

Abstract
Despite the fact that ionizing radiation (IR) is widely used as a standard treatment for breast cancer, much evidence suggests that IR paradoxically promotes cancer malignancy. However, the molecular mechanisms underlying radiation-induced cancer progression remain obscure. Here, we report that irradiation activates SRC signaling among SRC family kinase proteins, thereby promoting malignant phenotypes such as invasiveness, expansion of the cancer stem-like cell population, and resistance to anticancer agents in breast cancer cells. Importantly, radiation-activated SRC induced SLUG expression and caused epithelial–mesenchymal cell transition through phosphatidylinositol 3-kinase/protein kinase B and p38 MAPK signaling. In agreement, either inhibition of SRC or downstream signaling of p38 MAPK or protein kinase B effectively attenuated radiation-induced epithelial–mesenchymal cell transition along with an increase in the cancer stem-like cell population. In addition, downregulation of SRC also abolished radiation-acquired resistance of breast cancer cells to anticancer agents such as cisplatin, etoposide, paclitaxel, and IR. Taken together, our findings suggest that combining radiotherapy with targeting of SRC might attenuate the harmful effects of radiation and enhance the efficacy of breast cancer treatment.
Keywords: Cancer stem cells, epithelial–mesenchymal cell transition, ionizing radiation, resistance to anticancer agents, SRC
Ionizing radiation is an important adjuvant therapy for breast cancer patients. Clinically, because damage to normal tissues surrounding tumor is unavoidable in radiotherapy, a high dose of IR is often fractionated to reduce the side-effects of radiotherapy. However, many studies have reported that IR paradoxically promotes the malignant glioma cell phenotypes, allowing relapse after treatment.1,2 Notably, this effect is frequently observed after exposure to fractionated radiation that is not sufficient to eradicate the primary tumor. In parallel, several strong lines of evidence have suggested that irradiation promotes invasiveness of cancer cells through induction of EMT. However, the molecular signaling mechanisms underlying radiation-induced EMT in breast cancer cells remain obscure.
The most widely studied member of the SFKs is SRC. As a non-receptor kinase, SRC is an integrator of divergent signal transduction pathways that regulate numerous cellular processes such as cell proliferation, differentiation, migration, angiogenesis, and survival. Although transfection of normal fibroblasts with SRC alone does not induce cellular transformation,3 it is often elevated in multiple solid tumors.4–6 In particular, SRC kinase is highly activated in human malignant breast cancer tissues compared with benign breast tumors or adjacent normal breast tissues, and this elevated SRC activity is correlated with poor metastasis-free survival.7,8 In line with these observations, inhibition of SRC kinase effectively reduced the incidence of breast cancer metastasis and increased survival of mice,9 implicating SRC as a novel therapeutic target for the treatment of breast cancer cells.
In this study, we found that fractionated irradiation promotes migratory and invasive behavior of breast cancer cells through induction of the EMT program. Of note, irradiation caused activation of SRC which consequently activated PI3K, p38, and AKT, thereby promoting EMT through SLUG expression in breast cancer cells. Irradiation also increased resistance of cancer cells to anticancer treatments as well as the CD44+/CD24− cell population known as CSCs in breast cancer. However, we show that downregulation of SRC suppresses CSCs as well as EMT that were induced by irradiation. In addition, targeting SRC sensitized breast cancer cells to anticancer drugs such as cisplatin, etoposide, and paclitaxel as well as radiotherapy. Taken together, this study suggests that combining radiotherapy with targeting of SRC might attenuate radiation-induced EMT and enhance the efficacy of breast cancer treatment.
Materials and Methods
Chemical reagents and antibodies
Polyclonal antibody to CD44 (ab41478) was obtained from Abcam (Seoul, Korea). Polyclonal antibodies to AKT (9272S), p-AKT (S473) (9271S), p-AKT (T308) (9275S), SNAIL (3879), P38 (9121), and p-p38 (9211S) were obtained from Cell Signaling Technology (Beverly, MA, USA). Polyclonal antibodies to vimentin (sc-5565), SLUG (SC-10436), TWIST (sc-15393), c-SRC (sc-19), Lyn (sc-15), Fyn (sc-16), LCK (sc-13), and SOX2 (sc17319) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal antibody to ZEB1 (HPA027524) was purchased from Sigma (St. Louis, MO, USA). Polyclonal antibodies to N-cadherin (610920) and E-cadherin (612130) were purchased from BD Transduction Laboratory (Seoul, Korea). Monoclonal antibodies to β-actin and DAPI were purchased from Sigma. Etoposide (341205), cisplatin (232120), and LY294002 a chemical inhibitor specific to PI3K (440202), were purchased from Calbiochem (San Diego, CA, USA). Paclitaxel (T1912-5MG) was purchased from Sigma.
