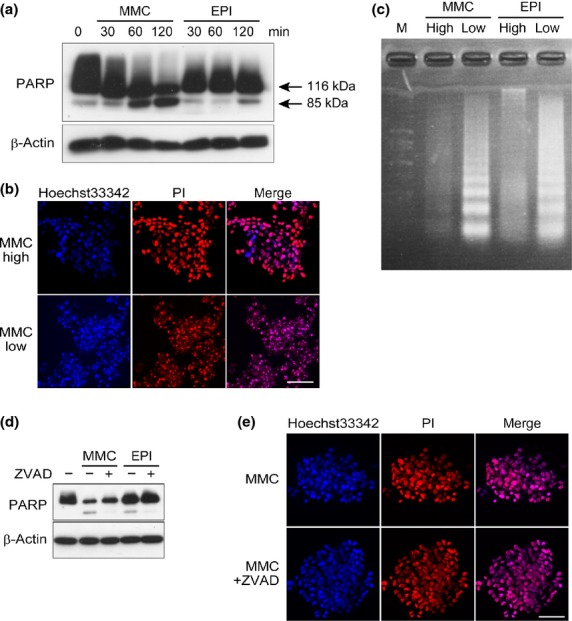
Absence of DNA fragmentation after exposure to high-dose chemotherapeutic agents despite activation of the apoptotic pathway. (a) Western blot of poly (ADP-ribose) polymerase (PARP) in cancer-tissue originated spheroids (CTOS) after exposure to mitomycin C (MMC) or epirubicin (EPI) (1 mg/mL) for the indicated time. (b) Propidium iodide (PI) (red) depicts morphology of nuclei in CTOS 48 h after a 2-h exposure to MMC (1 mg/mL, MMC high) or a 3-day exposure to MMC (0.01 mg/mL, MMC low). Scale bar, 50 μm. (c) DNA was electrophoresed in a 1% agarose gel. DNA was extracted from CTOS 48 h after a 2-h exposure to MMC or EPI (1 mg/mL), or a 3-day exposure to MMC or EPI (0.01 mg/mL). (d) Western blot of CTOS after 2 h exposure to EPI or MMC (1 mg/mL) with DMSO or Z-VAD-FMK. CTOS were pretreated with DMSO or Z-VAD-FMK for 1 h before treatment with EPI or MMC. (e) PI (red) depicts morphology of nuclei in CTOS 48 h after a 2-h exposure to MMC (1 mg/mL) with or without Z-VAD-FMK. CTOS were pretreated with DMSO or Z-VAD-FMK for 1 h before treatment with EPI or MMC. Scale bar, 50 μm.
We used CTOS prepared from primary patient samples to assess individual responses by measuring ATP levels after exposure to high-dose chemotherapy. Figure6a shows the clinicopathological characteristics of 19 patients from whom CTOS were prepared. The decrease of ATP levels varied among CTOS derived from 19 surgical specimens; it ranged from 22.5% to 83.1% in EPI-treated and from 16.9% to 88.4% in MMC-treated CTOS (Fig.6b). The mean reduction of ATP among 19 cases was 47.7% when treated with EPI and 50.6% when treated with MMC; the between-treatment difference was not statistically significant. No association was observed between clinicopathological factors and ATP decrease. There was no correlation in the ATP values between MMC and EPI after 2 h exposure to high-dose chemotherapeutics (Fig.6c), although it should be noted that one drug tended to have better effects than the other in some patients. The treatment protocols after TUR are too diverse and the number of patient samples is too small in this study to assess the correlation between the recurrence rate and the ATP decrease.
Figure 6.
