

Abstract
Cancer chemotherapy and radiotherapy are designed to kill cancer cells mostly by inducing DNA damage. DNA damage is normally recognized and repaired by the intrinsic DNA damage response machinery. If the damaged lesions are successfully repaired, the cells will survive. In order to specifically and effectively kill cancer cells by therapies that induce DNA damage, it is important to take advantage of specific abnormalities in the DNA damage response machinery that are present in cancer cells but not in normal cells. Such properties of cancer cells can provide biomarkers or targets for sensitization. For example, defects or upregulation of the specific pathways that recognize or repair specific types of DNA damage can serve as biomarkers of favorable or poor response to therapies that induce such types of DNA damage. Inhibition of a DNA damage response pathway may enhance the therapeutic effects in combination with the DNA-damaging agents. Moreover, it may also be useful as a monotherapy when it achieves synthetic lethality, in which inhibition of a complementary DNA damage response pathway selectively kills cancer cells that have a defect in a particular DNA repair pathway. The most striking application of this strategy is the treatment of cancers deficient in homologous recombination by poly(ADP-ribose) polymerase inhibitors. In this review, we describe the impact of targeting the cancer-specific aberrations in the DNA damage response by explaining how these treatment strategies are currently being evaluated in preclinical or clinical trials.
Keywords: Cancer therapy, DNA damage response, DNA repair, PARP inhibitors, synthetic lethality
The genome DNA is constantly exposed to various genotoxic insults. Among the variety of types of DNA damage, the most deleterious is the DNA double-strand break (DSB).(1) Double-strand breaks can be generated by endogenous sources such as reactive oxygen species produced during cellular metabolic processes and replication-associated errors, as well as by exogenous sources including ionizing radiation and chemotherapeutic agents. Double-strand breaks are also generated in a programmed manner during meiosis and during the V(D)J recombination and class switch recombination required for the development of lymphocytes. If left unrepaired, DSBs can result in cell death. If accurately repaired, DSBs can result in survival of cells with no adverse effects. If insufficiently or inaccurately repaired, DSBs can result in survival of cells showing genomic alterations that may contribute to tumor development.(2) In order to maintain genomic integrity, cells have evolved a well coordinated network of signaling cascades, termed the DNA damage response, to sense and transmit the damage signals to effector proteins, and induce cellular responses including cell cycle arrest, activation of DNA repair pathways, and cell death (Fig. 1).(1)
Fig. 1.

Overview of the diverse spectrum of DNA damage and the DNA damage response. The major repair pathways and key proteins used to process each type of damage are shown. In non-homologous end-joining (NHEJ), the Ku70/Ku80 complex binds to the DNA double-strand break ends and recruits the other indicated components. In base-excision repair (BER), poly(ADP-ribose) polymerase-1 (PARP-1) detects and binds to single-strand breaks and ensures accumulation of other repair factors at the breaks. Single-strand breaks containing modified DNA ends are recognized by damage-specific proteins such as apurinic/apyrimidinic endonuclease (APE1), which subsequently recruits Polβ and XRCC1-DNA ligase IIIα to accomplish the repair. All the molecules indicated here are aberrated in sporadic cancers. The proteins targeted for cancer therapy in the present clinical trials are marked with red asterisks. alt-NHEJ, alternative NHEJ; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related; FA, Fanconi anemia; HR, homologous recombination; MGMT, O6-methylguanine-DNA methyltransferase; MMR, mismatch repair; MRN, MRE11–RAD50–NBS1; NER, nucleotide excision repair; TLS, translesion synthesis.
Cancer chemotherapeutic agents and radiotherapy exert their cytotoxic effects by inducing DNA DSBs. As cancer cells often have specific abnormalities in the DNA damage response, therapeutic strategies based on such properties of cancer cells have been developed. Several inhibitors that block specific DNA damage responses or repair proteins have been tried not only as sensitizing agents in combination with DNA-damaging agents but also as single agents against cancers with defects in particular DNA repair pathways. The most prominent example of the latter is the killing effect of poly(ADP-ribose) polymerase (PARP) inhibitors on BRCA1- or BRCA2-defective tumors, which takes advantage of the defects in DNA repair in cancer cells.(3)
In this review, we will first outline the mechanism of the DNA damage response. Next, we will describe the aberrations in DNA damage responses in human cancers. Finally, we will explain how different DNA damage response pathways can be targeted for cancer therapy.
Mechanism of DNA Damage Response
DNA-damaging agents induce various types of DNA damage including modification of bases, intrastrand crosslinks, interstrand crosslinks (ICL), DNA–protein crosslinks, single-strand breaks (SSBs), and DSBs. Each type of DNA damage is recognized and processed by proteins involved in the DNA damage response (Fig. 1).
In response to DSBs, the MRE11–RAD50–NBS1 (MRN) complex senses and binds to DSB sites, and recruits and activates the ataxia telangiectasia mutated (ATM) kinase through its autophosphorylation.(4,5) Once activated, ATM phosphorylates a large number of downstream proteins.(6) Phosphorylation of Chk2 induces phosphorylation of the protein phosphatase CDC25A, leading to cell cycle arrest. Phosphorylation of BRCA1 leads to DSB repair as well as cell cycle arrest in the S phase, whereas activation of p53 triggers cell cycle arrest in the G1 phase or cell death. In the initiation of the response to SSBs or DNA replication fork collapse, the ataxia telangiectasia and Rad3-related (ATR) kinase is activated and recruited to the sites of DNA damage.(7) ATR phosphorylates and activates Chk1,(8) which plays a role in the S and G2/M cell checkpoints by regulating the stability of the CDC25 phosphatases. Activation of the 53BP1 protein, a mediator of the DNA damage response, contributes to the choice of the DSB repair pathways by promoting non-homologous end joining (NHEJ).(9)
The DNA repair pathways can either work independently or coordinately to repair different types of DNA damage (Fig. 1). Double-strand breaks are predominantly repaired by either NHEJ or homologous recombination (HR).(10) Non-homologous end joining is an error-prone repair pathway that is mediated by the direct joining of the two broken ends.(10) Factors involved in NHEJ include the Ku70/Ku80 complex, DNA-PK catalytic subunit (DNA-PKcs), the Artemis nuclease, XLF, XRCC4, and DNA ligase IV. Homologous recombination is an error-free repair pathway that requires a non-damaged sister chromatid to serve as a template for repair (Fig. 2).(10) Factors involved in HR include the MRN complex, CtIP, replication protein A (RPA), BRCA1, PALB2, BRCA2, and RAD51. In addition to NHEJ and HR, an alternative form of NHEJ, namely, alt-NHEJ, is also involved in DSB repair.(11) It exhibits a slower process than the classical NHEJ and can catalyze the joining of unrelated DNA molecules, leading to the formation of translocations as well as large deletions and other sequence alterations at the junction. Factors involved in this pathway include PARP-1, XRCC1, DNA ligase IIIα, polynucleotide kinase, and Flap endonuclease 1.
Fig. 2.
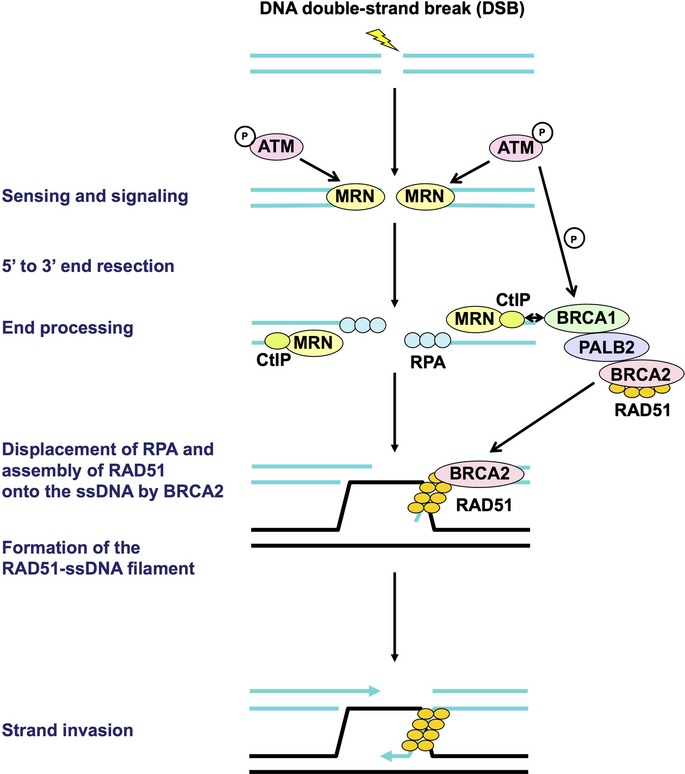
Early steps of homologous recombination. First, the DNA double-strand break is sensed by the MRE11–RAD50–NBS1 (MRN) complex, which subsequently recruits and activates the ataxia telangiectasia mutated (ATM) kinase. Then, the DNA ends are resected by the MRN complex and CtIP, resulting in generation of 3′ single-stranded DNA (ssDNA) overhangs on both sides of the break. These overhangs are coated and stabilized by replication protein A (RPA). Next, BRCA2, which forms the BRCA1–PALB2–BRCA2 complex, directly binds RAD51 and recruits it to the double-stranded DNA–ssDNA junction, and promotes the loading of RAD51 onto ssDNA. This step is followed by displacement of RPA from ssDNA ends and assembly of the RAD51–ssDNA filament, which is mediated by BRCA2, leading to strand invasion into an undamaged homologous DNA template. All the molecules indicated here are aberrated in sporadic cancers. None of the proteins indicated here are targeted for cancer therapy in the present clinical trials. P, phosphorylation.
Single-strand breaks and subtle changes to DNAs are repaired using base-excision repair (BER) proteins,(12) which include PARP-1, XRCC1, DNA ligase IIIα, and apurinic/apyrimidinic endonuclease (APE1). Bulky DNA lesions such as pyrimidine dimers caused by UV irradiation are processed by the nucleotide excision repair (NER) pathway,(13) which requires the excision repair cross-complementing protein 1 (ERCC1). Base mismatches arising as a result of replication errors can be repaired by the mismatch repair pathway.(14)
In the repair of ICL, ubiquitin-mediated activation of the Fanconi anemia (FA) pathway plays a key role.(15) The FA pathway is constituted by at least 15 FA gene products, whose germline defects result in FA, a cancer predisposition syndrome. Activation of the FA core complex, which is comprised of eight FA proteins (FANCA/B/C/E/F/G/L/M) and associated proteins, leads to monoubiquitination of FANCD2 and FANCDI, which subsequently coordinates three critical DNA repair processes, including nucleolytic incision by XPF-ERCC1 and SLX4 endonucleases, translesion DNA synthesis, and HR.
