

Abstract
Given that treatment options for patients with glioblastoma are limited, much effort has been made to clarify the underlying mechanisms of gliomagenesis. Recent genome-wide genomic and epigenomic analyses have revealed that mutations in epigenetic modifiers occur frequently in gliomas and that dysregulation of epigenetic mechanisms is closely associated with glioma formation. Given that epigenetic changes are reversible, understanding the epigenetic abnormalities that arise in gliomagenesis might be key to developing more effective treatment strategies for glioma. In this review, we focus on the recent advancements in epigenetic research with respect to gliomas, consider how epigenetic mechanisms dynamically regulate tumor cells, including the cancer stem cell population, and discuss perspectives and challenges for glioma treatment in the near future.
Keywords: Glioma stem cell, DNA methylation, epigenetics, histone modification, non-coding RNA
Glioblastoma multiforme (GBM) is the most lethal form of glioma. As the term glioblastoma “multiforme” suggests, the histopathology of this tumor is extremely variable. Both intertumor and intratumor heterogeneity are observed, in which tumor cells exist in multiple states of differentiation that show distinct properties.(1) Even with the most advanced treatments, which involve combinations of surgery, radiotherapy and chemotherapy with drugs such as temozolomide (TMZ), <5% of patients survive longer than 5 years post diagnosis.(2) Consequently, elucidating the underlying mechanisms that potentiate the aggressiveness of GBM is key to facilitating the development of new treatment strategies for this dreadful disease.
Alongside known genetic changes, aberrant epigenetic alterations have emerged as common hallmarks of many cancers, including glioblastoma.(3) Recent large-scale genomic and epigenomic profiling studies, such as The Cancer Genome Atlas, have yielded novel data and deeper insight into gliomagenesis.(4) One surprising discovery is the identification of a large number of inactivating mutations in genes that control the epigenome (Fig. 1). Exome sequencing studies of 291 GBM revealed that 46% of cases had at least one somatic mutation in genes associated with chromatin modification. The genes included those associated with DNA methylation (isocitrate dehydrogenase [IDH] 1, IDH2), histone modification (mixed lineage leukaemia 2 [MLL2], MLL3, MLL4, Enhancer of zeste 2 [EZH2] and histone deacetylase 2 [HDAC2]) and chromatin remodeling (a-thalassaemia/mental retardation syndrome X-linked [ATRX], death-domain associated protein [DAXX], CREB binding protein [CREBBP] and SWI/SNF-related matrix-associated, actin-dependent regulator of chromatin A2 [SMARCA2]).(5) These mutations result in the impairment of DNA methylation, histone modification and nucleosome positioning, and are associated with aberrant gene expression.(6) For instance, a subset of gliomas, normally low-grade gliomas or secondary high-grade gliomas (proneural type), are characterized by both mutations in IDH1 or IDH2 and hypermethylation of DNA. This subset is referred to as glioma CpG island methylator phenotype (G-CIMP).(4) Hence, clarification of the crosstalk between the genome and epigenome might suggest new molecular targets and possibilities for the treatment of GBM.
Fig. 1.
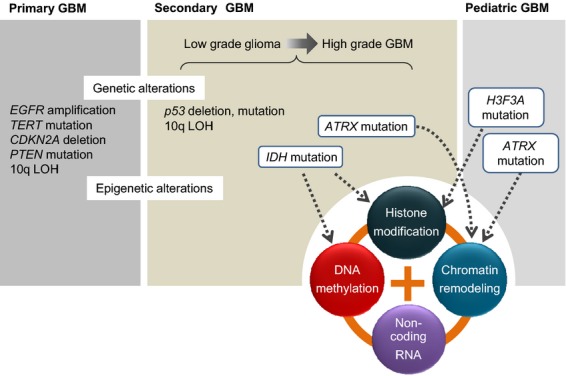
Genetic and epigenetic alterations in gliomas. TP53, IDH1 and ATRX are frequently mutated in low-grade gliomas and secondary GBM. Mutation of IDH1 leads to aberrant DNA methylation, whereas mutations in the important chromatin modifier ATRX affect chromatin structure. In pediatric GBM, mutations in H3F3A and ATRX are found frequently and associated closely with gliomagenesis.