Cell culture
Human breast epithelial cell lines MCF7 and SKBR3 were purchased from ATCC (Manassas, VA, USA). Cells were cultured in a humidified 5% CO2 atmosphere at 37°C. The hormone receptor status of MCF7 cells is ER+, PR+, and HER2−, and SKBR3 is ER− and HER2+. MCF7 cells were grown in minimum Eagle's medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 g/mL). SKBR3 cells were grown in DMEM supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 g/mL). The DMEM, FBS, penicillin, streptomycin, and trypsin were purchased from Gibco (Seoul, Korea).
Invasion and migration assays
For invasion assays, MCF7 or SKBR3 cancer cells (1 × 105) were loaded in the upper well of a Transwell chamber (8-μm pore size; Corning Glass, Seoul, Korea) that was precoated with 10 mg/mL growth factor-reduced Matrigel (BD Biosciences, Seoul, Korea) on the upper side of the chamber with the lower well filled with 0.8 mL growth medium. After incubation for 48 h, non-invaded cells on the upper surface of the filter were removed with a cotton swab, and cells that had migrated onto the lower surface of the filter were fixed and stained with a Diff-Quick kit (Fisher, Pittsburgh, PA, USA) and photographed (magnification, ×20). Invasiveness was determined by counting cells in five microscopic fields per well, and the extent of invasion was expressed as an average number of cells per microscopic field. Cells were imaged by phase contrast microscopy (Leica Microsystems, Bannockburn, IL, USA). For the migration assay, we used the Transwell chambers with inserts that contained the same type of membrane but without the Matrigel coating.
Transfection
Cells were transfected with DN-p38 pCMV5, control empty vector pCMV5, or siRNA duplexes (40 nM) by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), following the procedure recommended by the manufacturer. Western blot, immunocytochemistry, and FACS analysis or irradiation was carried out 48 h after transfection. All siRNAs targeting c-Src, AKT, and SLUG and a negative control siRNA were purchased from Genolution Pharmaceuticals (Seoul, Korea).
Western blot analysis
Cell lysates were prepared by incubating with lysis buffer (40 mM Tris-HCl, pH 8.0, 120 mM NaCl, 0.1% Nonidet-P40) supplemented with protease inhibitors. Proteins in whole-cell lysates were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Amersham, Arlington Heights, IL, USA). The membrane was blocked with 5% non-fat dry milk in TBS and incubated with primary antibodies overnight at 4°C. Blots were developed with HRP-conjugated secondary antibodies and proteins were visualized using ECL procedures (Amersham), according to the manufacturer's protocol. Secondary antibodies, anti-mouse IgG-HRP, anti-goat IgG-HRP, and anti-rabbit IgG-HRP were purchased from Santa Cruz Biotechnology.
Irradiation
Breast cancer cells were exposed to radiation using a 137Cs γ-ray source (Atomic Energy of Canada, Mississauga, Canada) at a dose rate of 3.81 Gy/min. Further analysis such as migration and invasion assay and Western blot and immunocytochemical analyses were carried out 48 h after fractionated irradiation (2 Gy ×3; 2 Gy/day for 3 days).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. Following fixation, cells were incubated at 4°C overnight with mouse polyclonal anti-human E-cadherin (1:200), rabbit polyclonal anti-human anti-vimentin (1:200), PE-mouse anti-human CD44 (1:200), and Goat polyclonal anti-human anti-SLUG (1:200) primary antibodies in PBS with 1% BSA and 0.1% Triton X-100. Immunostaining of proteins was visualized using Alexa Fluor 488-conjugated anti-rabbit and anti-mouse or anti-goat secondary antibodies (Molecular Probes, Seoul, Korea). Nuclei were counterstained with DAPI (Sigma). Immunostaining was observed with an Olympus IX71 fluorescence microscope (Olympus, Seoul, Korea).
Fluorescence-activated cell sorting analysis
Cells were stained with a PE-mouse anti-human CD44 (BD Pharmingen, Seoul, Korea) and FITC-mouse anti-human CD24 (BD Pharmingen) in PBS containing 0.5% BSA and 2 mM EDTA. As a control, cells were stained with PE mouse IgG1κ isotype or FITC mouse IgG2a, κ isotype (BD Pharmingen). The CD44+/CD24− cells were then analyzed by FACS using a BD FACSCalibur system equipped with Cell Quest software (BD Biosciences).
