

Abstract
We aimed to reveal the prevalence and pattern of human papillomavirus (HPV) infection and p53 mutations among Japanese head and neck squamous cell carcinoma (HNSCC) patients in relation to clinicopathological parameters. Human papillomavirus DNA and p53 mutations were examined in 493 HNSCCs and its subset of 283 HNSCCs. Oropharyngeal carcinoma was more frequently HPV-positive than non-oropharyngeal carcinoma (34.4% vs 3.6%, P < 0.001), and HPV16 accounted for 91.1% of HPV-positive tumors. In oropharyngeal carcinoma, which showed an increasing trend of HPV prevalence over time (P < 0.001), HPV infection was inversely correlated with tobacco smoking, alcohol drinking, p53 mutations, and a disruptive mutation (P = 0.003, <0.001, <0.001, and <0.001, respectively). The prevalence of p53 mutations differed significantly between virus-unrelated HNSCC and virus-related HNSCC consisting of nasopharyngeal and HPV-positive oropharyngeal carcinomas (48.3% vs 7.1%, P < 0.001). Although p53 mutations were associated with tobacco smoking and alcohol drinking, this association disappeared in virus-unrelated HNSCC. A disruptive mutation was never found in virus-related HNSCC, whereas it was independently associated with primary site, such as the oropharynx and hypopharynx (P = 0.01 and 0.03, respectively), in virus-unrelated HNSCC. Moreover, in virus-unrelated HNSCC, G:C to T:A transversions were more frequent in ever-smokers than in never-smokers (P = 0.04), whereas G:C to A:T transitions at CpG sites were less frequent in ever-smokers than in never-smokers (P = 0.04). In conclusion, HNSCC is etiologically classified into virus-related and virus-unrelated subgroups. In virus-related HNSCC, p53 mutations are uncommon with the absence of a disruptive mutation, whereas in virus-unrelated HNSCC, p53 mutations are common, and disruptive mutagenesis of p53 is related with oropharyngeal and hypopharyngeal carcinoma.
Keywords: Disruptive mutation, Epstein–Barr virus, head and neck squamous cell carcinoma, human papillomavirus, p53 mutation
Head and neck squamous cell carcinoma (HNSCC) is etiologically correlated with tobacco smoking and alcohol abuse, whereas a subset of oropharyngeal carcinoma is caused by infection of high risk human papillomavirus (HPV), predominantly HPV type 16 (HPV16).(1,2) The p53 protein, as a tumor suppressor, has a highly conserved role as a guardian of the genome and is central to cellular anticancer mechanisms.(3) One of the most common molecular alterations in HNSCC is the abrogation of p53 function, which is mediated by mutations of its gene, p53, loss of heterozygosity of p53, or interaction with HPV oncoprotein E6.(4–6) Tobacco smoking increases the frequency of p53 mutations in a dose-dependent manner,(7) whereas E6 binds and degrades p53.(8)
Crystal structural analyses of the effect of p53 mutations on DNA binding classifies p53 mutations into two distinct types, disruptive mutations and non-disruptive mutations, according to their ability to knock out p53 function.(9,10) Disruptive mutations are non-conservative mutations occurring within the core DNA binding domain L2/L3, stop mutations, or frameshift mutations, and thus entirely abrogate p53 function. All other mutations defined as non-disruptive mutations affect p53 function in a limited fashion. It has been previously shown that HPV-positive oropharyngeal carcinoma is less likely to harbor p53 mutations as compared with HPV-negative oropharyngeal carcinoma.(11) Recently, an inverse relationship between HPV infection and disruptive mutations in oropharyngeal carcinoma has been reported, though in a small number.(12)
Human papillomavirus-positive oropharyngeal carcinoma is an entity clinically and epidemiologically different from HPV-negative oropharyngeal carcinoma. Oropharyngeal carcinoma that is HPV-positive has risk factors related to sexual behavior, whereas HPV-negative oropharyngeal carcinoma is linked with tobacco smoking and alcohol drinking.(2) Patients with HPV-positive oropharyngeal carcinoma survive better than patients with HPV-negative oropharyngeal carcinoma.(13) Of note is an increasing HPV prevalence in oropharyngeal carcinoma over time, which currently exceeds 70% in the USA and 90% in Europe.(14,15) However, there is a report showing the presence of a racial disparity in HPV prevalence.(16) Human papillomavirus-positive tumors are significantly less frequent in black patients than in white patients. Despite these findings, the prevalence and pattern of HPV infection and p53 mutations in HNSCC among Japanese patients remain less well established. We designed the present study to address this issue in a large patient group and to give a further insight into the association between HPV status, p53 mutations, and clinicopathological parameters.
Materials and Methods
Patients
A consecutive series of 493 patients with newly diagnosed HNSCC at Osaka University Medical School Hospital (Suita, Japan) between October 1995 and December 2012 were examined for tumor HPV status, and its subset of 283 patients were also examined for tumor p53 mutations. All tumors were staged according to the 2002 International Union Against Cancer TNM staging system. The protocol of the present study was approved by the Institutional Review Board in December 2004, and thereafter patients provided written informed consent. The Institutional Review Board waived informed consent of patients before December 2004 due to the retrospective nature of the study, under the condition that information on the present study was disclosed on the website so that patients could apply for refusal of the enrolment.
DNA extraction
Formalin-fixed, paraffin-embedded biopsy specimens of untreated tumors were cut into 5-μm serial sections, one of which was stained with H&E, and examined using light microscopy. The other sections were used for DNA extraction by means of the DNeasy tissue kit (Qiagen, Valencia, CA, USA) when tumor cells accounted for at least 70% of the tissue sample. Tissues with <70% tumor cells were microdissected to enrich the tumor cell content of the specimen prior to DNA extraction.
Detection and typing of high-risk HPV DNA and assessment of HPV16 physical status
Human papillomavirus DNA from HPV subtypes 16, 18, 31, 33, 35, 52, or 58 was detected by PCR to amplify DNA fragments within the ORFs of E6 and E7 using consensus primers, pU-1M and pU-2R, as described previously.(17,18) The amplified products were typed by direct sequencing. Direct sequencing was carried out by the dideoxy chain termination method using the Big Dye Terminator cycle sequencing kit (Perkin-Elmer, Forester City, CA, USA). After ethanol precipitation, the samples were analyzed by the autoanalyzer (ABI PRISM 3100 genetic analyzer; Perkin-Elmer). The physical status of HPV16 was addressed according to ratio of E2 to E6 copy numbers, which was determined by real-time PCR amplification of the E2 and E6 ORFs as described previously.(18) Primers for the E2 ORF were designed to amplify the E2 hinge region, which is usually deleted after viral integration into the host genome.(19) The E2 and E6 ORFs were quantified with the ABI PRISM 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). CaSki cells, containing 500 integrated copies of HPV16 DNA per cell, were used as a positive control.(20)
Detection of p53 gene mutations
Polymerase chain reaction amplification followed by SSCP analysis and direct sequencing was carried out as previously described(21) to detect p53 mutations in exons 4–8, containing 94% of all mutations described in HNSCC (International Agency for Research on Cancer TP53 Database, http://p53.iarc.fr/). The PCR primer pairs for the amplification are described elsewhere.(22) The SSCP bands that showed altered mobility as compared to a wild-type control (human lymphocyte DNA), were extracted from the gel and reamplified by PCR to enrich mutated alleles. The mutated alleles were purified using the QIAquick PCR purification kit (Qiagen). Direct sequencing was carried out as described above.