Recent transcriptome analysis revealed that more than 90% of the human genome is transcribed and transcription is not limited to protein-coding regions.(7) The expression of significant numbers of non-coding RNA (ncRNA) is regulated during development in a cell-type specific manner and these RNA, which include microRNA (miRNA, miR) and long non-coding RNA (lncRNA), are associated with multiple cell functions.(8) A proportion of the ncRNA appear to be associated with epigenetic modifiers via direct interaction or are involved in the post-transcriptional regulation of such modifiers.(9,10) Recent studies have shown that ncRNA are not only potential key regulators of cellular differentiation and proliferation, but also have tumor suppressive or oncogenic functions in many types of cancer.(11) Thus, ncRNA and associated epigenetic regulators play important roles in a wide variety of physiological and pathological processes, including tumor formation.
It is likely that therapies for gliomas that target epigenetic molecules will be in clinical trials and use in the near future. However, the current limited knowledge about the mechanisms by which such molecules function must be considered. Consequently, understanding how the epigenome regulates certain genomic loci is of vital importance in enabling the epigenome to be exploited as a fruitful source of new therapeutic approaches for GBM. This review outlines recent advances in epigenetic research with respect to gliomas. In particular, it focuses on the dynamic regulation of glioma cells, and discusses the clinical implications of the development of novel therapies for this devastating disease.
Aberrant DNA Methylation in Glioblastoma Multiforme
Dysregulation of epigenetic transcriptional control owing to aberrant DNA methylation is a fundamental feature of human malignancies.(3) It has also been suggested that aberrant DNA methylation contributes to the development and progression of gliomas.(4,12) In general, cancer cells display global hypomethylation and regional hypermethylation simultaneously, with hypermethylation occurring especially at select gene-associated CpG islands that are normally unmethylated. In addition, comprehensive studies of colorectal cancers have revealed a subset of cases in which high rates of aberrant hypermethylation are present in promoters; these cases are described as CpG island methylator phenotype (CIMP) positive.(13) CIMP tumors exhibit distinct genetic and clinical features, including high rates of BRAF and KRAS mutations, a low frequency of TP53 mutation, specific histology (mucinous and poorly differentiated), a proximal location and characteristic clinical outcome.(14) Recent genome-wide analyses have demonstrated the existence of CIMP in breast cancers, lung cancers and gliomas.(4,15,16) These studies indicate that a specific pathological molecular pathway related to aberrant DNA hypermethylation is involved in the development of each CIMP tumor. Glioma-CIMP (G-CIMP) is subclassified on the basis of distinct genetic and clinical features, which include high rates of mutation in IDH1 and TP53, a young age at diagnosis, a better prognosis than other patients with glioma and identification as the proneural subtype. G-CIMP tumors are frequently found in secondary GBM, which shows a step-wise progression from low-grade glioma to high-grade GBM.(17)
Mutations in IDH1 have been shown to induce the accumulation of methylated DNA via the inhibition of DNA demethylation (Fig. 2).(18) IDH are NAD+ and NADP+-dependent enzymes that catalyze the third step of the tricarboxylic acid (TCA) cycle, and mutations in IDH1 cause a metabolite called 2-hydroxyglutarate (2-HG) to accumulate.(19) The accumulated 2-HG impairs the activity of ten-eleven translocation (TET) methylcytosine dioxygenase, which results in DNA hypermethylation.(18) This is supported by an in vitro study that demonstrates that the DNA methylation pattern is altered in human astrocytes expressing mutant IDH1 (R132H).(20) Collectively, these data indicate that mutation of IDH1 may result in G-CIMP through inhibition of the TET-mediated production of 5-hydroxymethylcytosine (5hmC), which is a primary mode of DNA demethylation. Although further investigations are required before it can be concluded that impaired TET activity is the major cause of aberrant DNA hypermethylation, these fascinating studies clearly demonstrate a link between an altered metabolite profile owing to the mutation of metabolic genes and an aberrant epigenome associated with cancer.