Quantification of cell death
Cancer cells were treated with anticancer agents cisplatin (50 μM), etoposide (75 μM), paclitaxel (500 nM), or IR (10 Gy). At 48 h after treatment, cell death was measured by FACS analysis using propidium iodide staining. Cells were harvested by trypsinization, washed in PBS, and then incubated in propidium iodide (50 ng/mL) for 5 min at room temperature. Cells (10 000 per sample) were analyzed by BD FACSCalibur, using Cell Quest software.
Statistical analysis
All experimental data are reported as means; error bars represent SD. Statistical analyses were carried out using non-parametric Student's t-tests.
Results
Irradiation promotes migratory and invasive properties of breast cancer cells through EMT
To study the harmful effects of radiotherapy, we examined whether IR causes breast cancer cells to acquire migratory and invasive properties. To this end, MCF7 and SKBR3 breast cancer cell lines were exposed to fractionated doses of radiation (2 Gy ×3; 2 Gy/day for 3 days). Cancer cells were then applied to migration and invasion assays that were carried out by counting migrated cells in Transwells that had been pre-coated with Matrigel (invasion assay) or left uncoated (migration assay). By these analysis, we observed that exposure to radiation enhances the migratory and invasive properties of breast cancer cells (Fig.1a). As the migratory and invasive behavior of cancer cells is often associated with EMT, we next examined the effect of radiation on expression levels of EMT markers such as E-cadherin, N-cadherin, and vimentin. Importantly, the expression of E-cadherin was decreased, whereas N-cadherin and vimentin were increased 48 h after irradiation (Fig.1b,c). We also examined expression levels of EMT transcription factors such as SNAIL, SLUG, ZEB1, and TWIST after irradiation of breast cancer cells. In line with the above data, irradiation caused an increase of SLUG; however, other EMT transcription factors were not altered by irradiation (Fig.1d). To test whether radiation-induced EMT is mainly caused by SLUG, we examined migratory and invasive properties of MCF7 breast cancer cells after treatment with siRNA targeting SLUG prior to irradiation. Of note, downregulation of SLUG effectively attenuated radiation-induced migratory and invasive properties of breast cancer cells Fig. S1a). In agreement with this result, downregulation of SLUG also blocked radiation-induced EMT markers (Fig. S1b,c). Taken together, these results suggest that irradiation causes breast cancer cells to acquire migratory and invasive properties by inducing SLUG and thereby triggering the EMT program.
Figure 1.
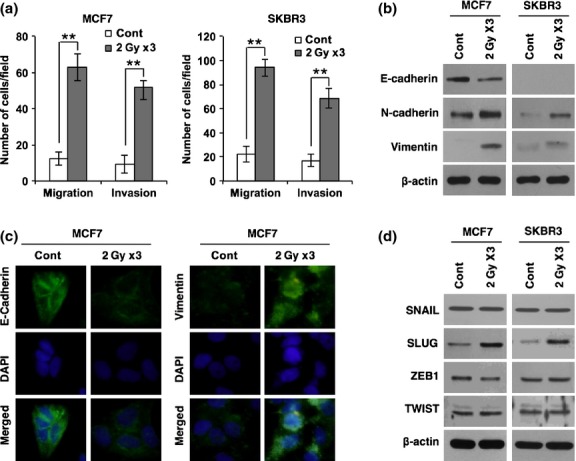
Fractionated radiation promotes invasiveness of breast cancer cells through epithelial–mesenchymal transition (EMT). (a) Migration and invasion assay of MCF7 and SKBR3 breast cancer cells in Transwells after fractionated irradiation. (b) Western blot for EMT markers E-cadherin, N-cadherin, and vimentin in MCF7 and SKBR3 breast cancer cells after irradiation. (c) Immunocytochemistry for EMT markers such as E-cadherin and vimentin in MCF7 cancer cells after irradiation. (d) Western blot for EMT transcription factors SNAIL, SLUG, ZEB1, and TWIST in MCF7 and SKBR3 breast cancer cells after irradiation. β-actin was used as a loading control. Error bars represent mean ± SD of triplicate samples. **P < 0.01. Cont, control.