Aberrations in DNA Damage Responses in Human Cancers
In sporadic cancers, both activation and inactivation of the DNA damage response are found in various cancers,(16–62) as summarized in Table 1.
Table 1.
Examples of aberrations in DNA damage responses in human sporadic cancers
| Molecule | Activation or inactivation | Type of aberrations | Type(s) of cancer | Frequency | Phenotypes | Reference(s) |
|---|---|---|---|---|---|---|
| ATM | Activation | Increased autophosphorylation | Bladder, breast cancers | 30–68% | Cancer barrier function | (16,18) |
| Increased copy number | Prostate cancers | ˜2% | (51) | |||
| Inactivation | Mutation | Pancreatic, lung, colon, endometrial, prostate, skin, kidney, breast, central nervous system, ovarian cancers | 1–7% | (49,50) | ||
| Hematopoietic and lymphoid malignancies | ˜11% | (49) | ||||
| Loss of heterozygosity, loss | Pancreatic cancers | ˜5% | (50) | |||
| Decreased copy number | Prostate cancers | ˜5% | (51) | |||
| Decreased expression | Breast, head and neck cancers | 25–75% | (54,55) | |||
| MRE11 | Inactivation | Decreased expression | Breast cancers | 7–31% | (19,54,56) | |
| Colorectal, gastric, pancreatic cancers with microsatellite instability | 67–100% | (19) | ||||
| RAD50 | Activation | Increased expression | Colorectal cancers | ˜24% | (21) | |
| Inactivation | Decreased expression | Breast cancers | 3–28% | (19,54,56) | ||
| Colorectal, gastric cancers with microsatellite instability | 28–71% | (19) | ||||
| NBS1 | Activation | Increased expression | Esophageal, head and neck, non-small-cell lung cancers, hepatomas | 40–52% | Poor prognosis | (19,20) |
| Inactivation | Decreased expression | Breast cancers | 10–46% | (19,54,56) | ||
| Chk1 | Activation | Increased phosphorylation | Cervical cancers | ˜25% | (27) | |
| Increased expression | Lung, liver, breast, colorectal, ovarian, cervical cancers | 46–100% | Resistance to chemotherapy, poor prognosis | (22–27) | ||
| Inactivation | Decreased expression | Lung, ovarian cancers, hetapocellular carcinomas | 9–32% | (22,23,26) | ||
| Chk2 | Activation | Increased phosphorylation | Bladder, colon, lung cancers, melanomas | 30–50% | Cancer barrier function | (16,17) |
| Increased expression | Ovarian cancers | ˜37% | (26) | |||
| Inactivation | Decreased expression | Breast, non-small cell lung cancers | 28–47% | (57,58) | ||
| p53 | Inactivation | Mutation | Solid tumors | ˜50% | (47) | |
| Hematopoietic malignancies | ˜10% | (47) | ||||
| Decreased expression | Solid and hematopoietic tumors | ˜50% | Resistance to chemotherapy, poor prognosis | (48) | ||
| CDC25A | Activation | Increased expression | Thyroid, breast, ovarian, liver, colorectal, laryngeal, esophageal cancers, non-Hodgkin's lymphomas | 17–70% | (28) | |
| CDC25B | Activation | Increased expression | Thyroid, breast, ovarian, liver, gastric, colorectal, laryngeal, esophageal, endometrial, prostate cancers, gliomas, non-Hodgkin's lymphomas | 20–79% | (28) | |
| CDC25C | Activation | Increased expression | Colorectal, endometrial cancers, non-Hodgkin's lymphomas | 13–27% | (28) | |
| DNA-PKcs | Activation | Increased expression | Glioblastoma, prostate cancers | ˜49% | Poor survival | (29,30) |
| RAD51 | Activation | Increased expression | Breast, head and neck, non-small-cell lung cell, pancreatic cancers, soft tissue sarcomas | 24–66% | Resistance to platinum agents, poor outcome | (31–35) |
| Inactivation | Decreased expression | Breast, colorectal cancers | ˜30% | (59,60) | ||
| BRCA1 | Activation | Increased expression | Lung cancers | ˜22% | Resistance to chemotherapy | (36) |
| Inactivation | Mutation | Breast, ovarian cancers | <10% | (52,53) | ||
| Decreased expression | Breast, ovarian, lung cancers | 9–30% | (60–62) | |||
| BRCA2 | Inactivation | Mutation | Breast, ovarian cancers | <10% | (52,53) | |
| Decreased expression | Ovarian cancers | 13% | (61) | |||
| ERCC1 | Activation | Increased expression | Colorectal, ovarian, gastric, head and neck, non-small-cell lung cancers | 14–70% | Resistance to platinum agents | (31,37–43) |
| Inactivation | Decreased expression | Colorectal, gastric, non-small-cell lung cancers | 30–77% | (37,38,42,43) | ||
| APE1 | Activation | Increased expression | Bladder, breast, cervical, head and neck, liver, non-small-cell lung cancers, ovarian cancers, medulloblastomas, gliomas, osteosarcomas, germ cell tumors | 19–99% | Resistance to chemotherapy and/or radiation | (44) |
| PARP | Activation | Increased expression | Breast cancers, germ cell tumors | 5–47% | (45,46) | |
| FANCA | Inactivation | Decreased expression/loss of expression | Acute myelogenous leukemias | 4–40% | (64,65) | |
| Mutation | Acute myelogenous leukemias | ˜7.6% | (64) | |||
| FANCC | Inactivation | Mutation, loss of heterozygosity | Pancreatic cancers | ˜9% | (64) | |
| FANCF | Inactivation | Decreased expression/loss of expression | Breast, cervical, head and neck, non-small-cell lung, ovarian cancers, acute myelogenous leukemias, germ cell tumors | 6.7˜30% | (64,65) | |
| FANCG | Inactivation | Loss of expression | Acute myelogenous leukemias | 27% | (65) |
Expression has been confirmed at mRNA and/or protein levels. Studies using cultured cancer cells are excluded.
Regarding activation of the DNA damage response proteins, increased autophosphorylation of ATM and ATM-dependent phosphorylation of Chk2 are reported in early-stage tumors, suggesting that the DNA damage response may serve as a barrier to the malignant progression of tumors.(16,17) In contrast, a recent study reports that ATM is hyperactive in late-stage breast tumor tissues, suggesting that the ATM-mediated DNA damage response also plays a role in tumor progression and metastasis.(18) Increased expression of NBS1, RAD50, Chk1, Chk2, CDC25A, CDC25B, and CDC25C are also reported.(19–28) DNA-PK catalytic subunit is reported to be upregulated in radiation-resistant tumors or in tumors with poor survival.(29,30) Overexpression of RAD51, BRCA1, ERCC1, APE1, and PARP1 is also observed in various cancers and is associated with resistance to chemotherapy.(31–46)
However, inactivation of DNA damage response proteins is also observed in various cancers. The p53 gene is one of the most frequently mutated genes in human sporadic cancers. Although the reported frequencies of p53 mutation vary among the types of cancer, it is estimated that more than half of cancers might have inactivated p53 due to mutations, deletion, loss of heterozygosity of the gene, or decreased expression.(47,48) Although inactivating mutations in ATM, BRCA1, or BRCA2 are less frequent than those in the p53 gene,(49–53) decreased expression of ATM, the MRN complex, Chk2, RAD51, BRCA1, BRCA2, and ERCC1 is frequently observed, suggesting that aberration of the DNA damage response is common in sporadic cancers.(19,22,23,26,54–62) Promoter hypermethylation of the BRCA1 gene is frequently observed and may be one of the predominant mechanisms for deregulation of the BRCA1 gene.(62) Furthermore, our group reported the functional inactivation of BRCA2 in cancer cells aberrantly expressing SYCP3, a cancer-testis antigen.(63) Disruption of the FA pathway resulting from mutations or decreases or loss of expression due to promoter hypermethylation has been also described in various cancers.(64,65)
As described above, both activation and inactivation of the DNA damage response are observed in cancers, and are expected to determine important properties of the DNA damage response machinery present in each cancer. The status of BRCA has been adopted as an important condition factor in current clinical trials, however, the status of other DNA damage response proteins have not yet been translated into clinical trials. In the next section, we will introduce various approaches for taking advantage of these cancer-specific properties of the DNA damage response in cancer therapy.
How Can Different DNA Damage Response Pathways be Targeted for Cancer Therapy?
Because the efficacy of cancer chemotherapy and radiotherapy relies on generation of DNA damage that will be recognized and repaired by intrinsic DNA repair pathways, aberrant expression of a particular DNA damage response protein should be a biomarker of resistance or favorable response to therapies that induce the corresponding types of DNA damage.(66) For example, patients with surgically treated non-small-cell lung cancer whose tumors lacked expression of ERCC1 were shown to benefit from cisplatin-based adjuvant chemotherapy in a clinical study.(38) Another example is the case of RAD51, whose expression can serve as a marker of cisplatin resistance in non-small-cell lung cancer, which is consistent with the role of HR in the repair of ICL.(31)
In contrast, many inhibitors of the DNA damage response have been developed and some of them have been tested for their potential to enhance DNA damage-induced tumor cell killing in preclinical studies and clinical trials (Tables 2 and 3).
Table 2.