Fig. 2.
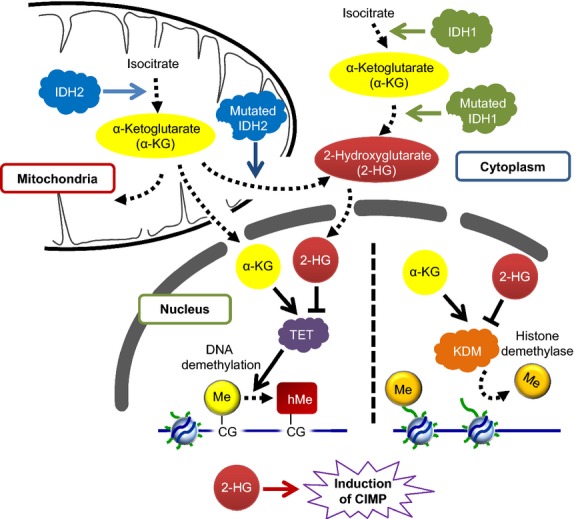
IDH mutations induce G-CIMP. Mutations in IDH1 (a cytoplasmic enzyme) and IDH2 (a mitochondrial enzyme) are found frequently in proneural glioblastoma multiforme (GBM). IDH1 mutations are more common than IDH2 mutations. Mutated IDH1 and IDH2 gain the ability to produce the metabolite, 2-hydroxyglutarate (2-HG), which inhibits α-ketoglutarate (α-KG)-dependent dioxygenases, including histone demethylases and the TET protein family. Therefore, mutation of IDH1 is the mechanistic cause of G-CIMP through inhibition of the TET-mediated production of 5hmC, which is a primary mode of DNA demethylation.
Dysregulation of Histone Modifications in Glioblastoma Multiforme
Histone modifications have long been thought to have a functional influence on the regulation of transcription. It is now apparent that they influence a variety of DNA-templated processes.(6) Among the different possible histone modifications, trimethylation of histone H3 lysine 27 (H3K27me3) has been identified as a key epigenetic modification during development, including neural cell differentiation. Aberrant H3K27me3 is frequently observed in many types of cancer.(21) Our study and other studies have revealed that H3K27me3-mediated gene silencing is mechanistically distinct from gene silencing mediated by DNA methylation.(22,23) Indeed, H3K27me3 and DNA methylation have been shown to be mutually exclusive in CpG islands in a precise genome-wide analysis.(24) A significant difference between the two mechanisms relates to the stability of repression. The pattern of H3K27me3-mediated gene silencing can change dynamically during differentiation due to the existence of H3K27 methylases (EZH2 and EZH1) and demethylases (UTX and JMJD3). In contrast, DNA methylation within CpG islands is highly stable, although recently the presence of TET-mediated DNA demethylation machinery has been indicated in a certain context.(25–27)
EZH2, which is the catalytic subunit of Polycomb Repressive Complex 2 (PRC2), has a histone methyltransferase activity with substrate specificity for H3K27 and produces dimethylated H3K27 (H3K27me2) or H3K27me3 (Fig. 3).(21) H3K27me3 serves as a signal for specific binding of the chromodomain of another polycomb repressor complex, PRC1.(28) In general, PRC1 and PRC2 work collaboratively to repress transcription. However, recent analyses have revealed that PRC1 and PRC2 do not share all their targets: a substantial proportion of PRC2 targets are not occupied by PRC1, and vice versa, which implies that PRC1 and PRC2 can act independently.(29,30) Intriguingly, a component of PRC1, BMI1, sometimes plays an opposing role to PRC2 in the context of stem cell regulation.(31) BMI1 maintains the self-renewal of hematopoietic and neural stem cells as well as stemness in cancer stem cells (CSC),(32,33) whereas PRC2 restricts hematopoietic stem cell/progenitor activities.(31) Studies have also demonstrated that PRC2 not only acts to promote self-renewal, but also controls fate choices within the multipotent lineage during neural and muscle development.(21,34,35) Indeed, PRC2 is a key regulator of the differentiation of glioma stem cells (GSC).(36) The precise molecular mechanisms by which PRC1 and PRC2 control stemness and stem cell differentiation remain elusive. However, independent action of PRC1 and PRC2 might enable the precise and complex regulation of gene function during development.