Irradiation triggers EMT through activation of SRC in breast cancer cells
To investigate the molecular mechanisms underlying radiation-induced EMT in breast cancer cells, we examined the activation status of SFKs. Of note, we observed that irradiation specifically increases the phosphorylation of SRC kinase protein among SFKs (Fig.2a). As irradiation promoted the activation of SRC, we next examined whether SRC activation is responsible for radiation-triggered EMT in breast cancer cells. To this end, we analyzed the migratory and invasive behavior of MCF7 cells that are pretreated with siRNA targeting SRC and then irradiated. Of note, when SRC is downregulated by treatment with siRNA, radiation effects on migration and invasion were inhibited in MCF7 cancer cells (Fig.2b). In parallel with these results, siRNA-mediated downregulation of SRC also attenuated radiation-induced EMT markers (Fig.2c,d). Downregulation of SRC recovered the expression levels of E-cadherin, N-cadherin, and vimentin in irradiated cancer cells to the levels of non-irradiated cells. In agreement, downregulation of SRC blocked the radiation-induced SLUG expression, whereas other EMT regulators were not altered (Fig.2e,f). To further confirm that radiation-activated SRC contributes to EMT, we overexpressed SRC in breast cancer cells and analyzed the migratory and invasive properties as well as EMT markers. As phosphorylation of Try527 inactivates SRC through the interaction of p-Tyr527 with the SH2 domain, we also used a mutant form of SRC Y527F that is constitutively active. Overexpression of either WT SRC or mutant form Y527F enhanced the migratory and invasive properties of MCF7 breast cancer cells (Figs2g,h, S2a). In agreement, SRC overexpression also increased N-cadherin and decreased E-cadherin, although the effect was weak compared to the effect of SRC downregulation (Fig.2i). Collectively, these results suggest that radiation triggers the EMT program through activation of SRC in breast cancer cells.
Figure 2.

Irradiation promotes epithelial–mesenchymal transition (EMT) through activation of SRC in breast cancer cells. (a) Kinase assay for SFK proteins (SRC, LYN, FYN, and LCK) using enolase as a substrate in MCF7 breast cancer cells after exposure to fractionated radiation. (b) Migration and invasion assay in MCF7 cancer cells transfected with siRNA targeting SRC or scrambled control siRNA (si-Cont) prior to irradiation. (c, d) Western blot analysis (c) and immunocytochemistry (d) for EMT markers E-cadherin, N-cadherin, and vimentin in MCF7 cancer cells transfected with siRNA targeting SRC or scrambled control siRNA prior to irradiation. (e, f) Western blot analysis for EMT transcription factors SLUG, SNAIL, ZEB1, and TWIST (e), and immunocytochemistry for EMT transcription factor SLUG (f) in MCF7 cancer cells transfected with siRNA targeting SRC or scrambled control siRNA prior to irradiation. (g, h) Migration and invasion assay in MCF7 (g) and SKBR3 (h) cancer cells transfected with SRC WT, mutant form SRC Y527F, or control vector pcDNA3.1. (i) Western blot analysis for E-cadherin and N-cadherin in MCF7 cells transfected with SRC WT, mutant form SRC Y527F, or control vector pcDNA3.1. β-actin was used as a loading control. Error bars represent mean ± SD of triplicate samples. *P < 0.05; **P < 0.01. Cont, control; IP, Immunoprecipitation.
Radiation-activated SRC transduces intracellular signaling pathways PI3K/AKT and p38 MAPK to increase migratory and invasive behavior
In the above data, we showed that irradiation induces activation of SRC, thereby increasing SLUG expression and triggering EMT in breast cancer cells. Thus, we examined the intracellular signaling pathways that are triggered by SRC and are responsible for radiation-induced EMT in breast cancer cells. Importantly, we found that PI3K/AKT and p38 signaling pathways are activated by irradiation; however, treatment with siRNA targeting SRC attenuated the radiation-induced activation of PI3K/AKT and p38 (Fig.3a). We next examined the phosphorylation status of AKT after transfection with the DN mutant form of p38. Of note, transfection with DN-p38 effectively inhibited the phosphorylation of AKT on Ser473, whereas DN-p38 had no effect on the phosphorylation of Thr308, suggesting that p38 is a downstream effector of SRC, promoting phosphorylation of AKT on Ser473 (Fig.3b). Also, transfection with DN-p38 attenuated radiation-induced EMT markers such as N-cadherin and vimentin, and EMT transcription factor SLUG (Fig. S2b). In line with these results, treatment with siRNA targeting AKT effectively mitigated the radiation-induced migratory and invasive properties of MCF7 breast cancer cells (Fig.3c). In agreement, treatment with PI3K specific inhibitor LY294002 attenuated radiation-induced EMT markers and regulator SLUG (Fig. S2c). Taken together, these results suggest that radiation promotes the EMT program by SRC-mediated activation of PI3K and p38 MAPK that consequently activates AKT signaling in breast cancer cells.