Statistical analysis
Associations between HPV infection, p53 mutations, and a disruptive mutation with clinicopathological parameters were assessed by univariate analyses involving logistic regression model or Clopper–Pearson's exact test, as appropriate. Multivariate analyses were made by forward stepwise logistic regression model. Akaike's information criterion(23) was used to evaluate the relative usefulness of the model. Differences in HPV prevalence, p53 mutation prevalence, and pattern of p53 mutations between groups were examined by Fisher's exact test. Trend in HPV prevalence over calendar time was evaluated by the Cochran–Armitage test. All statistical tests were two-tailed at a significant level of 0.05. All data were analyzed using sas Version 9.3 (SAS Institute, Cary, NC, USA).
Patients were classified as light and heavy smokers if their lifetime cigarette exposure was below and equal to or above the median pack-years in each analyzed group, respectively. Alcohol abuse was estimated by alcohol index, which was calculated by the formula: number of alcoholic units per day multiplied by year-duration. Twenty-eight grams of alcohol contained in alcoholic beverages corresponded to one unit of alcohol. Patients were classified as light and heavy drinkers if their lifetime alcohol consumption was below and equal to or above the median alcohol index in each analyzed group, respectively. Ever smokers and drinkers were defined as former plus current smokers and drinkers, respectively.
Results
Prevalence and pattern of HPV in HNSCCs
There was a striking difference in HPV prevalence between oropharyngeal carcinoma and non-oropharyngeal carcinoma (Table 1). Fifty-six of 163 (34.4%) oropharyngeal carcinomas were HPV-positive, whereas 12 of 330 (3.6%) non-oropharyngeal carcinomas were HPV-positive (P < 0.001). Prevalence of HPV in oral, hypopharyngeal, and laryngeal carcinomas was drastically decreased compared with that in oropharyngeal carcinoma (P < 0.001). Of note is nasopharyngeal carcinoma, 12.0% of which was HPV-positive, although the prevalence was still significantly less than in oropharyngeal carcinoma (odds ratio (OR), 0.3; 95% confidence interval (CI), 0.05–0.9; P = 0.04). As summarized in Table 2, HPV16 was the most prevalent, accounting for 62 of 68 (91.1%) HPV-positive tumors. All HPV-positive tumors were positive for a single type of high-risk HPV. An E2 to E6 transcript ratio consistent with complete viral integration was detected in 24 of 62 (38.7%) HPV16-positive tumors. The remaining 38 (61.3%) HPV16-positive tumors showed the presence of both episomal and integrated viral forms.
Table 1.
Prevalence of human papillomavirus (HPV) in head and neck squamous cell carcinomas according to primary tumor site (n = 493)
| Primary site | Total no. | HPV-positive n (%) | Odds ratio† | 95% CI | P-value |
|---|---|---|---|---|---|
| Oropharynx | 163 | 56 (34.4) | Reference | ||
| Non-oropharynx (whole) | 330 | 12 (3.6) | 0.1 | 0.0400–0.1000 | <0.001 |
| Oral cavity | 57 | 0 (0.0) | 0.0 | 0.0008–0.2000 | <0.001 |
| Nasopharynx | 25 | 3 (12.0) | 0.3 | 0.0500–0.9000 | 0.035 |
| Hypopharynx | 171 | 6 (3.5) | 0.1 | 0.0200–0.2000 | <0.001 |
| Larynx | 77 | 3 (3.9) | 0.1 | 0.0200–0.3000 | <0.001 |
Univariate analysis was made by logistic regression model, except for oral cavity that was made by the Clopper–Pearson method. CI, confidence interval.
Table 2.
Subtype of human papillomavirus (HPV) and physical status of HPV16 (n = 68)
| Type of HPV | No. | % | Physical status |
||
|---|---|---|---|---|---|
| Integrated no. (%) | Mixed no. (%) | Episomal no.(%) | |||
| HPV16 | 62 | 91.1 | 24 (38.7) | 38 (61.3) | 0 (0.0) |
| HPV18 | 1 | 1.5 | |||
| HPV33 | 1 | 1.5 | |||
| HPV35 | 3 | 4.4 | |||
| HPV58 | 1 | 1.5 | |||
Association of HPV status with clinicopathological factors in oropharyngeal carcinoma
As HPV-positive tumors were highly predominant in oropharyngeal carcinoma, we addressed the relationship between HPV status and clinicopathological factors in the single group of patients with oropharyngeal carcinoma (Table 3). Older patients, heavy smokers, and heavy drinkers were less likely to have HPV-positive tumors than younger patients (OR, 0.4; 95% CI, 0.2–0.8; P = 0.009), light smokers (OR, 0.4; 95% CI, 0.2–0.7; P = 0.003), and light drinkers (OR, 0.3; 95% CI, 0.1–0.6; P < 0.001), respectively. The prevalence of HPV infection was significantly decreased in the posterior wall and soft palate (OR, 0.1; 95% CI, 0.02–0.4; P < 0.001) as compared with the palatine tonsil. In contrast, poorly differentiated or undifferentiated tumors were more likely to be HPV-positive than well differentiated tumors (OR, 3.7; 95% CI, 1.3–11.0; P = 0.01). In the multivariate analysis, age, alcohol drinking, and subsite remained as significant determinants of HPV infection.
Table 3.