Fig. 3.
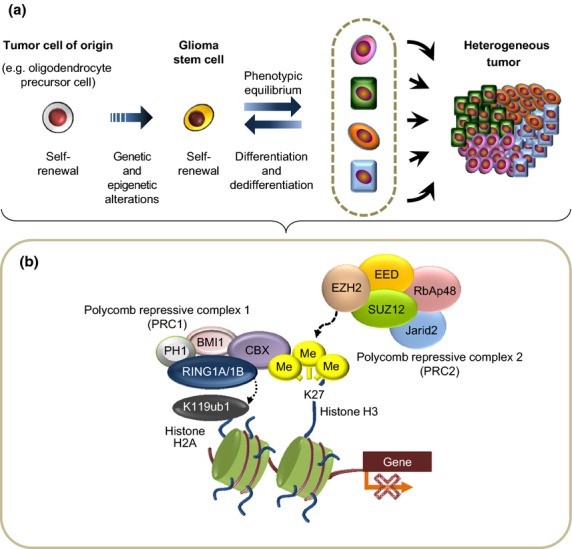
Roles of PRC in the formation of heterogeneous tumors. (a) Glioma stem cells (GSC) are considered able to differentiate aberrantly into diverse cell types. The process of differentiation of GSC into non-GSC is reversible and shows phenotypic equilibrium. (b) PRC2 has a histone methyltransferase activity with substrate specificity for H3K27 and produces H3K27me3, which is thought to recruit PRC1 via proteins of the CBX family. The RING1A/1B complex in PRC1 induces the mono-ubiquitination of histone 2A lysine 119 (H2AK119), which is thought to affect chromatin structure and block the recruitment of transcriptional activation factors (bottom panel). We have demonstrated that PRC2 is required for the self-renewal of GSCs as well as GSC differentiation in response to oncogenic cues, which leads to the establishment of heterogenous tumors.
Recently a cogent study showed that recurrent heterozygous mutations in H3F3A, which encodes the replication-independent histone H3 variant H3.3, result in amino acid substitutions at two critical positions within the histone tail (K27M, G34R/G34V) in infant and adolescent GBM.(37,38) The presence of these mutations is mutually exclusive and is linked to a distinct gene expression pattern. Cases of GBM with the H3F3A G34 mutation show characteristic features that include high rates of mutation in TP53, ATRX and DAXX, high levels of alternative lengthening of telomeres (ALT) activity, DNA hypomethylation and a hemispheric location, whereas cases of GBM with the H3F3A K27 mutation show high frequencies of TP53 mutation, DNA hypomethylation, a midline location and diffuse intrinsic pontine glioma, and a poor prognosis. Given that ATRX and DAXX are essential for the incorporation of H3.3 at pericentromeric heterochromatin and at telomeres, mutations in these genes are associated strongly with alternative lengthening of telomeres and specific gene expression profiles that result in gliomagenesis.(37)
Histone H3.3 that carries the K27 mutation acts in a dominant-negative manner and leads to a global reduction in levels of the repressive histone modification H3K27me3.(39) The reduced levels of H3K27me3 might also affect the global DNA methylation status and result in DNA hypomethylation that activates gene expression.