Examples of DNA damage response inhibitors in preclinical studies
| Pathway | Target(s) | Name(s) | Preclinical evidence |
|---|---|---|---|
| DNA damage | MRE11 | Mirin, telomelysin | Sensitization to ionizing radiation |
| sensors and mediators | ATM | KU55933, KU60019, CP466722 | Sensitization to ionizing radiation and topoisomerase inhibitors |
| ATR | Schisandrin B | Sensitization to UV treatment | |
| NU6027, VE-821 | Sensitization to ionizing radiation and a variety of chemotherapy | ||
| Cell cycle checkpoints | Chk1 | SAR-020106 | Sensitization to irinotecan and gemcitabine |
| Chk2 | VRX0466617 | Sensitization to ionizing radiation | |
| Non-homologous end joining | DNA-PK | NU7026, NU7441 | Sensitization to ionizing radiation and topoisomerase II inhibitors |
| DNA-PK and PI3K | KU-0060648 | Sensitization to etoposide and doxorubicin | |
| DNA ligase IV | SCR7 | Sensitization to ionizing radiation and etoposide | |
| Alternative non-homologous end joining | DNA ligases I and IIIα | L67 | Sensitization to ionizing radiation and methyl methanesulfonate |
| Homologous recombination (HR) | RAD51 | B02, A03, A10 | Identified by high-throughput screenings of RAD51 inhibitors |
Table 3.
Examples of DNA damage response inhibitors in clinical trials
| Pathway | Target(s) | Name | Combination | Type of cancer | Clinical trial number | Stage | Trial periods |
|---|---|---|---|---|---|---|---|
| Cell cycle checkpoints | Chk1 | UCN-01 | Combination therapy | ||||
| Carboplatin | Advanced solid tumor | NCT00036777 | Phase I | Completed | |||
| Irinotecan | Metastatic or unresectable solid tumor, triple negative breast cancer | NCT00031681 | Phase I | Completed | |||
| Cytarabine | Refractory or relapsed acute myelogenous leukemia, myelodysplastic syndrome | NCT00004263 | Phase I | Completed | |||
| Perifosine | Relapsed or refractory acute leukemia, chronic myelogenous leukemia, high risk myelodysplastic syndrome | NCT00301938 | Phase I | Completed | |||
| Gemcitabine | Unresectable or metastatic pancreatic cancer | NCT00039403 | Phase I | Completed | |||
| Topotecan | Relapsed or progressed small-cell lung cancer | NCT00098956 | Phase II | Completed | |||
| Cisplatin | Advanced malignant solid tumor | NCT00012194 | Phase I | Terminated | |||
| Fluorouracil | Metastatic pancreatic cancer | NCT00045747 | Phase II | Completed | |||
| Prednisone | Refractory solid tumor, lymphoma | NCT00045500 | Phase I | Completed | |||
| Irinotecan | Advanced solid tumor | NCT00047242 | Phase I | Completed | |||
| Fluorouracil, leucovorin | Metastatic or unresectable solid tumor | NCT00042861 | Phase I | Completed | |||
| Topotecan | Advanced ovarian epithelial, primary peritoneal, fallopian tube cancer | NCT00072267 | Phase II | Completed | |||
| Fludarabine | Recurrent or refractory lymphoma or leukemia | NCT00019838 | Phase I | Completed | |||
| Fluorouracil | Advanced or refractory solid tumor | NCT00004059 | Phase I | Completed | |||
| Cisplatin | Advanced or metastatic solid tumor | NCT00006464 | Phase I | Completed | |||
| Topotecan | Recurrent ovarian epithelial cancer, fallopian tube cancer, primary peritoneal cavity cancer | NCT00045175 | Phase I | Completed | |||
| Fludarabine | Chronic lymphocytic leukemia or lymphocytic lymphoma | NCT00045513 | Phase I, II | Active, not recruiting | |||
| Monotherapy | |||||||
| Relapsed or refractory T-cell lymphoma | NCT00082017 | Phase II | Completed | ||||
| Metastatic melanoma | NCT00072189 | Phase II | Completed | ||||
| Breast cancer, lymphoma, prostatic neoplasm | NCT00001444 | Phase I | Completed | ||||
| Leukemia/lymphoma/unspecified adult solid tumor | NCT00003289 | Phase I | Completed | ||||
| Advanced or metastatic kidney cancer | NCT00030888 | Phase II | Active, not recruiting | ||||
| SCH900776 | Combination therapy | ||||||
| Cytarabine | Relapsed acute myeloid leukemia | NCT01870596 | Phase II | Until January, 2016 | |||
| Cytarabine | Acute myelogenous leukemia/acute lymphocytic leukemia | NCT00907517 | Phase I | Terminated | |||
| Gemcitabine | Solid tumor/lymphoma | NCT00779584 | Phase I | Completed | |||
| Hydroxyurea | Advanced solid tumors | NCT01521299 | Phase I | Withdrawn | |||
| LY2603618 | Combination therapy | ||||||
| Desipramine, pemetrexed, gemcitabine | Cancer | NCT01358968 | Phase I | Completed | |||
| Pemetrexed, gemcitabine | Advanced or metastatic solid tumor | NCT01296568 | Phase I | Completed | |||
| Pemetrexed,cisplatin | NSCLC | NCT01139775 | Phase I, II | Until March, 2014 | |||
| Gemcitabine | Pancreatic cancer | NCT00839332 | Phase I, II | Completed | |||
| Gemcitabine | Solid tumor | NCT01341457 | Phase I | Until December, 2014 | |||
| Pemetrexed | Cancer | NCT00415636 | Phase I | Completed | |||
| Pemetrexed | NSCLC | NCT00988858 | Phase II | Until April, 2014 | |||
| Chk1 and Chk2 | XL844 | Combination therapy | |||||
| Gemcitabine | Advanced cancer, lymphoma | NCT00475917 | Phase I | Terminated | |||
| Monotherapy | |||||||
| Advanced cancer, lymphoma | NCT00475917 | Phase I | Terminated | ||||
| Chronic lymphocytic leukemia | NCT00234481 | Phase I | Terminated | ||||
| AZD7762 | Combination therapy | ||||||
| Gemcitabine | Solid tumor | NCT00413686 | Phase I | Completed | |||
| Gemcitabine | Solid tumor | NCT00937664 | Phase I | Terminated | |||
| Irinotecan | Solid tumor | NCT00473616 | Phase I | Terminated | |||
| PF-00477736 | Combination therapy | ||||||
| Gemcitabine | Advanced solid tumor | NCT00437203 | Phase I | Terminated | |||
| Non-homologous end joining | DNA-PK and mTOR | CC-115 | Monotherapy | ||||
| Multiple myeloma, non-Hodgkin's lymphoma, glioblastoma, squamous cell carcinoma of head and neck, | NCT01353625 | Phase I | Until April, 2015 | ||||
| prostate cancer, Ewing's osteosarcoma, chronic lymphocytic leukemia | |||||||
| Base excision repair | APE1 | TRC102 | Combination therapy | ||||
| Pemetrexed | Neoplasm | NCT00692159 | Phase I | Completed | |||
| Temozolomide | Lymphoma, solid tumor | NCT01851369 | Phase I | Until February, 2015 | |||
| Fludarabine | Relapsed or refractory hematologic malignancy | NCT01658319 | Phase I | Until January, 2015 | |||
| Lucanthone | Combination therapy | ||||||
| Radiotherapy | Brain metastases from NSCLC | NCT02014545 | Phase II | Until Decemcer, 2017 | |||
| Temozolomide and radiation | Glioblastoma multiforme | NCT01587144 | Phase II | Terminated | |||
| PARP | Rucaparib (AG014688) | Combination therapy | |||||
| Cisplatin | Triple negative breast cancer or ER/PR+, HER2− breast cancer with known BRCA1/2 mutations | NCT01074970 | Phase II | Until May, 2014 | |||
| Carboplatin | Advanced solid tumor | NCT01009190 | Phase I | Until Dec, 2013 | |||
| Monotherapy | |||||||
| Platinum-sensitive, relapsed, high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer | NCT01891344 | Phase II | Until December, 2015 | ||||
| Solid tumor (Phase I), ovarian cancer with germline BRCA mutations (Phase II) | NCT01482715 | Phase I, II | Until March, 2014 | ||||
| Platinum-sensitive, high-grade serous or endometrioid epithelial ovarian, primary peritoneal or fallopian tube cancer | NCT01968213 | Phase III | Until November, 2016 | ||||
| BRCA-mutated locally advanced or metastatic breast cancer or advanced ovarian cancer | NCT00664781 | Phase II | Until September, 2014 | ||||
| Olaparib (AZD2281) | Combination therapy | ||||||
| Cediranib | Recurrent ovarian, fallopian tube, peritoneal cancer or recurrent triple-negative breast cancer | NCT01116648 | Phase I, II | Until May, 2014 | |||
| Abiraterone, prednisone, or prednisolone | Metastatic castration-resistant prostate cancer | NCT01972217 | Phase II | Until July, 2018 | |||
| Bkm120 | Recurrent triple-negative breast cancer or recurrent high-grade serous ovarian cancer | NCT01623349 | Phase I | Until Dec, 2014 | |||
| Radiotherapy | Esophageal cancer | NCT01460888 | Phase I | Until August, 2018 | |||
| Paclitaxel | Recurrent or metastatic gastric cancer | NCT01063517 | Phase II | Completed | |||
| Radiotherapy with or without cisplatin | Locally advanced NSCLC | NCT01562210 | Phase I | Until March, 2015 | |||
| Irinotecan, cisplatin, mitomycin C | Advanced pancreatic cancer | NCT01296763 | Phase I, II | Until January, 2016 | |||
| Temozolomide | Relapsed glioblastoma | NCT01390571 | Phase I | Until September, 2015 | |||