Association of human papillomavirus (HPV) status with clinicopathological factors in oropharyngeal carcinoma (n = 163)
| Factor and level | Total no. | HPV-positive | Univariate analysis† |
Multivariate analysis† |
||||
|---|---|---|---|---|---|---|---|---|
| No. (%) | Odds ratio | 95% CI | P-value | Odds ratio | 95% CI | P-value | ||
| Sex | ||||||||
| Male | 140 | 46 (32.9) | Reference | |||||
| Female | 23 | 10 (43.5) | 1.6 | 0.6–3.8 | 0.327 | |||
| Age at diagnosis‡ | ||||||||
| <65 years | 73 | 33 (45.2) | Reference | Reference | ||||
| ≥65 years | 90 | 23 (25.6) | 0.4 | 0.2–0.8 | 0.009 | 0.4 | 0.2–0.7 | 0.005 |
| Per 10 years | 0.7 | 0.5–1.0 | 0.030 | |||||
| Cigarrete smoking‡,§ | ||||||||
| <42 pack-years | 81 | 37 (45.7) | Reference | |||||
| ≥42 pack years | 81 | 19 (23.5) | 0.4 | 0.2–0.7 | 0.003 | |||
| Per 5 pack-years | 0.9 | 0.8–1.0 | <0.001 | |||||
| Never | 22 | 13 (59.1) | Reference | |||||
| Ever | 140 | 43 (30.7) | 0.3 | 0.1–0.8 | 0.011 | |||
| Alcohol drinking‡,§ | ||||||||
| <69 units of sake index | 81 | 39 (48.2) | Reference | Reference | ||||
| ≥69 units of sake index | 81 | 17 (21.0) | 0.3 | 0.1–0.6 | <0.001 | 0.3 | 0.1–0.6 | 0.001 |
| Per 5 units of sake index | 0.9 | 0.9–1.0 | <0.001 | |||||
| Never | 37 | 20 (54.1) | Reference | |||||
| Ever | 125 | 36 (28.8) | 0.3 | 0.2–0.7 | 0.005 | |||
| Subsite | ||||||||
| Palatine tonsil | 93 | 42 (45.2) | Reference | Reference | ||||
| Lingual tonsil | 43 | 12 (27.9) | 0.5 | 0.2–1.0 | 0.053 | 0.4 | 0.2–0.9 | 0.040 |
| Posterior wall and soft palate | 27 | 2 (7.4) | 0.1 | 0.02–0.4 | <0.001 | 0.1 | 0.02–0.4 | <0.001 |
| Tumor stage | ||||||||
| T1–T2 | 96 | 36 (37.5) | Reference | |||||
| T3–T4 | 67 | 20 (29.9) | 0.7 | 0.4–1.4 | 0.310 | |||
| Nodal stage | ||||||||
| N0–N1 | 59 | 17 (28.8) | Reference | |||||
| N2–N3 | 104 | 39 (37.5) | 1.5 | 0.8–3.0 | 0.300 | |||
| Cell differentiation | ||||||||
| Well | 34 | 7 (20.6) | Reference | |||||
| Moderate | 72 | 23 (31.9) | 1.8 | 0.7–5.1 | 0.214 | |||
| Poor–undifferentiated | 39 | 19 (48.7) | 3.7 | 1.3–11.0 | 0.011 | |||
| Unknown | 18 | 7 (38.9) | 2.5 | 0.7–8.9 | 0.163 | |||
Univariate analysis was carried out using the logistic regression model; multivariate analysis was by the forward stepwise logistic regression model.
Median of age at diagnosis, cigarette smoking, and alcohol abuse was 65 year, 42 pack-years, and 69 units of sake index, respectively.
One pack-year and one unit of sake index are defined as the equivalent of smoking one pack of cigarettes per day for 1 year and drinking 28 g alcohol per day for 1 year, respectively. CI, confidence interval.
Trend of HPV prevalence in oropharyngeal carcinoma
As HPV-positive tumors were specific to the palatine and lingual tonsils among subsites of the oropharynx, we addressed the chronological change of HPV prevalence in oropharyngeal carcinoma originating from these subsites. Prevalence of HPV was increased from 36.8% during 1995 to 1999, 39.0% during 2000 to 2009, to 48.3% during 2010 to 2012 (Fig. 1). This increasing trend across calendar periods was statistically significant (P < 0.001).
Fig. 1.
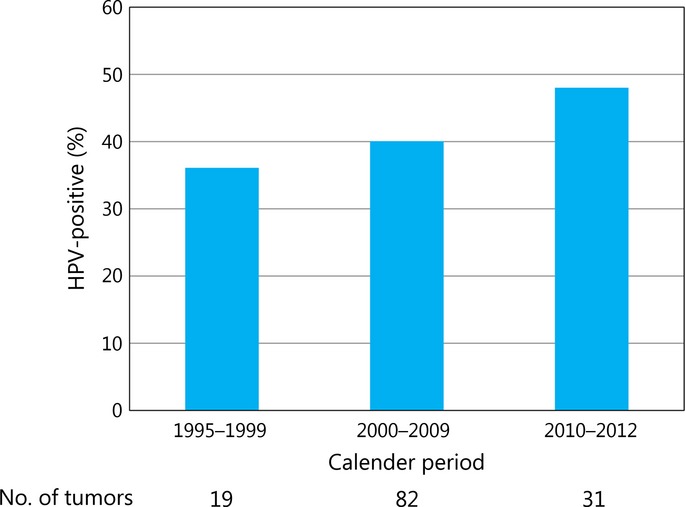
Prevalence of human papillomavirus infection in carcinomas of the palatine and lingual tonsils across three calendar periods (1995–1999, 2000–2009, and 2010–2012). An increasing trend was statistically significant (P < 0.001). The number of specimens evaluable for each assay is shown below the x-axis.
Association of HPV status with p53 mutations
In oropharyngeal carcinoma, 28 of 57 (49.1%) tumors carrying wild-type p53 were HPV-positive, whereas 2 of 25 (8.0%) tumors harboring any p53 mutation and none of 10 tumors harboring a disruptive mutation were HPV-positive (Table 4). In turn, 2 of 30 (6.7%) HPV-positive tumors harbored p53 mutations. Accordingly, tumors harboring any p53 mutation, and particularly a disruptive mutation, were less likely to be HPV-positive as compared with tumors carrying wild-type p53 (for any p53 mutation: OR, 0.09; 95% CI, 0.01–0.3; P < 0.001; for a disruptive mutation: OR, 0.05; 95% CI, 0.003–0.9; P < 0.001), indicating an inverse relationship between HPV infection and any p53 mutation and a disruptive mutation. After adjustment for tobacco smoking and alcohol drinking in the multivariate analysis, p53 mutations remained inversely associated with HPV infection (P = 0.005). As none of the tumors harboring a disruptive mutation was HPV-positive, multivariate analysis of a disruptive mutation at a risk of HPV infection was prohibited. We also examined whether there was an inverse relationship between HPV infection and p53 mutations in non-oropharyngeal carcinoma as observed in oropharyngeal carcinoma. As shown in Table 5 of 117 (4.3%) tumors carrying wild-type p53 and 4 of 84 (4.8%) tumors harboring any p53 mutation were HPV-positive, indicating the absence of the inverse relationship. Of note, all the mutations observed in HPV-positive tumors were non-disruptive. Human papillomavirus infection and p53 disruptive mutation did not coexist even in non-oropharyngeal carcinoma.
Table 4.