The K27 mutant and consequent reduced levels of H3K27me3 have the same effect as the loss of EZH2 function, which suggests that EZH2 might have a tumor suppressor function. However, these findings are inconsistent with the evidence that increased EZH2 activity increases the level of H3K27me3 and that EZH2 might act as an oncogene via the repression of tumor suppressor genes.(40–43) The possible bifunctional role of EZH2 as both an oncogene and tumor suppressor in human malignancies indicates that signaling outcomes downstream of dysregulated EZH2 activity depend on the context. Given that cancer is a disease of faulty differentiation, dysregulation of EZH2-H3K27me3 is involved in different steps of carcinogenesis. Therefore, it might be rational to determine whether PRC2 is required for the differentiation and dedifferentiation of CSC (i.e. tumor cell plasticity), or for the epithelial–mesenchymal transition, rather than modulating EZH2 levels to assess whether it is acting as an oncogene or a tumor suppressor in a defined cell context.(21)
Interestingly, mutations in H3F3A occur in a mutually exclusive manner with mutations in IDH1.(44) As mentioned above, IDH1 mutations induce the accumulation of 2-HG, which acts as an inhibitor of multiple a-ketoglutarate (a-KG)-dependent dioxygenases, including histone demethylases and the TET protein family.(18,45,46) This inhibition results in characteristic genome-wide changes in patterns of histone and DNA methylation. The different mutations are associated with GBM with distinct clinical characters, probably due to distinct molecular features that include differences in gene expression and DNA methylation profiles.
Dysregulation of Non-Coding RNA in Glioblastoma Multiforme
Recent advancements in the technology to identify ncRNA using microarrays or next generation sequencing technologies have provided an extraordinary abundance of novel data on a genome-wide scale, which, in turn, have led to deeper insights into the biology of ncRNA.(7) Some ncRNA play key regulatory and functional roles.
miRNA are short non-coding endogenous RNA that post-transcriptionally regulate the expression of a large number of genes.(47) They downregulate gene expression by binding to the 3′-untranslated regions of specific target mRNA to induce direct mRNA degradation or translational inhibition. miRNA play important roles in a wide variety of physiological and pathological processes, including tumor formation. Aberrant expression of miRNA can induce tumor suppression or have an oncogenic effect that results in tumor formation.(48)
Studies have shown that miRNA play an important regulatory role during tumor formation in GBM.(49,52–54,58) miR-9/9* is highly abundant in CD133+ cells and inhibition of miR-9/9* leads to reduced formation of neurospheres (Fig. 4).(49) Conceivably, CD133 is a stem cell marker that might be able to distinguish the small subpopulations of cancer cells with strong tumorigenic activity that are found in some GBM.(50) However, recent reports, including ours, suggest that expression of CD133 is a feature of GSC rather than a defined marker that distinguishes cells at different hierarchical levels in tumors.(36,51) The robustness of some markers of cancer stem cells remains to be determined.
Fig. 4.
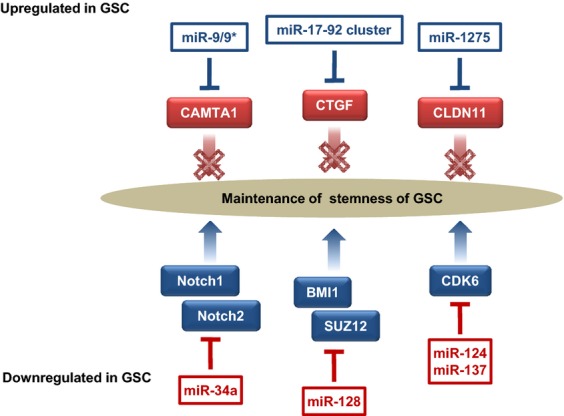
miRNA associated with the maintenance of glioma stem cells (GSC). miR-9/9*, the miR17-92 cluster, and miR-1275 are upregulated in GSC. Targets of these miRNA, namely calmodulin-binding transcription activator 1 (CAMTA1), connective tissue growth factor (CTGF) and CLDN11, act as tumor suppressors. In contrast, miR-34a, miR-128, miR-124 and miR-137 are downregulated in GSC. miR-34a inhibits the expression of Notch1 and Notch2, which are receptors of notch signaling molecules. miR-128 inhibits GSC self-renewal by directly targeting BMI1 and SUZ12, components of PRC1 and PRC2, respectively. miR-124 and miR-137 induce G0/G1 cell cycle arrest by targeting cyclin-dependent kinase 6 (CDK6).