Figure 3.
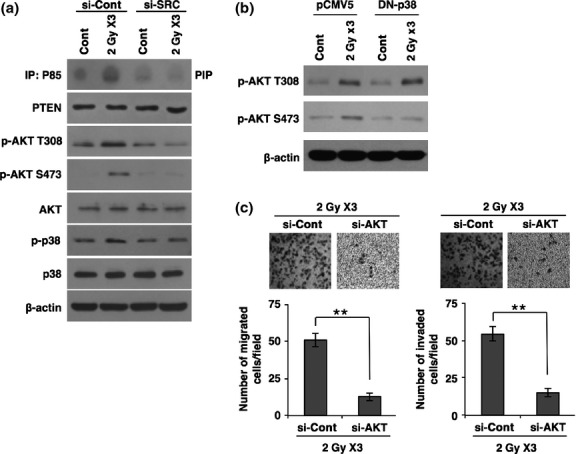
Radiation-activated SRC transduces intracellular signaling of phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and p38 MAPK to increase migratory and invasive behavior. (a) PI3 kinase assay and Western blot analysis for phosphorylation status of AKT and p38 MAPK in MCF7 cancer cells transfected with siRNA targeting SRC or scrambled control siRNA (si-Cont) prior to irradiation. (b) Western blot analysis for phosphorylation status of AKT in MCF7 cancer cells transfected by dominant negative (DN)-p38 or control pCMV5 prior to irradiation. (c) Migration and invasion assay in MCF7 breast cancer cells transfected with siRNA targeting AKT (si-AKT) or scrambled control siRNA prior to irradiation. β-actin was used as a loading control. Error bars represent mean ± SD of triplicate samples. **P < 0.01. PTEN, phosphatase and tensin homolog. Cont, control.
Radiation-activated SRC promotes CSCs
As EMT is often associated with the CSC population, we examined whether radiation-activated SRC is also involved in expansion of the CSC population. To this end, we analyzed the CD44+/CD24− cell population in both MCF7 and SKBR3 breast cancer cells after exposure to a single dose (6 Gy) or fractionated dose (2 Gy ×3) of radiation. Notably, irradiation caused an increase in the CD44+/CD24− cell population, well-known as CSCs in breast cancer (Fig.4a). Intriguingly, fractionated irradiation had more effect on the increase of CD44+/CD24− cells compared to single irradiation, although irradiated doses were equal. However, treatment with siRNA targeting SRC suppressed the radiation-induced expansion of CD44+ cells to basal levels (Fig.4b).
Figure 4.

Irradiation promotes breast cancer stem cell populations through SRC signaling. (a) Quantification of CD44+/CD24− cell population by FACS analysis in MCF7 and SKBR3 cancer cells after irradiation. (b) Quantification of CD44+ cell population by FACS analysis in MCF7 cancer cells transfected with siRNA targeting SRC (si-SRC) or scrambled control siRNA (si-Con) prior to irradiation. (c, d) Western blot analysis for CD44, SOX2, Notch-1, and Notch-2 (c), and immunocytochemistry for CD44 (d) in MCF7 cancer cells transfected with siRNA targeting SRC or scrambled control siRNA prior to irradiation. (e, f) Quantification of the CD44+ cell population by FACS analysis in MCF7 cancer cells transfected with siRNA targeting AKT (si-AKT) (e) or p38 MAPK (si-p38) (f) prior to irradiation. (g, h) Quantification of the CD44+ cell population by FACS analysis in MCF7 (g) and SKBR3 (h) cancer cells 48 h after transfection with SRC WT, mutant form SRC Y527F, or control vector pcDNA3.1. (i) Western blot analysis for SOX2 in MCF7 cells 48 h after transfection with SRC WT, mutant form SRC Y527F, or control vector pcDNA3.1. β-actin was used as a loading control. Error bars represent mean ± SD of triplicate samples. *P < 0.05; **P < 0.01. Cont, control.
Much evidence has suggested that SOX2 and Notch are also critical proteins in the maintenance of breast CSCs.10–12 To further confirm that radiation-activated SRC promotes breast CSCs, we examined the expression levels of SOX2, Notch-1, and Notch-2 as well as CD44 in MCF7 cells after irradiation and treatment with siRNA targeting SRC. In agreement with the above data, irradiation increased the expression of CD44, SOX2, and Notch-2; however, treatment with siRNA against SRC attenuated those expression levels that were analyzed by Western blot and immunocytochemistry (Fig.4c,d).