Relationship between human papillomavirus (HPV) status and p53 mutations in head and neck squamous cell carcinomas (n = 283)
| p53 mutations | Total no. | HPV-positive | Univariate analysis† |
Multivariate analysis‡,§ |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. (%) | Odds ratio | 95% CI | P-value | Odds ratio | 95% CI | P-value | ||||
| Oropharyngeal carcinoma | p53 status | Wild-type | 57 | 28 (49.1) | Reference | Reference | ||||
| Mutant | 25 | 2 (8.0) | 0.09 | 0.01–0.3 | <0.001 | 0.1 | 0.02–0.5 | 0.005 | ||
| Mutation category | Wild-type | 57 | 28 (49.1) | Reference | ||||||
| Non-disruptive | 15 | 2 (13.3) | 0.2 | 0.02–0.6 | 0.008 | |||||
| Disruptive | 10 | 0 (0.0) | 0.05 | 0.003–0.9 | <0.001 | |||||
| Non-oropharyngeal carcinoma | p53 status | Wild-type | 117 | 5 (4.3) | Reference | |||||
| Mutant | 84 | 4 (4.8) | 1.1 | 0.3–4.4 | 0.868 | |||||
| Mutation category | Wild-type | 117 | 5 (4.3) | Reference | ||||||
| Non-disruptive | 61 | 4 (6.6) | 1.6 | 0.4–6.2 | 0.516 | |||||
| Disruptive | 23 | 0 (0.0) | 0.4 | 0.02–7.4 | 0.176 | |||||
Univariate analysis was carried out using the logistic regression model for p53 status and by the Clopper–Pearson method for mutation category.
Multivariate analysis was carried out using the forward stepwise logistic regression model.
Ajusted for cigarette smoking and alcohol drinking. CI, confidence interval.
Table 5.
Association between p53 mutations and clinicopathological factors in Japanese patients with head and neck squamous cell carcinoma (n = 283)
| Factor and level | Total no. |
p53 status |
Mutation category |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutant No. (%) |
Univariate analysis† |
Disruptive No. (%) |
Univariate analysis† |
||||||
| Odds ratio | 95% CI | P-value | Odds ratio | 95% CI | P-value | ||||
| Sex | |||||||||
| Male | 240 | 94 (39.2) | Reference | 31 (33.0) | Reference | ||||
| Female | 43 | 15 (34.9) | 0.8 | 0.4–1.6 | 0.593 | 2 (13.3) | 0.3 | 0.05–1.2 | 0.101 |
| Age at diagnosis‡ | |||||||||
| <63 years | 131 | 45 (34.4) | Reference | 19 (42.2) | Reference | ||||
| ≥63 years | 152 | 64 (42.1) | 1.4 | 0.9–2.3 | 0.181 | 14 (21.9) | 0.4 | 0.2–0.9 | 0.023 |
| Per 10 years | 1.2 | 0.9–1.5 | 0.140 | 0.7 | 0.5–1.1 | 0.120 | |||
| Cigarrete smoking‡,§ | |||||||||
| <45 pack-years | 131 | 43 (32.8) | Reference | 11 (25.6) | Reference | ||||
| ≥45 pack years | 146 | 64 (43.8) | 1.6 | 1.0–2.6 | 0.060 | 21 (32.8) | 1.4 | 0.6–3.4 | 0.421 |
| Unknown | 6 | 2 (33.3) | 1.0 | 0.2–5.8 | 0.979 | 1 (50.0) | 2.9 | 0.1–77.8 | 0.472 |
| Per 5 pack-years | 1.0 | 1.0–1.1 | 0.040 | 1.0 | 1.0–1.1 | 0.310 | |||
| Never | 47 | 15 (31.9) | Reference | 1 (6.7) | Reference | ||||
| Ever | 230 | 92 (40.0) | 1.4 | 0.7–2.8 | 0.295 | 31 (33.7) | 7.1 | 1.3–131.8 | 0.017 |
| Alcohol drinking†,§ | |||||||||
| <80 units of sake index | 135 | 41 (30.4) | Reference | 13 (31.7) | Reference | ||||
| ≥80 units of sake index | 142 | 66 (46.5) | 2.0 | 1.2–3.3 | 0.006 | 19 (28.8) | 0.9 | 0.4–2.1 | 0.749 |
| Unknown | 6 | 2 (33.3) | 1.1 | 0.2–6.1 | 0.878 | 1 (50.0) | 2.2 | 0.08–57.4 | 0.601 |
| Per 5 units of sake index | 1.0 | 1.0–1.0 | 0.210 | 1.0 | 1.0–1.0 | 0.240 | |||
| Never | 59 | 14 (23.7) | Reference | 3 (21.4) | Reference | ||||
| Ever | 218 | 93 (42.7) | 2.4 | 1.3–4.8 | 0.007 | 29 (31.2) | 1.7 | 0.5–7.7 | 0.445 |
| Primary site | |||||||||
| Oral cavity | 37 | 18 (48.7) | Reference | 2 (11.1) | Reference | ||||
| Nasopharynx | 21 | 1 (4.8) | 0.1 | 0.003–0.3 | <0.001 | 0 (0.0) | 2.2 | 0.1–70.4 | 0.632 |
| HPV-negative ooropharynx | 52 | 23 (44.2) | 0.8 | 0.4–2.0 | 0.680 | 10 (43.5) | 6.2 | 1.3–44.9 | 0.019 |
| HPV-positive oropharynx | 30 | 2 (6.7) | 0.1 | 0.01–0.3 | <0.001 | 0 (0.0) | 1.3 | 0.05–36.3 | 0.505 |
| Hypopharynx | 108 | 49 (45.4) | 0.9 | 0.4–1.9 | 0.730 | 18 (36.7) | 4.6 | 1.1–31.5 | 0.030 |
| Larynx | 35 | 16 (45.7) | 0.9 | 0.3–2.2 | 0.803 | 3 (18.8) | 1.8 | 0.3–15.7 | 0.526 |
| Tumor stage | |||||||||
| T1–T2 | 138 | 55 (39.9) | Reference | 15 (27.3) | Reference | ||||
| T3–T4 | 145 | 54 (37.2) | 0.9 | 0.6–1.4 | 0.652 | 18 (33.3) | 1.3 | 0.6–3.1 | 0.491 |
| Nodal stage | |||||||||
| N0–N1 | 132 | 51 (38.6) | Reference | 12 (23.5) | Reference | ||||
| N2–N3 | 151 | 58 (38.4) | 1.0 | 0.6–1.6 | 0.969 | 21 (36.2) | 1.8 | 0.8–4.4 | 0.148 |
| Cell differentiation | |||||||||
| Well | 79 | 37 (46.8) | Reference | 12 (32.4) | Reference | ||||
| Moderate | 119 | 50 (42.0) | 0.8 | 0.5–1.5 | 0.504 | 16 (32.0) | 1.0 | 0.4–2.5 | 0.966 |
| Poor–undifferentiated | 69 | 19 (27.5) | 0.4 | 0.2–0.9 | 0.015 | 4 (21.1) | 0.6 | 0.1–1.9 | 0.364 |
| Unknown | 16 | 3 (18.8) | 0.3 | 0.1–0.9 | 0.031 | 1 | 1.0 | 0.05–11.9 | 0.975 |
| HPV status | |||||||||
| Negative | 244 | 103 (42.2) | Reference | 33 (32.0) | Reference | ||||
| Positive | 39 | 6 (15.4) | 0.2 | 0.09–0.6 | <0.001 | 0 (0.0) | 0.2 | 0.009–0.8 | 0.034 |
Univariate analysis was carried out using the logistic regression model.