| Paclitaxel | Advanced gastric cancer | NCT01924533 | Phase III | Until December, 2017 | |||
| Carboplatin and paclitaxel | Stage III, stage IV relapsed ovarian cancer or uterine cancer | NCT01650376 | Phase I, II | Until February, 2015 | |||
| Radiation therapy and cetuximab | Advanced squamous cell carcinoma of the head/neck with heavy smoking histories | NCT01758731 | Phase I | Until July, 2016 | |||
| Gefitinib | EGFR mutation-positive advanced NSCLC | NCT01513174 | Phase I, II | Until June, 2015 | |||
| Temozolomide | Advanced Ewing's sarcoma | NCT01858168 | Phase I | Until July, 2017 | |||
| Carboplatin | Mixed muellerian cancer, cervical cancer, ovarian cancer, breast cancer, primary peritoneal cancer, fallopian tube cancer, | NCT01237067 | Phase I | Until September, 2014 | |||
| endometrial cancer, carcinosarcoma | |||||||
| Carboplatin and paclitaxel | Advanced ovarian cancer | NCT01081951 | Phase II | Until June, 2013 | |||
| Cisplatin-based chemoradiotherapy | Locally advanced squamous cell caricinoma of the head and neck | NCT01491139 | Phase I | Withdrawn | |||
| Irinotecan | Triple-negative metastatic breast cancer, advanced ovarian cancer | NCT00535353 | Phase I | Until December, 2013 | |||
| Carboplatin and/or paclitaxel | Locally advanced or metastatic colorectal cancer | NCT00516724 | Phase I | Until December, 2014 | |||
| Dacarbazine | Advanced melanoma | NCT00516802 | Phase I | Completed | |||
| Paclitaxel | Metastatic triple negative breast cancer | NCT00707707 | Phase I | Until December, 2012 | |||
| Liposomal doxorubicin | Advanced solid tumor | NCT00819221 | Phase I | Until August, 2013 | |||
| Topotecan | Advanced solid tumor | NCT00516438 | Phase I | Completed | |||
| Gemcitabine | Pancreatic cancer | NCT00515866 | Phase I | Completed | |||
| Bevacizumab | Advanced solid tumor | NCT00710268 | Phase I | Completed | |||
| Cisplatin | Advanced solid tumor | NCT00782574 | Phase I | Until December, 2014 | |||
| Carboplatin | Breast and ovarian cancer with BRCA mutations or family histories | NCT01445418 | Phase I | Recruiting | |||
| Monotherapy | |||||||
| Advanced solid tumor | NCT01900028 | Phase I | Until February, 2015 | ||||
| Advanced solid tumor | NCT01921140 | Phase I | Until March, 2015 | ||||
| Advanced solid tumor | NCT01929603 | Phase I | Until May, 2015 | ||||
| Advanced solid tumor | NCT01851265 | Phase I | Until July, 2014 | ||||
| Advanced solid tumor with normal or impaired liver function | NCT01894243 | Phase I | Until December, 2015 | ||||
| Advanced solid tumor normal or impaired kidney function | NCT01894256 | Phase I | Until December, 2015 | ||||
| Metastatic breast cancer with germline BRCA1/2 mutations | NCT02000622 | Phase III | Until February, 2021 | ||||
| Advanced castration-resistant prostate cancer | NCT01682772 | Phase II | Until July, 2015 | ||||
| Advanced solid tumor | NCT01813474 | Phase I | Until November, 2014 | ||||
| BRCA-mutated ovarian cancer after a complete or partial response following platinum-based chemotherapy | NCT01874353 | Phase III | Until June, 2020 | ||||
| BRCA-mutated advanced cancer | NCT01078662 | Phase II | Until December, 2013 | ||||
| BRCA-mutated advanced ovarian cancer following first line platinum based chemotherapy | NCT01844986 | Phase III | Until January, 2022 | ||||
| Advanced Ewing's sarcoma | NCT01583543 | Phase II | Until April, 2015 | ||||
| Stage IV colorectal cancer with microsatellite instability | NCT00912743 | Phase II | Completed | ||||
| BRCA-deficient ovarian, peritoneal, fallopian tube cancer | NCT01661868 | Phase II | Withdrawn | ||||
| Advanced NSCLC | NCT01788332 | Phase II | Until May, 2015 | ||||
| BRCA-positive advanced breast cancer | NCT00494234 | Phase II | Until December, 2013 | ||||
| BRCA-positive advanced ovarian cancer | NCT00494442 | Phase II | Until December, 2013 | ||||
| Platinum-sensitive relapsed serous ovarian cancer | NCT00753545 | Phase II | Completed | ||||
| Advanced solid tumor | NCT00572364 | Phase I | Completed | ||||
| Advanced or metastatic solid tumor | NCT00633269 | Phase I | Completed | ||||
| Ovarian cancer | NCT00516373 | Phase I | Until December, 2014 | ||||
| Advanced solid tumor | NCT00777582 | Phase I | Until March, 2014 | ||||
| High grade ovarian cancer, triple-negative breast cancer, BRCA-mutated breast cancer or ovarian cancer | NCT00679783 | Phase II | Until December, 2012 | ||||
| BRCA-positive advanced ovarian cancer | NCT00628251 | Phase II | Until December, 2013 | ||||
| Veliparib (ABT-888) | Combination therapy | ||||||
| Gemcitabine, cisplatin | Locally advanced or metastatic pancreatic cancer with BRCA or PALB2 mutations | NCT01585805 | Phase II | Until July, 2017 | |||
| Temozolomide or combination with carboplatin and paclitaxel | Locally recurrent or metastatic breast cancer with BRCA mutations | NCT01506609 | Phase II | Until May, 2015 | |||
| Radiotherapy and temozolomide | Newly diagnosed childhood diffuse pontine glioma | NCT01514201 | Phase I, II | Until August, 2019 | |||
| Radiotherapy | Advanced solid malignancies with peritoneal carcinomatosis | NCT01264432 | Phase I | Until April, 2014 | |||
| Bendamustine, rituximab | Advanced lymphoma, multiple myeloma, or solid tumors | NCT01326702 | Phase I, II | Until November, 2015 | |||
| Topotecan | Relapsed epithelial ovarian, primary fallopian tube, or primary peritoneal cancer with negative or unknown BRCA status | NCT01690598 | Phase I, II | Until April, 2015 | |||
| Gemcitabine and radiotherapy | Locally advanced, unresectable pancreatic cancer | NCT01908478 | Phase I | Until July, 2019 | |||
| Dinaciclib with or without carboplatin | Advanced solid tumors with BRCA mutations | NCT01434316 | Phase I | Until January, 2016 | |||
| Radiotherapy, carboplatin, paclitaxel | Stage III NSCLC that cannot be removed by surgery | NCT01386385 | Phase I, II | Until December, 2016 | |||
| Doxorubicin, carboplatin, bevacizumab | Recurrent ovarian cancer, primary peritoneal cancer, or fallopian tube cancer | NCT01459380 | Phase I | Until August, 2015 | |||
| Cisplatin, gemcitabine | Advanced biliary, pancreatic, urothelial, NSCLC | NCT01282333 | Phase I | Terminated | |||
| Cisplatin, vinorelbine | Recurrent and/or metastatic breast cancer with BRCA mutations, triple-negative breast cancer | NCT01104259 | Phase I | Until September, 2014 | |||
| Mitomycin C | Metastatic, unresectable, or recurrent solid tumor | NCT01017640 | Phase I | Until June, 2014 | |||
| Capecitabine, radiotherapy | Locally advanced rectal cancer | NCT01589419 | Phase I | Until June, 2015 | |||
| Cyclophosphamide | Locally advanced or metastatic HER2-negative breast cancer | NCT01351909 | Phase I, II | Until May, 2015 | |||
| Docetaxel, cisplatin, fluorouracil, radiotherapy, hydroxyurea, paclitaxel | Stage IV head and neck cancer Solid tumor | NCT01193140 | Phase II | Completed | |||
| Temozolomide | NCT01711541 | Phase I, II | Until October, 2014 | ||||
| Cisplatin, etoposide | Extensive stage small-cell lung cancer, metastatic large cell neuroendocrine NSCLC, small-cell carcinoma of unknown primary or extrapulmonary origin | NCT01642251 | Phase I, II | Until January, 2018 | |||
| Paclitaxel, carboplatin | Metastatic, unresectable solid tumor with liver or kidney dysfunction | NCT01366144 | Phase I | Until July, 2015 | |||
| Oxaliplatin, capecitabine | BRCA-related malignancy, metastatic colorectal cancer, metastatic ovarian cancer, | NCT01233505 | Phase I | Until July, 2014 | |||
| metastatic gastrointestinal malignancies in which oxaliplatin has shown some activity | |||||||
| Carboplatin | Stage III or stage IV breast cancer with BRCA mutations | NCT01149083 | Phase II | Until June, 2014 | |||
| Temozolomide | Acute leukemia | NCT01139970 | Phase I | Until October, 2013 | |||
| Carboplatin, paclitaxel | Solid tumor | NCT01617928 | Phase I | Completed | |||
| Topotecan | Recurrent ovarian epithelial cancer, primary peritoneal cavity cancer, unspecified solid tumor | NCT01012817 | Phase I, II | Until June, 2018 | |||
| Carboplatin, paclitaxel | Advanced NSCLC | NCT01560104 | Phase II | Until September, 2014 | |||
| Carboplatin | HER2-negative metastatic or locally advanced breast cancer | NCT01251874 | Phase I | Until September, 2013 | |||