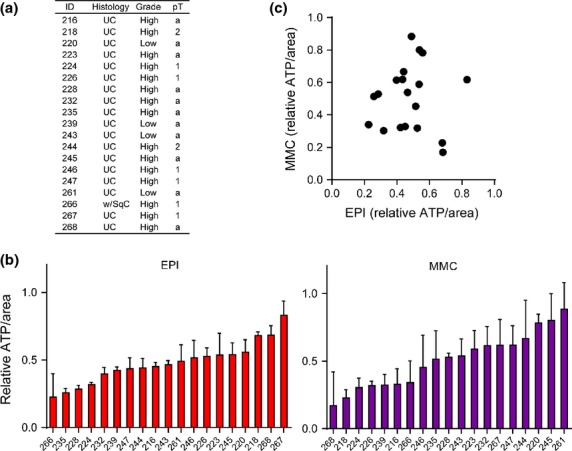
Individual responses of primary cancer-tissue originated spheroids (CTOS) to high-dose chemotherapeutic agents. (a) Clinicopathological characteristics of 19 primary specimens of bladder cancer. (b) Bar plot shows ATP levels in CTOS after 2 h exposure to epirubicin (EPI) or mitomycin C (MMC) (1 mg/mL). (c) Each dot represents ATP levels after 2 h exposure to EPI or MMC in CTOS prepared from each case. R2 = 0.000822 (linear regression analysis).
Discussion
In this study, we investigated the responses of bladder cancer cells to high doses of chemotherapeutics used in clinical IVC. We utilized CTOS, which retain the characteristics of original cancer cells compared with conventional cell lines.22–24 As a result, we expected the responses of CTOS to chemotherapeutics to reflect the responses of patient tumors. When CTOS of bladder cancer were exposed to high-dose chemotherapeutics in vitro, cell death was not accompanied by DNA fragmentation, although the apoptotic pathway was activated. Many chemotherapeutics reportedly induce apoptosis,12,32 characterized by DNA fragmentation with the activation of caspases. Indeed, we observed typical DNA fragmentation at relatively low chemotherapeutic doses. Thus, the mode of cell death enacted by high-dose chemotherapeutics was not typical apoptosis, indicating that the therapeutic effects of IVC differ from those of systemic chemotherapy.
Other types of programmed cell death include necrosis and autophagy.12,33,34 Programmed necrosis is a defined molecular process distinct from passive necrosis, which is caused by mechanical injury to the cells.13,35 Necrosis is morphologically characterized by a lack of DNA fragmentation and swelling of organelles with loss of membrane integrity.27,29,36–38 Mitochondrial dysfunction is characterized by decreased Δψm directly followed by ATP depletion, which is a key event in programmed necrosis.27,29 Thus, some characteristics of the cell death induced by high-dose drugs in this study are compatible with programmed necrosis; however, other characteristics are inconsistent with programmed necrosis. First, Ca2+ overload and production of ROS are typical inducers of programmed necrosis.29,39 ROS levels were decreased in this study. Second, inhibitors of cyclophilin D suppress necrosis by inhibiting the mitochondrial membrane permeability transition and the subsequent Δψm decrease and ATP depletion.30,39 Cyclosporin A, an inhibitor of cyclophilin D, suppressed neither ATP depletion nor cell death in this study. Third, alkylating DNA damage agents activate the DNA repair protein PARP, which is followed by depletion of NAD and ATP, and subsequently by necrotic cell death.31 In addition, release of cytochrome c from mitochondria is observed in the study. Initiation of the apoptotic pathway is compatible with our results, although the PARP inhibitor DPQ failed to suppress ATP depletion or cell death in this study. Taken together, cell death due to high-dose chemotherapeutics shares some of the characteristics of programmed necrosis; however, unknown factors other than ROS, cyclophilin D and PARP must be involved.
The success rate of CTOS preparation from NMIBC was higher than that from MIBC (74.6% vs 42.1%). E-cadherin is required to maintain CTOS derived from human colorectal cancer cells.22 As NMIBC reportedly expresses higher E-cadherin levels than MIBC,40 the different rates of success in CTOS preparation might be due to the differences in E-cadherin expression.
We analyzed multiple patient samples to assess variations in the response to high-dose chemotherapy. Burgues et al.21 used cultured cancer spheroids from clinical samples to examine the effects of high-dose chemotherapy. Using a trypan blue assay, they detected substantial cell death at 2 h. We did not observe clear evidence of increased cell death at that time point. In addition, their spheroids shrunk with time, while CTOS from NMIBC grew well in culture conditions.24 Suboptimal culture conditions might have caused early cell death in their assay.