Median of age at diagnosis, cigarette smoking, and alcohol abuse was 63 years, 45 pack-years, and 80 units of sake index, respectively.
One pack-year and one unit of sake index are defined as the equivalent of smoking one pack of cigarettes per day for 1 year and drinking 28 g alcohol per day for 1 year, respectively. CI, confidence interval; HPV, human papillomavirus.
Prevalence and pattern of p53 mutations in HNSCCs
One hundred and nine of 283 (38.5%) tumors were found to harbor p53 mutations. Of 109 tumors with p53 mutations, 76 (69.7%) and 33 (30.3%) were classified to harbor a non-disruptive mutation and a disruptive mutation, respectively. Table 5 summarizes the prevalence of any p53 mutation and a disruptive mutation in relation to clinicopathological factors and HPV status. Tumors in heavy smokers and heavy drinkers were more likely to harbor p53 mutations than tumors in light smokers (OR, 1.6; 95% CI, 1.0–2.6; P = 0.06) and light drinkers (OR, 2.0; 95% CI, 1.2–3.3; P = 0.006), respectively. Although the difference between light and heavy smokers was marginal, there was an increasing trend of p53 mutations in proportion to the amount of tobacco smoking (OR, 1.0; 95% CI, 1.0–1.1; P = 0.04, per 5 pack-years). Of note is a striking difference between virus-unrelated HNSCCs and virus-related HNSCCs. Human papillomavirus-positive oropharyngeal carcinoma and nasopharyngeal carcinoma that is caused by Epstein–Barr virus(24) constitute virus-related HNSCCs. Only one of 21 (4.8%) nasopharyngeal carcinomas and 2 of 30 (6.7%) HPV-positive oropharyngeal carcinomas harbored p53 mutations. In contrast, the frequency of p53 mutations ranged from 44.2% to 48.7% in virus-unrelated HNSCCs. The frequency of any p53 mutation was significantly higher in virus-unrelated HNSCCs than in virus-related HNSCCs (45.7% vs 5.9%, P < 0.001). Compared with oral carcinoma, nasopharyngeal and HPV-positive oropharyngeal carcinomas showed a significantly lower prevalence of p53 mutations (for nasopharyngeal carcinoma: OR, 0.1; 95% CI, 0.003–0.3; P < 0.001; for HPV-positive oropharyngeal carcinoma: OR, 0.1; 95% CI, 0.01–0.3; P < 0.001). Moreover, as in HPV-positive oropharyngeal carcinoma, no disruptive mutation was found in nasopharyngeal carcinoma.
Association of p53 mutations with clinicopathological parameters in virus-unrelated HNSCCs
As p53 mutations were extremely uncommon in virus-related HNSCCs, we sought to address an association of p53 mutations and a disruptive mutation with clinicopathological parameters in virus-unrelated HNSCCs (Table 6). Association of p53 mutations with tobacco smoking and alcohol drinking, which was evident in the whole study group, disappeared in the subpopulation of virus-unrelated HNSCCs. No clinicopathological parameter was associated with p53 mutations, whereas a disruptive mutation significantly correlated with tobacco exposure and primary site. The proportion of tumors with a disruptive mutation in tumors with any mutation was more frequent in ever-smoker patients than in never-smoker patients (7.1% vs 34.1%; OR, 6.7; 95% CI, 1.2–124.8; P = 0.02). As compared with oral carcinoma, HPV-negative oropharyngeal carcinoma and hypopharyngeal carcinoma were more likely to harbor a disruptive mutation (for HPV-negative oropharyngeal carcinoma: OR, 6.2; 95% CI, 1.3–44.9; P = 0.02; for hypopharyngeal carcinoma: OR, 4.6; 95% CI, 1.1–31.5; P = 0.03). After adjustment for age and tobacco exposure, HPV-negative oropharyngeal carcinoma and hypopharyngeal carcinoma remained independently associated with a disruptive mutation compared with oral carcinoma (for HPV-negative oropharyngeal carcinoma: OR, 7.6; 95% CI, 1.5–60.0; P = 0.01; for hypopharyngeal carcinoma: OR 4.9; 95% CI, 1.1–34.7; P = 0.03). Tobacco exposure also independently correlated with a disruptive mutation after adjustment for age and primary site (OR, 6.0; 95% CI, 1.0–115.3; P = 0.04).
Table 6.