| Paclitaxel, cisplatin | Advanced, persistent, or recurrent cervical cancer | NCT01281852 | Phase I, II | Until March, 2020 | |||
| Topotecan with or without carboplatin | Relapsed or refractory acute leukemia, high-risk myelodysplasia, or aggressive myeloproliferative disorders | NCT00588991 | Phase I | Until December, 2012 | |||
| Abiraterone, prednisone | Metastatic hormone-resistant prostate cancer | NCT01576172 | Phase II | Until February, 2014 | |||
| Topotecan and filgrastim or pegfilgrastim | Persistent or recurrent cervical cancer | NCT01266447 | Phase II | Until November, 2016 | |||
| Gemcitabine | Solid tumor | NCT01154426 | Phase I | Until October, 2013 | |||
| Modified FOLFOX6 | Metastatic pancreatic cancer | NCT01489865 | Phase I, II | Until December, 2014 | |||
| FOLFIRI | Advanced gastric cancer | NCT01123876 | Phase I | Until December, 2014 | |||
| Temozolomide | Recurrent or refractory childhood central nervous system tumor | NCT00946335 | Phase I | Until October, 2011 | |||
| Temozolomide | Hepatocellular carcinoma | NCT01205828 | Phase II | Until December, 2013 | |||
| Carboplatin, paclitaxel | Advanced solid tumor | NCT01281150 | Phase I | Until December, 2013 | |||
| Carboplatin, paclitaxel, doxorubicin, cyclophosphamide | Stage IIb-IIIc triple-negative breast cancer | NCT01818063 | Phase II | Until April, 2018 | |||
| Floxuridine | Metastatic epithelial ovarian, primary peritoneal cavity, or fallopian tube cancer | NCT01749397 | Phase I | Until March, 2016 | |||
| Liposomal doxorubicin | Recurrent ovarian cancer, fallopian tube cancer, or primary peritoneal cancer or metastatic triple-negative breast cancer | NCT01145430 | Phase I | Until March, 2014 | |||
| Bortezomib, dexamethasone | Relapsed refractory multiple myeloma | NCT01495351 | Phase I | Until October, 2013 | |||
| Temozolomide | Recurrent small-cell lung cancer | NCT01638546 | Phase II | Until June, 2017 | |||
| Cyclophosphamide, doxorubicin | Metastatic or unresectable solid tumor, non-Hodgkin's lymphoma | NCT00740805 | Phase I | Until December, 2013 | |||
| Whole brain radiation | Brain metastases from NSCLC | NCT01657799 | Phase II | Until November, 2014 | |||
| Temozolomide | Recurrent high grade serous ovarian, fallopian tube, or primary peritoneal cancer | NCT01113957 | Phase II | Completed | |||
| Temozolomide | Metastatic or locally advanced breast cancer and BRCA1/2-associated breast cancer | NCT01009788 | Phase II | Until December, 2014 | |||
| Carboplatin, paclitaxel | Advanced cancer with liver or kidney problems | NCT01419548 | Phase I | Withdrawn | |||
| Whole brain radiation | Cancer with brain metastases | NCT00649207 | Phase I | Completed | |||
| Radiotherapy | Inflammatory or loco-regionally recurrent breast cancer | NCT01477489 | Phase I | Until December, 2016 | |||
| Carboplatin, paclitaxel, bevacizumab | Newly diagnosed ovarian epithelial cancer, fallopian tube cancer, or primary peritoneal cancer | NCT00989651 | Phase I | Until July, 2014 | |||
| Carboplatin, paclitaxel | Advanced solid tumor or BRCA1/2-associated advanced solid tumor | NCT00535119 | Phase I | Until October, 2012 | |||
| Temozolomide | Colorectal cancer | NCT01051596 | Phase II | Until December, 2013 | |||
| Cyclophosphamide | Refractory BRCA-positive ovarian, primary peritoneal or ovarian high-grade serous carcinoma, fallopian tube cancer, triple-negative breast cancer, and low-grade non-Hodgkin's lymphoma | NCT01306032 | Phase II | Until November, 2014 | |||
| Irinotecan | Metastatic or unresectable solid tumor, lymphoma | NCT00576654 | Phase I | Until December, 2013 | |||
| Temozolomide | Recurrent or refractory childhood central nervous system tumor | NCT00994071 | Phase I | Completed | |||
| Cyclophosphamide | Refractory solid tumor or lymphoma | NCT01445522 | Phase I | Completed | |||
| Temozolomide | Recurrent high-grade glioma | NCT01026493 | Phase I, II | Until February, 2014 | |||
| Cyclophosphamide | Solid tumor or lymphoma that did not respond to previous therapy | NCT00810966 | Phase I | Active, not recruiting | |||
| Radiotherapy, temozolomide | Grade IV astrocytoma | NCT00770471 | Phase I, II | Completed | |||
| Temozolomide | Metastatic prostate cancer | NCT01085422 | Phase I | Completed | |||
| Temozolomide | Advanced non-hematologic tumor | NCT00526617 | Phase I | Completed | |||
| Topotecan | Refractory solid tumor or lymphoma | NCT00553189 | Phase I | Completed | |||
| Temozolomide | Metastatic melanoma | NCT00804908 | Phase II | Until March, 2014 | |||
| Carboplatin, gemcitabine | Advanced solid tumor | NCT01063816 | Phase I | Until September, 2014 | |||
| Radiotherapy | Breast cancer | NCT01618357 | Phase I | Until April, 2016 | |||
| Monotherapy | |||||||
| Solid tumor | NCT01199224 | Phase I | Completed | ||||
| Locally advanced or metastatic pancreatic cancer | NCT01585805 | Phase II | Until July, 2017 | ||||
| Metastatic, unresectable, or recurrent solid tumors | NCT01017640 | Phase I | Until June, 2014 | ||||
| Stage III or Stage IV breast cancer with BRCA mutations | NCT01149083 | Phase II | Until June, 2014 | ||||
| BRCA-mutated metastatic or unresectable malignancy, high grade serous ovarian, fallopian tube, or peritoneal cancer | NCT01853306 | Phase I | Until January, 2015 | ||||
| BRCA-mutated epithelial ovarian, fallopian tube, or primary peritoneal cancer | NCT01540565 | Phase II | Until April, 2014 | ||||
| Advanced solid tumor | NCT02009631 | Phase I | Until December, 2014 | ||||
| BRCA-related malignancy, platinum-refractory ovarian, fallopian tube, or primary peritoneal cancer or basal-like breast cancer, advanced solid tumor | NCT00892736 | Phase I | Until December, 2013 | ||||
| Relapsed epithelial ovarian, primary fallopian or primary peritoneal cancer with BRCA mutations | NCT01472783 | Phase I, II | Until December, 2015 | ||||
| Chronic lymphocytic leukemia, follicular lymphoma, unspecified solid tumor | NCT00387608 | Phase I | Completed | ||||
| Invasive breast cancer | NCT01042379 | Phase II | Until November, 2014 | ||||
| Advanced solid tumor | NCT01827384 | Phase II | Until March, 2017 | ||||
| INO-1001 | Combination therapy | ||||||
| Temozolomide | Unresectable melanoma | NCT00272415 | Phase I | Terminated | |||
| MK4827 | Combination therapy | ||||||
| Liposomal doxorubicin | Advanced solid tumor, platinum-resistant high grade serous ovarian cancer | NCT01227941 | Phase I | Terminated | |||
| Temozolomide | Advanced solid tumor, glioblastoma multiforme, melanoma | NCT01294735 | Phase I | Completed | |||
| Carboplatin, paclitaxel, liposomal doxorubicin | Advanced solid tumor | NCT01110603 | Phase I | Terminated | |||
| Monotherapy | |||||||
| Advanced solid tumor | NCT01226901 | Phase I | Terminated | ||||
| Mantle cell lymphoma | NCT01244009 | Phase II | Withdrawn | ||||
| Advanced solid tumors, chronic lymphocytic leukemia, T-cell-prolymphocytic leukemia | NCT00749502 | Phase I | Completed | ||||
| Advanced HER2-negative, germline BRCA mutation-positive breast cancer | NCT01905592 | Phase III | Until October, 2015 | ||||
| CEP-9722 | Combination therapy | ||||||
| Gemcitabine, cisplatin | Advanced solid tumor or mantle cell lymphoma | NCT01345357 | Phase I | Completed | |||
| Temozolomide | Advanced solid tumor | NCT00920595 | Phase I | Completed | |||
| Monotherapy | |||||||
| Advanced solid tumor | NCT01311713 | Phase I, II | Terminated | ||||
| Advanced solid tumor | NCT00920595 | Phase I | Completed | ||||
| E7016 | Combination therapy | ||||||
| Temozolomide | Advanced solid tumor | NCT01127178 | Phase I | Completed | |||
| Temozolomide | Wild-type BRAF stage IV melanoma, unresectable stage III melanoma | NCT01605162 | Phase II | Until March, 2014 | |||
| BMN673 | Monotherapy | ||||||
| Acute myeloid leukemia, myelodysplastic syndrome, chronic lymphocytic leukemia, mantle cell lymphoma | NCT01399840 | Phase I | Until June, 2013 | ||||
| Advanced or recurrent solid tumor | NCT01286987 | Phase I | Until June, 2013 | ||||
| Advanced solid tumor with deleterious BRCA mutations | NCT01989546 | Phase I, II | Until August, 2016 | ||||
| Advanced breast cancer with BRCA mutations | NCT01945775 | Phase III | Until June, 2016 | ||||
For current status and information of clinical trials, refer to http://clinicaltrials.gov/, a service of the US National Institutes of Health. NSCLC, non-small-cell lung cancer.