We found that the extent to which ATP levels were decreased differed with each drug among patients, and some CTOS exhibited very little decrease of ATP levels. A clinical study must be performed to prove the correlation between decreased ATP levels and recurrence. The assay itself is ready to be applied in a clinical setting. First, CTOS can be prepared quickly from TUR samples of NMIBC. Second, the assay in this study can be performed within 24 h after acquiring surgical samples, so that it meets the necessity of starting IVC within 24 h after TUR. Although a meta-analysis of IVC revealed no difference in the overall incidence of recurrence between drugs,6 the sensitivity assay using CTOS might help in selecting the more suitable drug for each patient.
Aside from heterogeneous sensitivity between patients, recurrence in the clinical setting after TUR with IVC might be also explained by the implantation of cancer cells into the bladder wall. Indeed, the cells implant and are covered by extracellular matrix within several hours after TUR.10,41–43 This is supported by the fact that IVC is more effective when it is started immediately after TUR.44 Interactions between cancer cells and the extracellular matrix might also contribute to cell survival. Additional experiments with CTOS will help to elucidate the process.
Acknowledgments
We thank Drs K. Kakimoto, Y. Arai, Y. Yamaguchi, K. Takeda and Y. Ishizuya for preparing clinical samples. We thank A. Mizukoshi and T. Yasuda for technical assistance, and M. Izutsu for secretarial assistance.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Levels of reactive oxygen species in cancer-tissue originated spheroids (CTOS) treated with high dose mitomycin C (MMC). Representative images of CTOS exposed to control medium or MMC (1 mg/mL). CTOS were labeled with a ROS-sensitive dye, DCFDA. CTOS treated with hydrogen peroxide were used as a positive control. DMSO was used as a vehicle control. DCFDA fluorescence was detected at 5 min after the treatments. Scale bar, 200 μm.
Fig. S2. Activation of apoptotic pathways after exposure to high-dose chemotherapeutics. Western blot of cleaved caspase-3, caspase-8 and caspase-9 in cancer-tissue originated spheroids (CTOS) after exposure to mitomycin C (MMC) or EPI (1 mg/mL) for the indicated time.
Fig. S3. Time course of cell viability in cancer-tissue originated spheroids (CTOS) exposed to low-dose mitomycin C (MMC). ATP levels in CTOS exposed to MMC of 0.01 mg/mL were measured at the indicated time points. Relative ATP values were calculated by dividing by the initial area of CTOS, and the values relative to the control are shown.
Table S1. Growth rate of cancer-tissue originated spheroids (CTOS) prepared from bladder cancer specimens.
References
- American Cancer Society. Cancer Facts and Figures 2014. [Cited 21 May 2014.] Available from URL: http://www.cancer.org/acs/groups/content/@research/documents/webcontent/acspc-042151.pdf.
- Parekh DJ, Bochner BH, Dalbagni G. Superficial and muscle-invasive bladder cancer: principles of management for outcomes assessments. J Clin Oncol. 2006;24:5519–27. doi: 10.1200/JCO.2006.08.5431. [DOI] [PubMed] [Google Scholar]
- Botteman MF, Pashos CL, Redaelli A, Laskin B, Hauser R. The health economics of bladder cancer: a comprehensive review of the published literature. Pharmacoeconomics. 2003;21:1315–30. doi: 10.1007/BF03262330. [DOI] [PubMed] [Google Scholar]
- Avritscher EB, Cooksley CD, Grossman HB, et al. Clinical model of lifetime cost of treating bladder cancer and associated complications. Urology. 2006;68:549–53. doi: 10.1016/j.urology.2006.03.062. [DOI] [PubMed] [Google Scholar]
- Sievert KD, Amend B, Nagele U, et al. Economic aspects of bladder cancer: what are the benefits and costs? World J Urol. 2009;27:295–300. doi: 10.1007/s00345-009-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvester RJ, Oosterlinck W, van der Meijden APM. A single immediate postoperative instillation of chemotherapy decreases the risk of recurrence in patients with stage ta t1 bladder cancer: a meta-analysis of published results of randomized clinical trials. J Urol. 2004;171:2186–90. doi: 10.1097/01.ju.0000125486.92260.b2. [DOI] [PubMed] [Google Scholar]