Association between p53 mutations and clinicopathological factors in virus-unrelated head and neck squamous cell carcinomas (n = 232)
| Factor and level |
p53 status |
Mutation category |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutant |
Univariate analysis† |
Disruptive | Univariate analysis† |
Multivariate analysis† |
||||||||
| Total no. | No. (%) | Odds ratio | 95% CI | P-value | No. (%) | Odds ratio | 95% CI | P-value | Odds ratio | 95% CI | P-value | |
| Sex | ||||||||||||
| Male | 202 | 93 (46.0) | Reference | 31 (33.3) | Reference | |||||||
| Female | 30 | 13 (43.3) | 0.9 | 0.4–1.9 | 0.781 | 2 (15.4) | 0.4 | 0.05–1.5 | 0.166 | |||
| Age at diagnosis‡ | ||||||||||||
| <63 years | 102 | 45 (44.1) | Reference | 19 (42.2) | Reference | Reference | ||||||
| ≥63 years | 130 | 61 (46.9) | 1.1 | 0.7–1.9 | 0.670 | 14 (23.0) | 0.4 | 0.2–0.9 | 0.035 | 0.4 | 0.1–1.0 | 0.042 |
| per 10 years | 1.1 | 0.8–1.4 | 0.710 | 0.7 | 0.5–1.2 | 0.180 | ||||||
| Cigarrete smoking‡,§ | ||||||||||||
| <45 pack-years | 95 | 42 (44.2) | Reference | 11 (26.2) | Reference | |||||||
| ≥45 pack years | 135 | 63 (46.7) | 1.1 | 0.7–1.9 | 0.713 | 21 (33.3) | 1.4 | 0.6–3.4 | 0.434 | |||
| Unknown | 2 | 1 (50.0) | 1.3 | 0.08–22.2 | 0.831 | 1 (100.0) | ||||||
| Per 5 pack-years | 1.0 | 1.0–1.0 | 0.670 | 1.0 | 1.0–1.1 | 0.340 | ||||||
| Never | 31 | 14 (45.2) | Reference | 1 (7.1) | Reference | Reference | ||||||
| Ever | 199 | 91 (45.7) | 1.0 | 0.5–2.2 | 0.953 | 31 (34.1) | 6.7 | 1.2–124.8 | 0.023 | 6.0 | 1.0–115.3 | 0.045 |
| Alcohol drinking‡,§ | ||||||||||||
| <80 units of sake index | 97 | 40 (41.2) | Reference | 13 (32.5) | Reference | |||||||
| ≥80 units of sake index | 133 | 65 (48.9) | 1.4 | 0.8–2.3 | 0.250 | 19 (29.2) | 0.9 | 0.4–2.0 | 0.724 | |||
| Unknown | 2 | 1 (50.0) | 1.4 | 0.08–1.4 | 0.830 | 1 (100.0) | ||||||
| Per 5 units of sake index | 1.0 | 1.0–1.0 | 0.740 | 1.0 | 1.0–1.0 | 0.260 | ||||||
| Never | 36 | 13 (36.1) | Reference | 3 (23.1) | Reference | |||||||
| Ever | 194 | 92 (47.4) | 1.6 | 0.8–3.4 | 0.207 | 29 (31.5) | 1.5 | 0.4–7.2 | 0.527 | |||
| Primary site | ||||||||||||
| Oral cavity | 37 | 18 (48.7) | Reference | 2 (11.1) | Reference | Reference | ||||||
| HPV-negative oropharynx | 52 | 23 (44.2) | 0.8 | 0.4–2.0 | 0.680 | 10 (43.5) | 6.2 | 1.3–44.9 | 0.019 | 7.6 | 1.5–60.0 | 0.014 |
| Hypopharynx | 108 | 49 (45.4) | 0.9 | 0.4–1.9 | 0.730 | 18 (36.7) | 4.6 | 1.1–31.5 | 0.030 | 4.9 | 1.1–34.7 | 0.033 |
| Larynx | 35 | 16 (45.7) | 0.9 | 0.3–2.2 | 0.803 | 3 (18.8) | 1.8 | 0.3–15.7 | 0.530 | 1.4 | 0.1–14.1 | 0.763 |
| Tumor stage | ||||||||||||
| T1–T2 | 107 | 54 (50.5) | Reference | 15 (27.8) | Reference | |||||||
| T3–T4 | 125 | 52 (41.6) | 0.7 | 0.4–1.2 | 0.175 | 18 (34.6) | 1.4 | 0.6–3.2 | 0.447 | |||
| Nodal stage | ||||||||||||
| N0–N1 | 106 | 50 (47.2) | Reference | 12 (24.0) | Reference | |||||||
| N2–N3 | 126 | 56 (44.4) | 0.9 | 0.5–1.5 | 0.678 | 21 (37.5) | 1.9 | 0.8–4.5 | 0.132 | |||
| Cell differentiation | ||||||||||||
| Well | 73 | 37 (50.7) | Reference | 12 (32.4) | Reference | |||||||
| Moderate | 106 | 49 (46.2) | 0.8 | 0.5–1.5 | 0.557 | 16 (32.7) | 1.0 | 0.4–2.5 | 0.981 | |||
| Poor–undifferntiated | 41 | 17 (41.5) | 0.7 | 0.3–1.5 | 0.343 | 4 (23.5) | 0.6 | 0.2–2.3 | 0.500 | |||
| Unknown | 12 | 3 (25.0) | 0.3 | 0.07–1.2 | 0.091 | 1 (33.3) | 1.0 | 0.05–11.9 | 0.975 | |||
Univariate analysis was carried out using the logistic regression model; multivariate analysis was carried out using the forward stepwise logistic regression model.
Median of age at diagnosis, cigarette smoking, and alcohol abuse was 64 years, 49 pack-years, and 100 units of sake index, respectively.
One pack-year and one unit of sake index are defined as the equivalent of smoking one pack of cigarettes per day for 1 year and drinking 28 g alcohol per day for 1 year, respectively. CI, confidence interval.
Pattern of p53 mutations in virus-unrelated HNSCCs
The prevalence of any p53 mutation was not associated with tobacco smoking in virus-unrelated HNSCCs, as shown in Table 6, whereas tobacco carcinogen is known to link with a specific type of mutagenesis.(25) We addressed the pattern of p53 mutations in virus-unrelated HNSCCs, in relation to tobacco exposure. One hundred and twelve of 232 (48.3%) tumors harbored a total of 165 p53 mutations (22 tumors with two distinct mutations, 6 with three, 2 with four, 2 with five, and 1 with six), including 118 missense mutations, 12 nonsense mutations, 8 frame-shifts, and 27 silent mutations (detail of the mutations in Table S1), although six tumors harbored only silent mutations. The codon distribution of p53 mutations differed depending on tobacco exposure. In ever-smokers, 19 of 143 (13.2%) mutations were detected at codons described as “hotspots” for transversions (codon 157, one mutation; codon 158, six mutations; codon 245, six mutations; codon 248, five mutations; codon 273, one mutation),(26) whereas, in never-smokers, mutations at hotspots were found only at codon 245, accounting for 2 of 22 (9.1%) mutations. Figure 2 shows the types of mutations according to smoking status. Transversions accounted for 39.2% of mutations in ever-smokers, whereas the vast majority (81.8%) of mutations in never-smokers were transitions. Two types of mutations showed consistent difference in relation to smoking. G:C to T:A transversions were more prevalent in ever-smokers than in never-smokers (22.4% vs 4.5%, P = 0.04), whereas G:C to A:T transitions at CpG sites were less prevalent in ever-smokers than in never-smokers (10.5% vs 27.3%, P = 0.04).
Fig. 2.
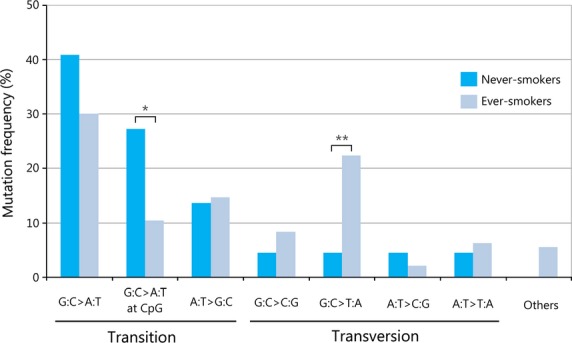
Pattern of p53 mutations in human papillomavirus-unrelated head and neck tumors from never- and ever-smokers. One hundred sixty-five mutations are represented in 112 patients. Never-smokers showed a higher prevalence of G:C to A:T transitions at CpG sites than ever-smokers (*P = 0.04). Ever-smokers showed a higher prevalence of G:C to T:A transversions than never-smokers (**P = 0.04). Others represent insertion and deletion.