The miR-17-92 cluster is thought to be involved in the regulation of GSC differentiation, apoptosis and proliferation.(52) The level of miR-17-92 transcripts is significantly higher in primary astrocytic tumors than in normal brain tissue and increases significantly with tumor progression. A high-level amplification of the miR-17-92 locus has also been found in glioblastoma specimens. Inhibition of miR-17-92 induces apoptosis and decreases cell proliferation in GSC. It has also been shown that both miR-124 and miR-137 are downregulated in high-grade gliomas and upregulated during adult neural stem cell differentiation.(53) Transfection of miR-124 or miR-137 inhibits the proliferation of GSC, via the suppression of cyclin-dependent protein kinase 6 (CDK6), and induces morphological changes in human GSC and the expression of neuronal differentiation markers. miR-34a directly inhibits the expression of c-Met, Notch1 and Notch2 in GSC.(54) Notch is a critical regulator of cell fate during development and also of normal stem cell maintenance.(55) Activation of the Notch pathway enhances the stemness, proliferation and radioresistance of GSC.(56)
The existence of a regulatory network between PRC and miRNA has been shown in GBM. One study demonstrates that miR-128 has a tumor-suppressive function and is downregulated in glioblastoma tissue. The induction of miR-128 expression significantly reduces the proliferation of glioma cells both in vitro and in vivo via downregulation of the oncogenic protein Bmi-1, which is a component of PRC1.(57) In addition, miR-128 inhibits GSC self-renewal. Another study shows that miR-128 directly targets SUZ12, a key component of PRC2. Ectopic expression of miR-128 in GSC significantly increases their radiosensitivity.(58) Thus, the deregulation of miRNA expression affects the activity of downstream tumor suppressors, oncogenes and other signaling molecules in a variety of steps in glioma progression, including the maintenance of GSC.
Plastic Epigenetic Regulation in Glioma Stem Cells
We have summarized here the recent advances in epigenetic research with respect to gliomas. These epigenetic changes are not only important for tumor formation but also play a pivotal role in the plasticity of tumor cells (Fig. 3). The dynamic plasticity of tumor cells might be required for the adaptation of their microenvironment, which, in turn, might contribute to the establishment of intratumor heterogeneity.(59–61) Functional and morphological heterogeneity characterize aggressive glioblastoma and contribute to invasion, metastasis and drug resistance.(36,62) Consequently, understanding the molecular mechanisms that underlie tumor heterogeneity and the associated plasticity is critical to address the current limitations in the treatment of GBM.
The emerging insights into gliomagenesis have revealed that GSC have the potential to initiate and maintain the growth of gliomas and might be crucial for their resistance to conventional therapies.(50,63) The early studies, which involved in vivo transplantation assays, revealed a hierarchy of tumor cells in GBM that comprised a strongly tumorigenic CSC population and a weaker tumorigenic non-CSC population. Later, a mouse model identified a putative endogenous GSC located at the apex of a cellular hierarchy that was involved in tumor maintenance and recurrence after chemotherapy.(64) GSC and normal neural stem cells appear to share common features that include self-renewal and the capability to differentiate into multiple lineages.(65) In response to signals within the tumor microenvironment, GSC are considered able to differentiate aberrantly into diverse cell types through dynamic epigenetic regulation. Consequently, the existence of GSC and plastic epigenetic regulation might be linked to the morphological and lineage heterogeneity that is observed in GBM. Given that PRC2-mediated H3K27me3 confers stemness and controls the development of organisms by regulating the expression of developmental genes,(66,67) tumor cells might usurp this epigenetic process to mediate adaptation to tumor environments.