Because radiation-activated SRC promoted EMT through activation of AKT, we next examined whether radiation-activated SRC increases the CSC population through AKT. As expected, downregulation of AKT abolished the radiation-induced expansion of the CD44+ cell population (Fig.4e). In parallel, downregulation of p38 that was activated by radiation-induced SRC also decreased CD44+ cells (Fig.4f).
To further confirm that radiation-activated SRC increases CSCs, we overexpressed SRC and analyzed CD44+ cells. Consistent with the effect of SRC downregulation, overexpression of either WT SRC or mutant form Y527F caused an increase in CD44+ cells (Fig.4g). The protein level of CD44 was also increased by overexpression of SRC WT or SRC Y527F (Fig.4h). Collectively, these results suggest that radiation-activated SRC promotes CSCs as well as EMT in breast cancer cells.
Radiation-activated SRC confers resistance to anticancer agents on breast cancer cells
As CSCs are reported to be resistant to chemo- and radiotherapy,13,14 we next examined whether irradiation increases the resistance of cancer cells to anticancer treatments through activation of SRC. To this end, breast cancer cells were exposed to fractionated radiation, and on the following day cells were treated with anticancer agents cisplatin, etoposide, or paclitaxel. When cell death was analyzed by FACS, we found that breast cancer cells exposed to fractionated radiation showed more resistance to cisplatin, etoposide, and paclitaxel compared to non-irradiated cells (Fig.5a–c). To examine whether pretreatment with fractionated radiation also confers radio-resistance on cancer cells, MCF7 cells were exposed to fractionated radiation and followed exposure to IR (10 Gy). Consistently, we observed that pretreatment with fractionated irradiation caused breast cancer cells to become more resistant to IR (10 Gy), compared to non-irradiated control cells (Fig.5d). However, pretreatment with siRNA targeting SRC prevented breast cancer cells from acquiring radiation-induced resistance to cisplatin, etoposide, paclitaxel, and IR (Fig.5). Taken together, these results suggest that radiation-activated SRC is necessary for the acquisition of resistance to chemo- and radiotherapy in breast cancer cells.
Figure 5.
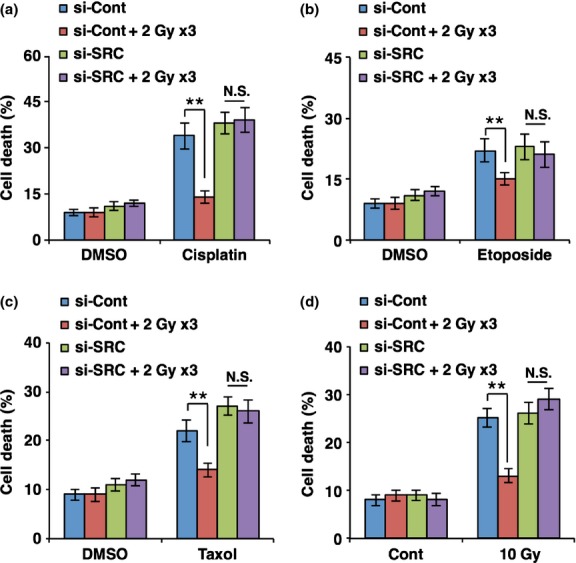
Downregulation of SRC mitigates radiation-enhanced resistance of breast cancer cells to anticancer agents. Quantification of cell death by FACS analysis using propidium iodide staining. MCF7 breast cancer cells transfected with siRNA targeting SRC (si-SRC) or scrambled control siRNA (si-Cont) were exposed to fractionated radiation and then treated with cisplatin (50 μM) (a), etoposide (b), paclitaxel (500 nM) (c), or exposure to ionizing radiation (10 Gy) (d). **P < 0.01. Cont, control; N.S., Not significant.
Discussion
Despite the fact that radiotherapy exerts its therapeutic effect by inducing apoptosis of tumor cells, emerging clinical evidence suggests that irradiation promotes the metastatic behavior of many cancers, including breast cancer.1,2 In this regard, rapidly accumulating evidence suggests that IR promotes cancer metastatic ability by triggering EMT that has a central role in cancer metastasis and has become the subject of intense investigation.15–17 However, the signaling molecular mechanisms underlying radiation-induced EMT remain obscure.