- Perlis N, Zlotta AR, Beyene J, Finelli A, Fleshner NE, Kulkarni GS. Immediate post-transurethral resection of bladder tumor intravesical chemotherapy prevents non-muscle-invasive bladder cancer recurrences: an updated meta-analysis on 2548 patients and quality-of-evidence review. Eur Urol. 2013;64:421–30. doi: 10.1016/j.eururo.2013.06.009. [DOI] [PubMed] [Google Scholar]
- Brocks CP, Buttner H, Bohle A. Inhibition of tumor implantation by intravesical gemcitabine in a murine model of superficial bladder cancer. J Urol. 2005;174:1115–8. doi: 10.1097/01.ju.0000168657.51551.49. [DOI] [PubMed] [Google Scholar]
- Akaza H, Kurth KH, Williams R, et al. Intravesical chemotherapy and immunotherapy for superficial tumors Basic mechanism of action and future direction. Urol Oncol. 1998;4:121–9. doi: 10.1016/s1078-1439(99)00015-0. [DOI] [PubMed] [Google Scholar]
- Pan JS, Slocum HK, Rustum YM, Greco WR, Gaeta JF, Huben RP. Inhibition of implantation of murine bladder tumor by thiotepa in cauterized bladder. J Urol. 1989;142:1589–93. doi: 10.1016/s0022-5347(17)39174-7. [DOI] [PubMed] [Google Scholar]
- Oosterlinck W, Kurth KH, Schroder F, Bultinck J, Hammond B, Sylvester R. A prospective European Organization for Research and Treatment of Cancer Genitourinary Group randomized trial comparing transurethral resection followed by a single intravesical instillation of epirubicin or water in single stage Ta, T1 papillary carcinoma of the bladder. J Urol. 1993;149:749–52. doi: 10.1016/s0022-5347(17)36198-0. [DOI] [PubMed] [Google Scholar]
- Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012;19:75–86. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli W, Ricotti L, Tesei A, et al. Schedule-dependent cytotoxic interaction between epidoxorubicin and gemcitabine in human bladder cancer cells in vitro. Clin Cancer Res. 2004;10:1500–7. doi: 10.1158/1078-0432.ccr-1107-03. [DOI] [PubMed] [Google Scholar]
- Chen SK, Chung CA, Cheng YC, et al. Hydrostatic pressure enhances mitomycin C induced apoptosis in urothelial carcinoma cells. Urol Oncol. 2014;32:e17–24. doi: 10.1016/j.urolonc.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Higuchi Y, Suzuki T, Qiu J, Yamamoto S, Shima H. Double short-time exposure to pirarubicin produces higher cytotoxicity against T24 bladder cancer cells. J Infect Chemother. 2011;17:11–6. doi: 10.1007/s10156-010-0088-y. [DOI] [PubMed] [Google Scholar]
- Gazzaniga P, Silvestri I, Gradilone A, et al. Gemcitabine-induced apoptosis in 5637 cell line: an in-vitro model for high-risk superficial bladder cancer. Anticancer Drugs. 2007;18:179–85. doi: 10.1097/CAD.0b013e328010ef47. [DOI] [PubMed] [Google Scholar]
- Yeung TK, Germond C, Chen X, Wang Z. The mode of action of taxol: apoptosis at low concentration and necrosis at high concentration. Biochem Biophys Res Commun. 1999;263:398–404. doi: 10.1006/bbrc.1999.1375. [DOI] [PubMed] [Google Scholar]
- Torres K, Horwitz SB. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res. 1998;58:3620–6. [PubMed] [Google Scholar]
- Bolenz C, Ikinger EM, Strobel P, et al. Topical chemotherapy in human urothelial carcinoma explants: a novel translational tool for preclinical evaluation of experimental intravesical therapies. Eur Urol. 2009;56:504–11. doi: 10.1016/j.eururo.2008.07.042. [DOI] [PubMed] [Google Scholar]
- Burgues JP, Gomez L, Pontones JL, Vera CD, Jimenez-Cruz JF, Ozonas M. A chemosensitivity test for superficial bladder cancer based on three-dimensional culture of tumour spheroids. Eur Urol. 2007;51:962–9. doi: 10.1016/j.eururo.2006.10.034. discussion 9–70. [DOI] [PubMed] [Google Scholar]