Inhibitors of ATM/ATR and the MRN complex
As ATM and the MRN complex play central roles as sensors or mediators in the DNA damage response, these molecules have been considered to be promising targets for radiosensitization or chemosensitization.(67) Several promising ATM inhibitors have been developed (Table 2). KU55933, the first specific inhibitor of ATM, inhibits radiation-induced ATM-dependent phosphorylation events and sensitizes cancer cells to radiation and topoisomerase inhibitors.(67) KU60019, an improved analog of KU55933, inhibits the DNA damage response and effectively radiosensitizes human glioma cells.(68) Mirin is an inhibitor of the MRN complex, which prevents MRN-dependent activation of ATM without affecting ATM protein kinase activity and inhibits MRE11-associated exonuclease activity.(67) Telomelysin is another inhibitor that inhibits the MRN complex through the adenoviral E1B-55 kDa protein.(67) The therapeutic outcomes of these agents remain to be tested in clinical trials. Although the long search for selective inhibitors of ATR has not yet paid off, schisandrin B was recently identified as a moderate selective ATR inhibitor, although it will also affect ATM at high concentrations.(69) Recently, two novel ATR inhibitors, NU6027 and VE-821, were also shown to sensitize cells to a variety of DNA-damaging agents in preclinical studies.(70,71)
Inhibitors of Chk1/Chk2 and CDC25
As the triggering of cell cycle checkpoints is crucial in the DNA damage response, these checkpoints have also been widely investigated as a potential target for cancer therapy (Table 3).(72) Among the inhibitors for Chk1 and/or Chk2, UCN-01 was the first to enter clinical trials, but it was discontinued due to toxicities such as symptomatic hypotension and neutropenia and a lack of convincing efficacy after phase II trials.(72) Other Chk1/Chk2 inhibitors with improved specificities, including XL844 and AZD7762, also entered clinical trials but failed to achieve a good response.(72) The selective Chk1 inhibitor SCH900776 has been used in phase I trials for acute leukemia in combination with cytarabine and for solid tumors in combination with gemcitabine, and showed some partial responses and stable disease.(72) The Chk1 inhibitor LY2603618 and the dual Chk1/Chk2 inhibitor LY2606368 are also currently being tested in early clinical trials. CDC25 phosphatases, the key factors in cyclin-dependent kinase activation crucial for cell cycle regulation, are also considered to represent promising novel targets in cancer therapy. CDC25 inhibitors have also been developed, and some have entered into clinical trials, although the clinical data is limited.(73)
Inhibition of NHEJ by DNA-PK inhibitors
Regarding NHEJ, inhibitors of DNA-PK, including NU7026 and NU7441, were found to induce extreme sensitivity to ionizing radiation as well as DNA-damaging agents in preclinical studies (Table 2).(74) However, the therapeutic efficacy of DNA-PK inhibitors depends on the expression levels of DNA-PK in cancer cells versus normal cells, and their clinical application is currently restricted because of their toxicity to normal cells. The dual mTOR and DNA-PKcs inhibitor CC-115 is undergoing early clinical evaluation (Table 3). KU-0060648 is a potent dual inhibitor of DNA-PK and PI-3K, which has recently been reported to enhance etoposide and doxorubicin cytotoxicity (Table 2).(75)
Inhibition of NHEJ or alt-NHEJ by DNA ligase inhibitors
DNA ligases are required for both NHEJ and alt-NHEJ pathways as well as other DNA repair pathways such as BER and NER. Small molecule inhibitors of human DNA ligases have been identified and shown to be cytotoxic and also to enhance the cytotoxicity of DNA-damaging agents. SCR7 is an inhibitor of DNA ligase IV, which is involved in the NHEJ pathway. SCR7 reduces cell proliferation in a DNA ligase IV-dependent manner and increases the tumor-inhibitory effects of agents that cause DSBs.(76) L67 is an inhibitor of DNA ligases I and IIIα, which are involved in the alt-NHEJ pathway as well as BER and NER. The levels of the alt-NHEJ proteins such as DNA ligase IIIα and WRN are reported to be elevated in BCR-ABL-positive CML cell lines,(77) so inhibition of alt-NHEJ factors may be an additional therapeutic approach in BCR-ABL-positive CML, which is usually treated by tyrosine kinase inhibitors. Indeed, CML cell lines with increased alt-NHEJ were shown to be hypersensitive to the combination of L67 and PARP inhibitor.(78)
Inhibitors of RAD51 and tyrosine kinases regulating HR
With respect to HR, there are currently few inhibitors that directly target HR proteins. Along with the RAD51 inhibitors that were recently identified (Table 2,79) the molecules that indirectly regulate HR may also be candidate targets for inhibiting HR. For example, the non-receptor tyrosine kinase c-Abl is activated by ATM in response to DNA damage, and subsequently phosphorylates RAD51.(80) Oncogenic fusion tyrosine kinases, such as BCR-ABL, TEL-ABL, TEL-JAK2, TEL-PDGFβR, and NPM-ALK, enhance the expression levels and/or tyrosine phosphorylation of RAD51.(81,82) From these findings, inhibitors of oncogenic tyrosine kinases are expected to sensitize cancer cells to DNA-damaging agents. Consistent with this hypothesis, treatments with the tyrosine inhibitor imatinib have been shown to enhance sensitivity to DNA crosslinking agents and ionizing radiation in cancer cells.(81) Furthermore, targeting RAD51 was shown to overcome imatinib resistance in CML cells.(83)
Inhibitors of histone deacetylases, heat shock protein 90, and DSB repair
Histone deacetylases (HDACs) are powerful regulators of the stability of the genome, and many HDAC inhibitors are shown to downregulate multiple components of the DNA damage response and repair, including HR, NHEJ, the MRN complex, and ATM.(84) Thus, the use of HDAC inhibitors in combination with DNA-damaging agents may be an area of great interest with potential clinical utility. The HDAC inhibitor PCI-24781 caused increased apoptosis by inhibiting RAD51-mediated HR when used in combination with the PARP inhibitor PJ34 in a preclinical study.(85) The inhibitor of heat shock protein 90, 17-allylamino-17-demethoxygeldanamycin, radiosensitizes human tumor cell lines by inhibiting RAD51-mediated HR.(86) Curcumin is a natural product that has been tested for its chemosensitizing potential, and sensitizes cancer cells to PARP inhibitors by inhibiting NHEJ, HR, and the DNA damage checkpoint.(87)
Inhibitors of PARP and APE1 in combination with DNA-damaging agents
Inhibitors of PARP, which inhibit the BER and SSB repair pathways, are the most advanced and promising drugs that target DNA repair.(88) A number of clinical trials using PARP inhibitors are currently underway (Table 3). Inhibitors of PARP were first tried in combination with DNA-damaging agents. Some clinical responses were observed in the phase I and II trials of the PARP inhibitor rucaparib in combination with temozolomide.(89,90) Further clinical trials of PARP inhibitors have been carried out in combination with various DNA-damaging agents and/or ionizing radiation (Table 3). Inhibitors of another BER protein APE1 are also being tested in combination with DNA-damaging agents in clinical trials (Table 3).
Using PARP inhibitors as single agents in BRCA-deficient cancers based on the principle of synthetic lethality
In 2005, PARP inhibitors were shown to selectively inhibit the growth of cells with defects in either the BRCA1 or BRCA2 genes, suggesting a new use of PARP inhibitors as single agents.(91,92) A possible explanation for this lethality is as follows. The cancer cells with defects in the BRCA gene are defective in HR, as the wild-type BRCA allele is absolutely lost. However, HR is intact in normal cells of the same patients who carry one wild-type BRCA allele and one mutant BRCA allele. Inhibition of PARP1 results in the accumulation of SSBs, which are converted to lethal DSBs that require HR for their repair. Although such lesions would be repaired by HR in normal cells, they are not repaired in BRCA1- or BRCA2-deficient cancer cells because these cells are defective in HR repair, and thus the tumor cells are led to death. This concept is termed synthetic lethality, namely, the process by which defects in two different genes or pathways together result in cell death while defects in one of the two different genes or pathways do not affect viability (Fig. 3).(3) This attractive new therapeutic strategy based on the principle of synthetic lethality relies on the frequent defects in the DNA damage response observed in cancer as summarized in the previous chapter and Table 1, in which alternative DNA damage response pathways may be activated to allow cancer cells to survive in the presence of genotoxic stress. Because this strategy targets the cancer-specific aberrations in the DNA damage response, it will cause few or no toxicities on normal cells. The first report of a clinical trial of a PARP inhibitor as a single agent in patients with BRCA mutations was the phase I study of the oral PARP inhibitor olaparib.(93) It established the safety of olaparib as a single agent, and good responses were observed in patients with BRCA-mutated breast, ovarian, or prostate tumors. In subsequent phase II studies, approximately one-third of the patients with breast or ovarian cancer with germline BRCA mutations showed a favorable response to the drug with no severe toxicities.(94) Several other PARP inhibitors are currently being investigated in patients with germline BRCA mutations as single agents (Table 3). It is likely that PARP inhibitors have significant benefit to at least a subpopulation of cancer patients with defects in BRCA-mediated HR pathways.
Fig. 3.
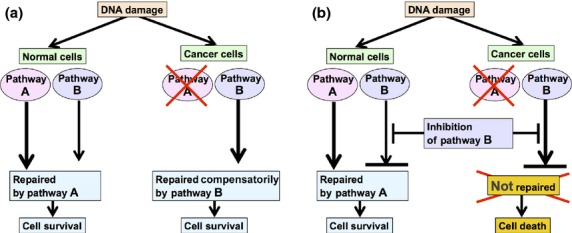
Principle of synthetic lethality. DNA damage is often processed by multiple DNA repair pathways. In the example shown here, pathways A and B are both intact in normal cells, whereas pathway A is defective in cancer cells. (a) In the absence of the pathway B inhibitor, cancer cells can survive, because the defect in pathway A is compensated by the alternative pathway B. (b) When the cells are treated with the pathway B inhibitor, both pathways will be blocked in cancer cells, which will result in cell death. However, normal cells will not be affected, because inhibition of pathway B will be compensated by pathway A.
Using PARP inhibitors as single agents in cancers with no BRCA mutations
The potential for PARP inhibitors as single agents has also been tested in clinical trials of cancers with no germline BRCA mutations, such as high-grade serous ovarian cancers and triple-negative breast cancers.(95) Inhibitors of PARP were also effective in a subset of cancers with no germline BRCA mutations, suggesting that there may be a subset of sporadic cancers that show features of “BRCAness,” which may show good response to PARP inhibitors.(96) Indeed, cancer cells expressing the cancer-testis antigen SYCP3, in which BRCA2 is functionally inactivated, as described above, show extreme hypersensitivity to a PARP inhibitor.(63) Defects in other HR-related proteins such as RAD51, RAD54, and RPA also confer selective sensitivity to PARP inhibition.(97) Moreover, defects in the DNA damage response proteins, such as NBS1, MRE11, ATR, ATM, FANCD2, FANCA, FANCC, Chk1, Chk2, and ERCC1, also confer selective sensitivity to PARP inhibition.(97,98)
Exploitation of other synthetic lethalities by DNA damage response
Taking advantage of the dysregulated DNA damage response in cancer using the synthetic lethality approach may be one of the most promising prospects for the future of cancer treatment. From this point of view, many efforts have been made to identify defects of two different DNA damage response genes or pathways that are synthetically lethal when combined. For example, ATM inhibition is shown to be synthetically lethal with FA pathway deficiency.(99) The suggested explanation for this lethality is as follows. The FA pathway-deficient cancer cells are defective in the repair of DNA replication fork stalling, which is normally repaired by ATR and the FA pathway. In FA pathway-deficient conditions, the stalled fork will collapse and form a DSB that will alternatively activate an ATM-dependent DNA damage response. Inhibition of ATM in such FA pathway-deficient cells will leave no alternative mechanism for repair, leading to cell death. The FA pathway-deficient cells are also hypersensitive to Chk1 silencing,(100) which may be explained by the hyperdependence of the FA pathway-deficient cells on G2/M checkpoint activation mediated by Chk1 for viability. Because defects in the FA pathway are frequently observed in a number of different types of cancer (Table 1,64,65) the use of ATM inhibitors or Chk1 inhibitors in FA pathway-deficient tumors will be a promising approach that should be evaluated in clinical trials in the future. In another example, RAD54B deficiency is shown to be synthetically lethal in cells with reduced Flap endonuclease 1 expression, but the mechanisms of this lethality remain to be elucidated.(101) Recently, inhibition of APE1 was shown to be synthetically lethal in BRCA- and ATM-deficient cells, presenting a novel model for APE inhibition as a synthetic lethal strategy in cells deficient in DSB repair.(102) Briefly, APE1 inhibition leads to AP site accumulation and results in indirect generation of SSBs that are eventually converted to toxic DSBs, which cannot be repaired in cells deficient in DSB repair. The APE1 inhibitors are being tested in combination with DNA-damaging agents in current clinical trials, and they may be evaluated further as a synthetic lethal strategy. More recently, inactivation of the HR protein RAD52 was shown to be synthetically lethal with deficiencies in BRCA2, BRCA1, and PALB2.(103,104) This lethal effect may be due to the loss of RAD51-dependent HR function mediated by the BRCA1–PALB2–BRCA2 complex, because human RAD52 is suggested to function in an independent and alternative repair pathway of RAD51-dependent HR when deficiencies exist in BRCA1, PALB2, or BRCA2. As no inactivating mutations of RAD52 have been documented in human sporadic cancers, inhibition of RAD52 could be an attractive strategy for improving cancer therapy in the BRCA- or PALB2-defective subgroup of cancers. Although no inhibitors of RAD52 have been developed yet, it would be of great interest to assess the effects of inhibition of RAD52 on cancer-specific killing of the cancers with “BRCAness” profiles and compare them with those of PARP inhibitors in future clinical trials. There might be additional synthetic lethalities to be discovered and exploited in future.