Recently, we examined GSC and found that biological interconversion between GSC and differentiated non-GSC is reversible and functionally plastic.(36) This interconversion is accompanied by gain or loss of PRC2-mediated H3K27me3 on genes associated with pluripotency or development (e.g. Nanog, Wnt1 and BMP5) together with alterations in the subcellular localization of EZH2 and other components of PRC2, such as SUZ12 and EED. Knockdown of EZH2 or pharmacological inhibition of EZH2 disrupts the morphological interconversion and impairs tumor cell invasion, which results in improved survival of GSC-bearing mice. In human GBM specimens, the proportion of tumor cells that contain nuclear EZH2 is higher around tumor blood vessels and the invasive front, which suggests that nuclear EZH2 might be involved in the process of reprogramming tumor cells that are in close proximity to surrounding environmental factors. Intriguingly, miR-1275, which has been newly identified as a target for regulation by PRC2-H3K27me3, regulates the expression of claudin 11 (CLDN11), which might be required for tumor cell differentiation, especially the oligodendroglial lineage, and contribute to the establishment of tissue heterogeneity in GBM.(68) These results suggest that miRNA involved in development are regulated via an epigenetic pathway that contributes to the phenotypic diversity of GBM.
Thus, the PRC2-miRNA epigenetic network contributes to intratumoral heterogeneity in GBM: some of the subpopulations of cells exhibit more differentiated features, whereas others have characteristics of stem-like cancer cells. Therapy targeted against PRC2 might reduce tumor cell plasticity and heterogeneity, metaphorically “freezing” the ability of tumor cells to adapt, and provide a new paradigm in the treatment of gliomas.
Perspectives
Epigenetic alterations play an important role in the molecular pathology of GBM. Given that inhibitors of histone deacetylases are currently in clinical trial for the treatment of GBM, it is important to understand the involvement of histone modifications during gliomagenesis.(69,70) However, knowledge about the role of dysregulation of epigenetic mechanisms in GBM is still very limited, especially how the epigenome is altered specifically at certain loci and how this affects the phenotypes of GBM. Following the discovery of GSC, therefore, it is important to elucidate the epigenetic mechanisms by which environmental cues control the differentiation of GSC into the diverse array of cell types that form GBM. Recent genome-wide studies have shown that the large number of ncRNA that are transcribed from the human genome include a group termed lncRNA.(7) LncRNA are known to regulate gene expression through providing a scaffold for chromatin modifying proteins, such as methylases, demethylases and deacetylases, and recruiting these proteins to target loci that can be situated close together in the genome (cis-regulation) or genomically distant (trans-regulation).(71–73) However, the scaffold function has been identified in only a few lncRNA. Further investigation is required to clarify the functional roles of lncRNA in order to elucidate the gene regulatory mechanisms that are important in gliomagenesis. Understanding the interactions between lncRNA and the genome, which are reversible changes, might suggest novel approaches for specific epigenome-targeted therapies for GBM.
Acknowledgments
This work was supported by a grant from PRESTO of Japan Science and Technology Agency (JST), and a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (25290048).
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–77. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8:413–23. doi: 10.1038/nrg2083. [DOI] [PubMed] [Google Scholar]
- 9.Guttman M, Donaghey J, Carey BW, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabbri M, Calore F, Paone A, Galli R, Calin GA. Epigenetic regulation of miRNAs in cancer. Adv Exp Med Biol. 2013;754:137–48. doi: 10.1007/978-1-4419-9967-2_6. [DOI] [PubMed] [Google Scholar]
- 11.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 12.Natsume A, Kondo Y, Ito M, Motomura K, Wakabayashi T, Yoshida J. Epigenetic aberrations and therapeutic implications in gliomas. Cancer Sci. 2010;101:1331–6. doi: 10.1111/j.1349-7006.2010.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–93. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 15.Fang F, Turcan S, Rimner A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3:75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shinjo K, Okamoto Y, An B, et al. Integrated analysis of genetic and epigenetic alterations reveals CpG island methylator phenotype associated with distinct clinical characters of lung adenocarcinoma. Carcinogenesis. 2012;33:1277–85. doi: 10.1093/carcin/bgs154. [DOI] [PubMed] [Google Scholar]