Discussion
In the present study we established the prevalence and pattern of HPV in Japanese patients with HNSCC. The most prevalent oropharyngeal carcinoma tumors were HPV-positive, especially those arising in the palatine and lingual tonsils, where HPV-positive tumors accounted for 45.2% and 27.9%, respectively, although this prevalence is still lower than in North America and Europe.(14,15) In the USA, HPV prevalence in oropharyngeal carcinoma significantly increased over calendar time, reaching up to 72% during 2000 to 2004.(14) We also showed the increasing trend in Japan. However, there is a racial disparity in HPV prevalence.(16) The prevalence is significantly lower in black patients than in white patients. Sexual behavior, which is significantly associated with the pathogenesis of HPV-positive oropharyngeal carcinoma,(2,26) might differ among races or nations. Additionally, the rate of smoking, which is the other major cause of oropharyngeal carcinogenesis, is higher in Japan than in the USA.(27) Given this knowledge, it remains uncertain whether HPV prevalence in oropharyngeal carcinoma among Japanese will reach as high as that among white populations. As for HPV subtypes, HPV16 commanded a vast majority (91.1%) of HPV-positive tumors in Japan, which is consistent with a worldwide systematic review.(28)
We addressed factors associated with HPV infection in oropharyngeal carcinoma. In accordance with previous reports,(11,29) we found that tobacco smoking, alcohol drinking, and older age were inversely related with HPV infection. Likewise, HPV-positive tumors were predominant in the palatine and lingual tonsils, and differentiation grade of HPV-positive tumors was poor. Although HPV-positive tumors present mostly at an early tumor stage and advanced nodal stage,(15) we failed to show such findings, the reason for which remains unknown. An additional discordance from previous reports was the absence of a gender gap. We did not find a higher rate of HPV-positive tumors in men than in women. The higher rate in men in North America and Europe is considered to be attributed to HPV prevalence in cervical rather than penile tissue, which might facilitate the chance of HPV infection when performing oral sex on a woman. Differences in oral sex behavior might be responsible for the absence of a gender gap in Japan.
Gillison et al.(11) examined HPV status and p53 mutations in 34 oropharyngeal carcinomas, and showed that HPV-positive tumors as compared with HPV-negative tumors were less likely to harbor p53 mutations. Westra et al.(12) showed that 12 of 20 (60.0%) tonsillar carcinomas without a disruptive mutation were HPV-positive, whereas none of 7 tonsillar carcinomas carrying a disruptive mutation was HPV-positive. In the present study we further verified these findings in a larger scale, and established the inverse relationship between HPV infection and any p53 mutation or a disruptive mutation in oropharyngeal carcinoma. However, we found the absence of that inverse relationship in non-oropharyngeal HNSCCs, as previously reported.(11) This finding, given the report that p53 mutations of HNSCCs are associated with poor outcome,(30) might partially explain why a prognostic advantage of HPV-positive tumors disappear in non-oropharyngeal HNSCCs.(31) We also found that HPV infection and a disruptive mutation did not coexist, irrespective of primary tumor site, indicating that the coexistence of HPV infection and p53 mutations occurs only when the mutations are non-disruptive. Disruptive mutations are selectively associated with allelic loss of wild-type p53,(32) and confer “gain of function” phenotype.(33,34) It seems likely that disruptive mutations lead directly to the acquisition of malignant phenotype, whereas non-disruptive mutations, in concert with other events targeting the p53 pathway such as transcriptionally active HPV, might produce a synergistic effect of p53 functional abrogation.
Of interest is the finding that, although there was a positive association between p53 mutations and tobacco smoking in the whole patient group, this association disappeared in the subpopulation of virus-unrelated HNSCCs. This is most probably because as high as 76.6% of virus-related HNSCC patients with known history of tobacco smoking were light or never-smokers (data not shown), and because no <94.1% of virus-related HNSCCs carried wild-type p53 (Table 5). There has been a series of studies showing that the frequency of p53 mutations in HNSCCs increases in proportion to the level of exposure to tobacco smoking.(35–37) In these studies, oropharyngeal carcinoma accounted for a substantial portion of the analyzed population, and HPV status of oropharyngeal carcinoma was unknown in spite of the inverse relationship of HPV infection with p53 mutations and tobacco smoking.(35–37) This background behind the studies might explain, at least in part, the divergence from our finding. Of note, in virus-unrelated HNSCCs a disruptive mutation was independently associated with primary site (the oropharynx and hypopharynx). This finding is potentially interesting because hypopharyngeal carcinoma is the most fatal among HNSCCs,(29) and because p53 disruptive mutations serve as an adverse prognostic factor in HNSCCs.(29) Another interesting finding is that in virus-unrelated HNSCCs, smoking was associated with increased prevalence of G:C to T:A transversions as reported for carcinomas of the lung(38) and upper aerodigestive tract.(35) This type of mutagenesis is caused by bulky carcinogen from tobacco smoke, such as polycyclic aromatic hydrocarbons.(25) Conversely, we found that smoking was associated with decreased prevalence of G:C to A:T transitions at CpG sites, a type of mutation commonly associated with endogenous mutagenesis processes, such as spontaneous deamination of methylated cytosine into thymine. This phenomenon has also been described in carcinomas of the upper aerodigestive tract.(35)
In conclusion, HNSCC is etiologically classified into virus-related and virus-unrelated subgroups. In virus-related HNSCC, p53 mutations are uncommon with the absence of a disruptive mutation. In virus-unrelated HNSCC, although p53 mutations are common, the prevalence is not associated with tobacco and alcohol exposure. Disruptive mutagenesis of p53 is closely related with primary site.
Acknowledgments
This work was supported in part by the Japan Society for the Promotion of Science, Kakenhi (Grant No. 19591966, to H.I.).
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Table S1 Details of p53 mutations in virus-unrelated head and neck squamous cell carcinomas.