- Kondo J, Endo H, Okuyama H, et al. Retaining cell–cell contact enables preparation and culture of spheroids composed of pure primary cancer cells from colorectal cancer. Proc Natl Acad Sci U S A. 2011;108:6235–40. doi: 10.1073/pnas.1015938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo H, Okami J, Okuyama H, et al. Spheroid culture of primary lung cancer cells with neuregulin 1/HER3 pathway activation. J Thorac Oncol. 2013;8:131–9. doi: 10.1097/JTO.0b013e3182779ccf. [DOI] [PubMed] [Google Scholar]
- Okuyama H, Yoshida T, Endo H, et al. Involvement of heregulin/HER3 in the primary culture of human urothelial cancer. J Urol. 2013;190:302–10. doi: 10.1016/j.juro.2012.12.106. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Roles of mitochondria in health and disease. Diabetes. 2004;53(Suppl 1):S96–102. doi: 10.2337/diabetes.53.2007.s96. [DOI] [PubMed] [Google Scholar]
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Schoenwaelder SM. Procoagulant platelets: are they necrotic? Blood. 2010;116:2011–8. doi: 10.1182/blood-2010-01-261669. [DOI] [PubMed] [Google Scholar]
- Lopez-Erauskin J, Galino J, Bianchi P, et al. Oxidative stress modulates mitochondrial failure and cyclophilin D function in X-linked adrenoleukodystrophy. Brain. 2012;135:3584–98. doi: 10.1093/brain/aws292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–82. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11:342–57. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- Kreuzaler P, Watson CJ. Killing a cancer: what are the alternatives? Nat Rev Cancer. 2012;12:411–24. doi: 10.1038/nrc3264. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. New Engl J Med. 2014;370:455–65. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y. Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997;4:429–34. doi: 10.1038/sj.cdd.4400262. [DOI] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–9. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–40. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Bryan RT, Tselepis C. Cadherin switching and bladder cancer. J Urol. 2010;184:423–31. doi: 10.1016/j.juro.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Gunther JH, Jurczok A, Wulf T, et al. Optimizing syngeneic orthotopic murine bladder cancer (MB49) Cancer Res. 1999;59:2834–7. [PubMed] [Google Scholar]
- Bohle A, Jurczok A, Ardelt P, et al. Inhibition of bladder carcinoma cell adhesion by oligopeptide combinations in vitro and in vivo. J Urol. 2002;167:357–63. [PubMed] [Google Scholar]
- Pode D, Alon Y, Horowitz AT, Vlodavsky I, Biran S. The mechanism of human bladder tumor implantation in an in vitro model. J Urol. 1986;136:482–6. doi: 10.1016/s0022-5347(17)44926-3. [DOI] [PubMed] [Google Scholar]
- Kaasinen E, Rintala E, Hellstrom P, et al. Factors explaining recurrence in patients undergoing chemoimmunotherapy regimens for frequently recurring superficial bladder carcinoma. Eur Urol. 2002;42:167–74. doi: 10.1016/s0302-2838(02)00260-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Levels of reactive oxygen species in cancer-tissue originated spheroids (CTOS) treated with high dose mitomycin C (MMC). Representative images of CTOS exposed to control medium or MMC (1 mg/mL). CTOS were labeled with a ROS-sensitive dye, DCFDA. CTOS treated with hydrogen peroxide were used as a positive control. DMSO was used as a vehicle control. DCFDA fluorescence was detected at 5 min after the treatments. Scale bar, 200 μm.
Fig. S2. Activation of apoptotic pathways after exposure to high-dose chemotherapeutics. Western blot of cleaved caspase-3, caspase-8 and caspase-9 in cancer-tissue originated spheroids (CTOS) after exposure to mitomycin C (MMC) or EPI (1 mg/mL) for the indicated time.
Fig. S3. Time course of cell viability in cancer-tissue originated spheroids (CTOS) exposed to low-dose mitomycin C (MMC). ATP levels in CTOS exposed to MMC of 0.01 mg/mL were measured at the indicated time points. Relative ATP values were calculated by dividing by the initial area of CTOS, and the values relative to the control are shown.
Table S1. Growth rate of cancer-tissue originated spheroids (CTOS) prepared from bladder cancer specimens.