In this study, we found that irradiation promotes the metastatic ability of breast cancer cells through EMT. Exposure to fractionated radiation increased expression of mesenchymal markers such as N-cadherin and vimentin, whereas it decreased epithelial marker E-cadherin. In parallel, irradiation induced EMT transcription factor SLUG but not other factors SNAIL, ZEB1, and TWIST, suggesting that radiation promotes EMT in breast cancer cells through SLUG induction. Very recently, several lines of evidence suggested that EMT is closely associated with acquisition of CSC phenotypes and resistance to anticancer agents, indicating that EMT is a complex cellular program driving to the multifaceted cancer progression.18,19 In agreement with these previous reports, we found that radiation-induced EMT was accompanied with an increase of CSCs. Also, irradiation caused breast cancer cells to acquire resistance to anticancer agents such as cisplatin, etoposide, and paclitaxel as well as IR. However, knockdown of SRC, which was activated by irradiation, efficiently attenuated the radiation-induced EMT, expansion of the CSC population, and resistance to anticancer agents, indicating that radiation-activated SRC is responsible for radiation-induced malignant phenotypes of breast cancer cells. In line with our results, many studies suggested that SRC could be a novel therapeutic target for treatment of breast cancer.9,20,21 Duffy et al.20 found that SRC is highly expressed in triple negative breast cancer cells, compared to non-triple negative cells and suggested SRC as a potential target for the treatment of triple negative breast cancer cells. As triple negative breast cancer cells do not have ER, PR, and HER2, these cancer cells are resistant to hormonal therapy such as tamoxifen, aromatase inhibitors, and therapies that target HER2 receptors such as herceptin. In this context, recent studies suggest that SRC represents a universal signaling node, and targeting of SRC could overcome multiple mechanisms of resistance against anticancer agents.21 Indeed, inhibition of SRC effectively reduced the incidence of breast cancer metastases and increased survival of tumor cell-inoculated mice.9
In this study, we observed that radiation-activated SRC promoted malignant phenotypes of breast cancer cells through the PI3K/AKT and p38 signaling pathways. We showed that downregulation of SRC attenuates p38 and AKT signaling that was activated by irradiation. Importantly, inhibition of p38 decreased the phosphorylation of AKT on Ser473 but not Thr308, suggesting that radiation-activated SRC promotes AKT signaling through activation of PI3K and p38 in breast cancer cells. Supporting our findings, previous studies have suggested that SRC signaling is often transduced through p38 and PI3K/AKT signaling components.22,23 In agreement, inhibition of p38 or AKT effectively attenuated radiation-induced invasiveness and stemness of breast cancer cells. Despite our observation that radiation activates the SRC signaling component among SFKs that is responsible for radiation-induced malignant phenotypes of breast cancer, the molecular mechanisms underlying radiation-induced activation of SRC remain unknown. Presumably, as SRC interacts with many receptor tyrosine kinases, we suppose that SRC could be activated by radiation-induced secretion factors that activate those receptors. In line with this hypothesis, previous studies have suggested that secretion of many cytokines is indeed stimulated by irradiation.24,25
In summary, our findings suggest that radiation promotes the EMT program by SRC-mediated activation of PI3K and p38 MAPK that consequently activates AKT signaling in breast cancer cells. These findings imply that the targeting of SRC might mitigate radiation-induced malignant phenotypes and increase the efficacy of radiotherapy for breast cancer treatment.
Acknowledgments
This work was supported by the National Research Foundation and the Korean Ministry of Science, ICT and Future Planning through its National Nuclear Technology Program (NRF-2012M2A2A7035878; NRF-2012M2B2B1055639).
Glossary
Abbreviations
- AKT
protein kinase B
- CSC
cancer stem cell
- DN
dominant negative
- EMT
epithelial–mesenchymal transition
- ER
estrogen receptor
- HER2
human epidermal growth factor receptor 2
- IR
ionizing radiation
- p-
phosphorylated
- PE
phycoerythrin
- PI3K
phosphatidylinositol 3-kinase
- PR
progesterone receptor
- SFK
SRC family kinase
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Ionizing radiation promotes epithelial–mesenchymal transition in breast cancer cells through SLUG.
Fig. S2. Overexpression of SRC and radiation-induced epithelial–mesenchymal transition by phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and p38 MAPK, downstream effectors of SRC in breast cancer cells.