Current Limitations and Future Perspectives
Although the data from clinical trials of the inhibitors of DNA damage response, including PARP inhibitors, seem encouraging, we should note that the use of PARP inhibitors also faces significant limitations.
The first limitation is the evolution of resistance. In the case of using PARP inhibitors in cancer cells carrying mutations in BRCA1 or BRCA2, the drug resistance can be caused by secondary mutations in the BRCA1 or BRCA2 gene that restore the open reading frame of the gene and enable the generation of functional BRCA proteins possessing the ability to repair DNA damage caused by PARP inhibitors.(105–107) Other suggested mechanisms underlying the resistance to PARP inhibitors include the loss of 53BP1 expression in BRCA-deficient cells and the upregulation of genes that encode P-glycoprotein efflux pumps,(108–111) although the importance of these factors in clinical resistance to PARP inhibitors has not been elucidated. In future clinical trials, it would be desirable to periodically monitor the sequences of BRCA1 and BRCA2 and the expression levels of the key proteins such as 53BP1 or P-glycoprotein efflux pumps.
The second limitation is the lack of reliable biomarkers of response or resistance to the inhibitors. There is a pressing need to identify biomarkers to predict the response to the inhibitors. Regarding the sensitivities to PARP inhibitors, elevated levels of PARP and CDK12 deficiency are suggested to be possible biomarkers for favorable responses.(45,112) We should also keep in mind that many factors might affect the DNA damage response and take into account the complexity of the networks regulating DNA repair. For instance, most cancer cells grow under hypoxia, a condition that activates hypoxia inducible factor-1 (HIF-1). Because HIF-1 contributes to therapy resistance, it is considered an attractive target molecule for cancer therapy. Diverse functional interactions between HIF-1 and the DNA damage response have also been described,(113) so the efficacy of the combination of HIF-1 inhibitors and inhibitors of the DNA damage response proteins should be examined in the future.
Conclusions
Defects or upregulation of the proteins involved in DNA damage response and repair are common in cancers, and may be induced by both genetic and epigenetic causes. Inhibition of the DNA damage response proteins can be used to enhance chemotherapy and radiotherapy, and also to selectively kill cancer cells showing deficiencies in particular DNA repair pathway(s) based on the principle of synthetic lethality. Inhibition of PARP in BRCA-defective cancers seemed effective in early clinical trials. Better understanding of the basic biology underlying the DNA damage response and the mechanisms responsible for its dysregulation in cancer will provide exciting opportunities for new and efficient cancer therapy targeting the DNA damage response.
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (Kakenhi) (grant nos. 23591836 and 25125705, to N. Hosoya) and by grants from the Takeda Science Foundation and from the Naito Foundation (to N. Hosoya).
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeijmakers JHJ. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 3.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–90. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 5.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 6.Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 7.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 8.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–39. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmermann M, de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014;24:108–17. doi: 10.1016/j.tcb.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–68. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dueva R, Iliakis G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl Cancer Res. 2013;2:163–77. [Google Scholar]
- 12.Dianov GL, Hübscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013;41:3483–90. doi: 10.1093/nar/gkt076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamileri I, Karakasilioti I, Garinis GA. Nucleotide excision repair: new tricks with old bricks. Trends Genet. 2012;28:566–73. doi: 10.1016/j.tig.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartkova J, Horejsí Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 17.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 18.Sun M, Guo X, Qian X, et al. Activation of the ATM-Snail pathway promotes breast cancer metastasis. J Mol Cell Biol. 2012;4:304–15. doi: 10.1093/jmcb/mjs048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dzikiewicz-Krawczyk A. The importance of making ends meet: mutations in genes and altered expression of proteins of the MRN complex and cancer. Mutat Res. 2008;659:262–73. doi: 10.1016/j.mrrev.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Yang M-H, Chiang W-C, Chou T-Y, et al. Increased NBS1 expression is a marker of aggressive head and neck cancer and overexpression of NBS1 contributes to transformation. Clin Cancer Res. 2006;12:507–15. doi: 10.1158/1078-0432.CCR-05-1231. [DOI] [PubMed] [Google Scholar]
- 21.Gao J, Zhang H, Arbman G, Sun XF. The different roles of hRAD50 in microsatellite stable and unstable colorectal cancers. Dis Markers. 2008;24:127–34. doi: 10.1155/2008/724796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grabauskiene S, Bergeron EJ, Chen G, et al. CHK1 levels correlate with sensitization to pemetrexed by CHK1 inhibitors in non-small cell lung cancer cells. Lung Cancer. 2013;82:477–84. doi: 10.1016/j.lungcan.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong J, Hu K, Yuan Y, et al. CHK1 targets spleen tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J Clin Invest. 2012;122:2165–75. doi: 10.1172/JCI61380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madoz-Gurpide J, Canamero M, Sanchez L, Solano J, Alfonso P, Casal JI. A proteomics analysis of cell signaling alterations in colorectal cancer. Mol Cell Proteomics. 2007;6:2150–64. doi: 10.1074/mcp.M700006-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Verlinden L, Van den Bempt I, Eelen G, et al. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor/progesterone receptor/HER-2 breast carcinomas. Cancer Res. 2007;67:6574–81. doi: 10.1158/0008-5472.CAN-06-3545. [DOI] [PubMed] [Google Scholar]
- 26.Ehlén Å, Nodin B, Rexhepaj E, et al. RBM3-regulated genes promote DNA integrity and affect clinical outcome in epithelial ovarian cancer. Transl Oncol. 2011;4:212–21. doi: 10.1593/tlo.11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, Li Y, Wang F, et al. Suppressed miR-424 expression via upregulation of target gene Chk1 contributes to the progression of cervical cancer. Oncogene. 2013;32:976–87. doi: 10.1038/onc.2012.121. [DOI] [PubMed] [Google Scholar]
- 28.Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 29.Kase M, Vardja M, Lipping A, Asser T, Jaal J. Impact of PARP-1 and DNA-PK expression on survival in patients with glioblastoma multiforme. Radiother Oncol. 2011;101:127–31. doi: 10.1016/j.radonc.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 30.Bouchaert P, Guerif S, Debiais C, Irani J, Fromont G. DNA-PKcs expression predicts response to radiotherapy in prostate cancer. Int J Radiat Oncol Biol Phys. 2012;84:1179–85. doi: 10.1016/j.ijrobp.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 31.Takenaka T, Yoshino I, Kouso H, et al. Combined evaluation of Rad51 and ERCC1 expressions for sensitivity to platinum agents in non-small cell lung cancer. Int J Cancer. 2007;121:895–900. doi: 10.1002/ijc.22738. [DOI] [PubMed] [Google Scholar]
- 32.Maacke H, Jost K, Opitz S, et al. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19:2791–5. doi: 10.1038/sj.onc.1203578. [DOI] [PubMed] [Google Scholar]
- 33.Hannay JAF, Liu J, Zhu Q-S, et al. Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: a role for p53/activator protein 2 transcriptional regulation. Mol Cancer Ther. 2007;6:1650–60. doi: 10.1158/1535-7163.MCT-06-0636. [DOI] [PubMed] [Google Scholar]
- 34.Maacke H, Opitz S, Jost K, et al. Over-expression of wild-type RAD51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer. 2000;88:907–13. doi: 10.1002/1097-0215(20001215)88:6<907::aid-ijc11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Connell PP, Jayathilaka K, Haraf DJ, Weichselbaum RR, Vokes EE, Lingen MW. Pilot study examining tumor expression of RAD51 and clinical outcomes in human head cancers. Int J Oncol. 2006;28:1113–9. [PubMed] [Google Scholar]
- 36.Taron M, Rosell R, Felip E, et al. BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Hum Mol Genet. 2004;13:2443–9. doi: 10.1093/hmg/ddh260. [DOI] [PubMed] [Google Scholar]
- 37.Squires MH, III, Fisher SB, Fisher KE, et al. Differential expression and prognostic value of ERCC1 and thymidylate synthase in resected gastric adenocarcinoma. Cancer. 2013;119:3242–50. doi: 10.1002/cncr.28175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–91. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 39.Dabholkar M, Bostick-Bruton F, Weber C, Bohr VA, Egwuagu C, Reed E. ERCC1 and ERCC2 expression in malignant tissues from ovarian cancer patients. J Natl Cancer Inst. 1992;84:1512–7. doi: 10.1093/jnci/84.19.1512. [DOI] [PubMed] [Google Scholar]
- 40.Steffensen KD, Waldstrøm M, Jakobsen A. The relationship of platinum resistance and ERCC1 protein expression in epithelial ovarian cancer. Int J Gynecol Cancer. 2009;19:820–5. doi: 10.1111/IGC.0b013e3181a12e09. [DOI] [PubMed] [Google Scholar]
- 41.Hayes M, Lan C, Yan J, et al. ERCC1 expression and outcomes in head and neck cancer treated with concurrent cisplatin and radiation. Anticancer Res. 2011;31:4135–9. [PubMed] [Google Scholar]
- 42.Shirota Y, Stoehlmacher J, Brabender J, et al. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J Clin Oncol. 2001;19:4298–304. doi: 10.1200/JCO.2001.19.23.4298. [DOI] [PubMed] [Google Scholar]
- 43.Kwon H-C, Roh MS, Oh SY, et al. Prognostic value of expression of ERCC1, thymidylate synthase, and glutathione S-transferase P1 for 5-fluorouracil/oxaliplatin chemotherapy in advanced gastric cancer. Ann Oncol. 2007;18:504–9. doi: 10.1093/annonc/mdl430. [DOI] [PubMed] [Google Scholar]