- 17.Lai A, Kharbanda S, Pope WB, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. 2011;29:4482–90. doi: 10.1200/JCO.2010.33.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–83. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gal-Yam EN, Egger G, Iniguez L, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A. 2008;105:12979–84. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kondo Y, Shen L, Cheng AS, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40:741–50. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 24.Brinkman AB, Gu H, Bartels SJ, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012;22:1128–38. doi: 10.1101/gr.133728.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondo Y, Shen L, Issa JP. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol Cell Biol. 2003;23:206–15. doi: 10.1128/MCB.23.1.206-215.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 27.Williams K, Christensen J, Helin K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2011;13:28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levine SS, Weiss A, Erdjument-Bromage H, Shao Z, Tempst P, Kingston RE. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol. 2002;22:6070–8. doi: 10.1128/MCB.22.17.6070-6078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ku M, Koche RP, Rheinbay E, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010;24:265–76. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majewski IJ, Ritchie ME, Phipson B, et al. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood. 2010;116:731–9. doi: 10.1182/blood-2009-12-260760. [DOI] [PubMed] [Google Scholar]
- 32.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–7. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lessard J, Sauvageau G. Bmi–1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 34.Pereira JD, Sansom SN, Smith J, Dobenecker MW, Tarakhovsky A, Livesey FJ. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci U S A. 2010;107:15957–62. doi: 10.1073/pnas.1002530107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palacios D, Mozzetta C, Consalvi S, et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7:455–69. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Natsume A, Ito M, Katsushima K, et al. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 2013;73:4559–70. doi: 10.1158/0008-5472.CAN-13-0109. [DOI] [PubMed] [Google Scholar]
- 37.Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 38.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–3. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24:660–72. doi: 10.1016/j.ccr.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107–17. doi: 10.1182/blood-2011-11-394932. [DOI] [PubMed] [Google Scholar]
- 42.Mallen-St Clair J, Soydaner-Azeloglu R, Lee KE, et al. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev. 2012;26:439–44. doi: 10.1101/gad.181800.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neff T, Sinha AU, Kluk MJ, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A. 2012;109:5028–33. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 45.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–8. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 48.Gangaraju VK, Lin H. MicroRNAs: key regulators of stem cells. Nat Rev. 2009;10:116–25. doi: 10.1038/nrm2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schraivogel D, Weinmann L, Beier D, et al. CAMTA1 is a novel tumour suppressor regulated by miR-9/9* in glioblastoma stem cells. EMBO J. 2011;30:4309–22. doi: 10.1038/emboj.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 51.Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–52. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ernst A, Campos B, Meier J, et al. De-repression of CTGF via the miR-17-92 cluster upon differentiation of human glioblastoma spheroid cultures. Oncogene. 2010;29:3411–22. doi: 10.1038/onc.2010.83. [DOI] [PubMed] [Google Scholar]
- 53.Silber J, Lim DA, Petritsch C, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Guessous F, Zhang Y, et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009;69:7569–76. doi: 10.1158/0008-5472.CAN-09-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fan X, Matsui W, Khaki L, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–52. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, Wakeman TP, Lathia JD, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28:17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Godlewski J, Nowicki MO, Bronisz A, et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–30. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 58.Peruzzi P, Bronisz A, Nowicki MO, et al. MicroRNA–128 coordinately targets Polycomb Repressor Complexes in glioma stem cells. Neuro Oncol. 2013;15:1212–24. doi: 10.1093/neuonc/not055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15:1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 60.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 61.Gupta PB, Fillmore CM, Jiang G, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 62.Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71:4055–60. doi: 10.1158/0008-5472.CAN-11-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 64.Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–52. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Azuara V, Perry P, Sauer S, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532–8. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 67.Lee TI, Jenner RG, Boyer LA, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katsushima K, Shinjo K, Natsume A, et al. Contribution of microRNA-1275 to Claudin11 protein suppression via a polycomb-mediated silencing mechanism in human glioma stem-like cells. J Biol Chem. 2012;287:27396–406. doi: 10.1074/jbc.M112.359109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–8. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Friday BB, Anderson SK, Buckner J, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2012;14:215–21. doi: 10.1093/neuonc/nor198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang KC, Yang YW, Liu B, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1:391–407. doi: 10.1158/2159-8290.CD-11-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu W, Alvarez-Dominguez JR, Lodish HF. Regulation of mammalian cell differentiation by long non-coding RNAs. EMBO Rep. 2012;13:971–83. doi: 10.1038/embor.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]