References
- Dirks PB. Brain tumor stem cells: bringing order to the chaos of brain cancer. J Clin Oncol. 2008;26:2916–24. doi: 10.1200/JCO.2008.17.6792. [DOI] [PubMed] [Google Scholar]
- Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell. 2010;18:619–29. doi: 10.1016/j.ccr.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalloway D, Coussens PM, Yaciuk P. Overexpression of the c-src protein does not induce transformation of NIH 3T3 cells. Proc Natl Acad Sci U. S. A. 1984;81:7071–5. doi: 10.1073/pnas.81.22.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–58. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- Jacobs C, Rubsamen H. Expression of pp60c-src protein kinase in adult and fetal human tissue: high activities in some sarcomas and mammary carcinomas. Cancer Res. 1983;43:1696–702. [PubMed] [Google Scholar]
- Hennipman A, van Oirschot BA, Smits J, Rijksen G, Staal GE. Tyrosine kinase activity in breast cancer, benign breast disease, and normal breast tissue. Cancer Res. 1989;49:516–21. [PubMed] [Google Scholar]
- Verbeek BS, Vroom TM, Adriaansen-Slot SS, et al. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J Pathol. 1996;180:383–8. doi: 10.1002/(SICI)1096-9896(199612)180:4<383::AID-PATH686>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Rucci N, Recchia I, Angelucci A, et al. Inhibition of protein kinase c-Src reduces the incidence of breast cancer metastases and increases survival in mice: implications for therapy. J Pharmacol Exp Ther. 2006;318:161–72. doi: 10.1124/jpet.106.102004. [DOI] [PubMed] [Google Scholar]
- Leis O, Eguiara A, Lopez-Arribillaga E, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–65. doi: 10.1038/onc.2011.338. [DOI] [PubMed] [Google Scholar]
- Lagadec C, Vlashi E, Alhiyari Y, Phillips TM, Bochkur Dratver M, Pajonk F. Radiation-induced Notch signaling in breast cancer stem cells. Int J Radiat Oncol Biol Phys. 2013;87:609–18. doi: 10.1016/j.ijrobp.2013.06.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnie G, Clarke RB. Mammary stem cells and breast cancer–role of Notch signalling. Stem Cell Rev. 2007;3:169–75. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- Zhang M, Atkinson RL, Rosen JM. Selective targeting of radiation-resistant tumor-initiating cells. Proc Natl Acad Sci U S A. 2010;107:3522–7. doi: 10.1073/pnas.0910179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frosina G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol Cancer Res. 2009;7:989–99. doi: 10.1158/1541-7786.MCR-09-0030. [DOI] [PubMed] [Google Scholar]
- Kawamoto A, Yokoe T, Tanaka K, et al. Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol Rep. 2012;27:51–7. doi: 10.3892/or.2011.1485. [DOI] [PubMed] [Google Scholar]
- Yan S, Wang Y, Yang Q, et al. Low-dose radiation-induced epithelial-mesenchymal transition through NF-kappaB in cervical cancer cells. Int J Oncol. 2013;42:1801–6. doi: 10.3892/ijo.2013.1852. [DOI] [PubMed] [Google Scholar]
- Liu W, Huang YJ, Liu C, et al. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3beta and repression of ZEB1. Lab Invest. 2014;94:362–70. doi: 10.1038/labinvest.2013.153. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tryfonopoulos D, Walsh S, Collins DM, et al. Src: a potential target for the treatment of triple-negative breast cancer. Ann Oncol. 2011;22:2234–40. doi: 10.1093/annonc/mdq757. [DOI] [PubMed] [Google Scholar]
- Zhang S, Huang WC, Li P, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461–9. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thobe BM, Frink M, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Src family kinases regulate p38 MAPK-mediated IL-6 production in Kupffer cells following hypoxia. Am J Physiol Cell Physiol. 2006;291:C476–82. doi: 10.1152/ajpcell.00076.2006. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Byun JY, Yun CH, Park IC, Lee KH, Lee SJ. c-Src-p38 mitogen-activated protein kinase signaling is required for Akt activation in response to ionizing radiation. Mol Cancer Res. 2008;6:1872–80. doi: 10.1158/1541-7786.MCR-08-0084. [DOI] [PubMed] [Google Scholar]
- Siva S, MacManus M, Kron T, et al. A pattern of early radiation-induced inflammatory cytokine expression is associated with lung toxicity in patients with non-small cell lung cancer. PLoS ONE. 2014;9:e109560. doi: 10.1371/journal.pone.0109560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller K, Meineke V. Radiation-induced alterations in cytokine production by skin cells. Exp Hematol. 2007;35:96–104. doi: 10.1016/j.exphem.2007.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Ionizing radiation promotes epithelial–mesenchymal transition in breast cancer cells through SLUG.
Fig. S2. Overexpression of SRC and radiation-induced epithelial–mesenchymal transition by phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and p38 MAPK, downstream effectors of SRC in breast cancer cells.