- 44.Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36:425–35. doi: 10.1016/j.ctrv.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 45.Klauke ML, Hoogerbrugge N, Budczies J, et al. Higher cytoplasmic and nuclear poly(ADP-ribose) polymerase expression in familial than in sporadic breast cancer. Virchows Arch. 2012;461:425–31. doi: 10.1007/s00428-012-1311-2. [DOI] [PubMed] [Google Scholar]
- 46.Mego M, Cierna Z, Svetlovska D, et al. PARP expression in germ cell tumours. J Clin Pathol. 2013;66:607–12. doi: 10.1136/jclinpath-2012-201088. [DOI] [PubMed] [Google Scholar]
- 47.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 48.Coutts AS, Adams CJ, La Thangue NB. p53 ubiquitination by Mdm2: a never ending tail? DNA Repair (Amst) 2009;8:483–90. doi: 10.1016/j.dnarep.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 49.Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. 2013 doi: 10.1038/onc.2013.275. doi: 10.1038/onc.2013.275. [DOI] [PubMed] [Google Scholar]
- 50.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banerjee S, Kaye S. PARP inhibitors in BRCA gene-mutated ovarian cancer and beyond. Curr Oncol Rep. 2011;13:442–9. doi: 10.1007/s11912-011-0193-9. [DOI] [PubMed] [Google Scholar]
- 54.Angele S, Treilleux I, Bremond A, Taniere P, Hall J. Altered expression of DNA double-strand break detection and repair proteins in breast carcinomas. Histopathology. 2003;43:347–53. doi: 10.1046/j.1365-2559.2003.01713.x. [DOI] [PubMed] [Google Scholar]
- 55.Ai L, Vo QN, Zuo C, et al. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol Biomarkers Prev. 2004;13:150–6. doi: 10.1158/1055-9965.epi-082-3. [DOI] [PubMed] [Google Scholar]
- 56.Bartkova J, Tommiska J, Oplustilova L, et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim DS, Kim MJ, Lee JY, et al. Epigenetic inactivation of checkpoint kinase 2 gene in non-small cell lung cancer and its relationship with clinicopathological features. Lung Cancer. 2009;65:247–50. doi: 10.1016/j.lungcan.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 58.Sullivan A, Yuille M, Repellin C, et al. Concomitant inactivation of p53 and Chk2 in breast cancer. Oncogene. 2002;21:1316–24. doi: 10.1038/sj.onc.1205207. [DOI] [PubMed] [Google Scholar]
- 59.Galamb O, Sipos F, Dinya E, Spisak S, Tulassay Z, Molnar B. mRNA expression, functional profiling and multivariate classification of colon biopsy specimen by cDNA overall glass microarray. World J Gastroenterol. 2006;12:6998–7006. doi: 10.3748/wjg.v12.i43.6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshikawa K, Ogawa T, Baer R, et al. Abnormal expression of BRCA1 and BRCA1-interactive DNA-repair proteins in breast carcinomas. Int J Cancer. 2000;88:28–36. [PubMed] [Google Scholar]
- 61.Hilton JL, Geisler JP, Rathe JA, et al. Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst. 2002;94:1396–406. doi: 10.1093/jnci/94.18.1396. [DOI] [PubMed] [Google Scholar]
- 62.Catteau A, Morris JR. BRCA1 methylation: a significant role in tumour development? Semin Cancer Biol. 2002;12:359–71. doi: 10.1016/s1044-579x(02)00056-1. [DOI] [PubMed] [Google Scholar]
- 63.Hosoya N, Okajima M, Kinomura A, et al. Synaptonemal complex protein SYCP3 impairs mitotic recombination by interfering with BRCA2. EMBO Rep. 2012;13:44–51. doi: 10.1038/embor.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taniguchi T, D'Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107:4223–33. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- 65.Xie Y, de Winter JP, Waisfisz Q, et al. Aberrant Fanconi anaemia protein profiles in acute myeloid leukaemia cells. Br J Haematol. 2000;111:1057–64. [PubMed] [Google Scholar]
- 66.Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12:587–98. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- 67.Kuroda S, Urata Y, Fujiwara T. Ataxia-telangiectasia mutated and the Mre11-Rad50-NBS1 complex: promising targets for radiosensitization. Acta Med Okayama. 2012;66:83–92. doi: 10.18926/AMO/48258. [DOI] [PubMed] [Google Scholar]
- 68.Golding SE, Rosenberg E, Adams BR, et al. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle. 2012;11:1167–73. doi: 10.4161/cc.11.6.19576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishida H, Tatewaki N, Nakajima Y, et al. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009;37:5678–89. doi: 10.1093/nar/gkp593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peasland A, Wang LZ, Rowling E, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer. 2011;105:372–81. doi: 10.1038/bjc.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prevo R, Fokas E, Reaper PM, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13:1072–81. doi: 10.4161/cbt.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308–16. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 73.Lavecchia A, Di Giovanni C, Novellino E. CDC25 phosphatase inhibitors: an update. Mini Rev Med Chem. 2012;12:62–73. doi: 10.2174/138955712798868940. [DOI] [PubMed] [Google Scholar]
- 74.Davidson D, Amrein L, Panasci L, Aloyz R. Small molecules, inhibitors of DNA-PK, targeting DNA repair, and beyond. Front Pharmacol. 2013;4:5. doi: 10.3389/fphar.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Munck JM, Batey MA, Zhao Y, et al. Chemosensitization of cancer cells by KU-0060648, a dual inhibitor of DNA-PK and PI-3K. Mol Cancer Ther. 2012;11:1789–98. doi: 10.1158/1535-7163.MCT-11-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Srivastava M, Nambiar M, Sharma S, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–87. doi: 10.1016/j.cell.2012.11.054. [DOI] [PubMed] [Google Scholar]
- 77.Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double- strand breaks. Blood. 2008;112:1413–23. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tobin LA, Robert C, Rapoport AP, et al. Targeting abnormal DNA double-strand break repair in tyrosine kinase inhibitor-resistant chronic myeloid leukemias. Oncogene. 2013;32:1784–93. doi: 10.1038/onc.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol. 2011;6:628–35. doi: 10.1021/cb100428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen G, Yuan S-SF, Liu W, et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J Biol Chem. 1999;274:12748–52. doi: 10.1074/jbc.274.18.12748. [DOI] [PubMed] [Google Scholar]
- 81.Slupianek A, Schmutte C, Tombline G, et al. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell. 2001;8:795–806. doi: 10.1016/s1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- 82.Slupianek A, Hoser G, Majsterek I, et al. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G2/M phase, and protection from apoptosis. Mol Cell Biol. 2002;22:4189–201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu J, Zhou L, Wu G, et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol Med. 2013;5:353–65. doi: 10.1002/emmm.201201760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer. 2013;108:748–54. doi: 10.1038/bjc.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci USA. 2007;104:19482–7. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Noguchi M, Yu D, Hirayama R, et al. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem Biophys Res Commun. 2006;351:658–63. doi: 10.1016/j.bbrc.2006.10.094. [DOI] [PubMed] [Google Scholar]
- 87.Ogiwara H, Ui A, Shiotani B, Zou L, Yasui A, Kohno T. Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor. Carcinogenesis. 2013;34:2486–97. doi: 10.1093/carcin/bgt240. [DOI] [PubMed] [Google Scholar]
- 88.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nature Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14:7917–23. doi: 10.1158/1078-0432.CCR-08-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Plummer R, Lorigan P, Steven N, et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013;71:1191–9. doi: 10.1007/s00280-013-2113-1. [DOI] [PubMed] [Google Scholar]
- 91.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 92.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 93.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 94.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 95.Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–61. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 96.Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer. 2004;4:814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 97.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 98.Postel-Vinay S, Bajrami I, Friboulet L, et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene. 2013;32:5377–87. doi: 10.1038/onc.2013.311. [DOI] [PubMed] [Google Scholar]
- 99.Kennedy RD, Chen CC, Stuckert P, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–9. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen CC, Kennedy RD, Sidi S, et al. CHK1 inhibition as a strategy for targeting Fanconi anemia (FA) DNA repair pathway deficient tumors. Mol Cancer. 2009;8:24. doi: 10.1186/1476-4598-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McManus KJ, Barrett IJ, Nouhi Y, Hieter P. Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc Natl Acad Sci USA. 2009;106:3276–81. doi: 10.1073/pnas.0813414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sultana R, McNeill DR, Abbotts R, et al. Synthetic lethal targeting of DNA double-strand break repair deficient cells by human apurinic/apyrimidinic endonuclease inhibitors. Int J Cancer. 2012;131:2433–44. doi: 10.1002/ijc.27512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Feng Z, Scott SP, Bussen W, et al. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci USA. 2011;108:686–91. doi: 10.1073/pnas.1010959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–8. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 106.Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Swisher EM, Sakai W, Karlan BY, et al. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581–6. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bunting SF, Callén E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA- mutated breast cancers. Nature Struct Mol Biol. 2010;17:688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bajrami I, Frankum JR, Konde A, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–97. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rohwer N, Zasada C, Kempa S, Cramer T. The growing complexity of HIF-1α's role in tumorigenesis: DNA repair and beyond. Oncogene. 2013;32:3569–76. doi: 10.1038/onc.2012.510. [DOI] [PubMed] [Google Scholar]