References
- 1.Hashibe M, Brennan P, Benhamou S, et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J Natl Cancer Inst. 2007;99:777–89. doi: 10.1093/jnci/djk179. [DOI] [PubMed] [Google Scholar]
- 2.Mork J, Lie AK, Glattre E, et al. Human papillomavirus infection as a risk factor for squamous-cell carcinoma of the head and neck. N Engl J Med. 2001;344:1125–31. doi: 10.1056/NEJM200104123441503. [DOI] [PubMed] [Google Scholar]
- 3.Efeyan A, Serrano M. p53: guardian of the genome and policeman of the oncogenes. Cell Cycle. 2007;6:1006–10. doi: 10.4161/cc.6.9.4211. [DOI] [PubMed] [Google Scholar]
- 4.Olshan AF, Weissler MC, Pei H, Conway K. p53 mutations in head and neck cancer: new data and evaluation of mutational spectra. Cancer Epidemiol Biomarkers Prev. 1997;6:499–504. [PubMed] [Google Scholar]
- 5.González MV, Pello MF, López-Larrea C, Suárez C, Menéndez MJ, Coto E. Loss of heterozygosity and mutation analysis of the p16 (9p21) and p53 (17p13) genes in squamous cell carcinoma of the head and neck. Clin Cancer Res. 1995;1:1043–9. [PubMed] [Google Scholar]
- 6.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 7.Blons H, Laurent-Puig P. TP53 and head and neck neoplasms. Hum Mutat. 2003;21:252–7. doi: 10.1002/humu.10171. [DOI] [PubMed] [Google Scholar]
- 8.Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and E7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J Natl Cancer Inst. 2009;101:412–23. doi: 10.1093/jnci/djp017. [DOI] [PubMed] [Google Scholar]
- 9.Friend S. p53: a glimpse at the puppet behind the shadow play. Science. 1994;265:334–5. doi: 10.1126/science.8023155. [DOI] [PubMed] [Google Scholar]
- 10.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 11.Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709–20. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 12.Westra WH, Taube JM, Poeta ML, Begum S, Sidransky D, Koch WM. Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2008;14:366–9. doi: 10.1158/1078-0432.CCR-07-1402. [DOI] [PubMed] [Google Scholar]
- 13.Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29:4294–301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Näsman A, Attner P, Hammarstedt L, et al. Incidence of human papillomavirus (HPV) positive tonsillar carcinoma in Stockholm, Sweden; an epidemic of viral-induced carcinoma? Int J Cancer. 2009;125:362–6. doi: 10.1002/ijc.24339. [DOI] [PubMed] [Google Scholar]
- 16.Settle K, Posner MR, Schumaker LM, et al. Racial survival disparity in head and neck cancer results from low prevalence of human papillomavirus infection in black oropharyngeal cancer patients. Cancer Prev Res (Phila) 2009;2:776–81. doi: 10.1158/1940-6207.CAPR-09-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujinaga Y, Shimada M, Okazawa K, Fukushima M, Kato I, Fujinaga K. Simultaneous detection and typing of genital human papillomavirus DNA using the polymerase chain reaction. J Gen Virol. 1991;72:1039–44. doi: 10.1099/0022-1317-72-5-1039. [DOI] [PubMed] [Google Scholar]
- 18.Ueda Y, Enomoto T, Miyatake T, et al. Analysis of clonality and HPV infection in benign, neoplastic, premalignant, and malignant lesions of the vulvar mucosa. Am J Clin Pathol. 2004;122:266–74. doi: 10.1309/65MK-PQT3-E2BD-M67E. [DOI] [PubMed] [Google Scholar]
- 19.Kalantari M, Karlsen F, Kristensen G, Holm R, Hagmar B, Johansson B. Disruption of the E1 and E2 reading frames of HPV 16 in cervical carcinoma is associated with poor prognosis. Int J Gynecol Pathol. 1998;17:146–53. doi: 10.1097/00004347-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 20.Badal V, Chuang LS, Tan EH, et al. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J Virol. 2003;77:6227–34. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hongyo T, Buzard GS, Calvert RJ, Weghorst CM. ‘Cold SSCP’: a simple, rapid and non-radioactive method for optimized single-strand conformation polymorphism analyses. Nucleic Acids Res. 1993;21:3637–42. doi: 10.1093/nar/21.16.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hongyo T, Hoshida Y, Nakatsuka S, et al. p53, K-ras, c-kit and beta-catenin gene mutations in sinonasal NK/T-cell lymphoma in Korea and Japan. Oncol Rep. 2005;13:265–71. [PubMed] [Google Scholar]
- 23.Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr. 1974;19:716–23. [Google Scholar]
- 24.Yoshizaki T, Ito M, Murono S, et al. Current understanding and management of nasopharyngeal carcinoma. Auris Nasus Larynx. 2012;39:137–44. doi: 10.1016/j.anl.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Smith LE, Denissenko MF, Bennett WP, et al. Targeting of lung cancer mutational hotspots by polycyclic aromatic hydrogens. J Natl Cancer Inst. 2000;92:803–11. doi: 10.1093/jnci/92.10.803. [DOI] [PubMed] [Google Scholar]
- 26.D'Souza G, Agrawal Y, Halpern J, Bodison S, Gillison ML. Oral sexual behaviors associated with prevalent oral human papillomavirus infection. J Infect Dis. 2009;199:1263–9. doi: 10.1086/597755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Organisation for Economic Co-Operation and Development. [Cited on 01 Mar 2014.] Available from URL: http://www.oecd.org/
- 28.Kreimer AR, Clifford GM, Boyle P, Franceschi S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev. 2005;14:467–75. doi: 10.1158/1055-9965.EPI-04-0551. [DOI] [PubMed] [Google Scholar]
- 29.Marur S, D'Souza G, Westra WH, Forastiere AA. HPV-associated head ans neck cancer: a virus-related cancer epidemic. Lancet Oncol. 2010;11:781–9. doi: 10.1016/S1470-2045(10)70017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poeta ML, Manola J, Goldwasser MA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–61. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isayeva T, Li Y, Maswahu D, Brandwein-Gensler M. Human papillomavirus in non-oropharyngeal head and neck cancers: a systematic literature review. Head Neck Pathol. 2012;6:S104–20. doi: 10.1007/s12105-012-0368-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erber R, Conradt C, Homann N, et al. TP53 DNA contact mutations are selectively associated with allelic loss and have a strong clinical impact in head and neck cancer. Oncogene. 1988;16:1671–9. doi: 10.1038/sj.onc.1201690. [DOI] [PubMed] [Google Scholar]
- 33.Lányi A, Deb D, Seymour RC, Ludes-Meyers JH, Subler MA, Deb S. “Gain of function” phenotype of tumor-derived mutant p53 requires the oligomerization/nonsequence-specific nucleic acid-binding domain. Oncogene. 1998;16:3169–76. doi: 10.1038/sj.onc.1201857. [DOI] [PubMed] [Google Scholar]
- 34.Dittmer D, Pati S, Zambetti G, et al. Gain of function mutations in p53. Nat Genet. 1993;4:42–6. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 35.Szymańska K, Levi JE, Menezes A, et al. TP53 and EGFR mutations in combination with lifestyle risk factors in tumors of the upper aerodigestive tract from South America. Carcinogenesis. 2010;31:1054–9. doi: 10.1093/carcin/bgp212. [DOI] [PubMed] [Google Scholar]
- 36.Liloglou T, Scholes AG, Spandidos DA, Vaughan ED, Jones AS, Field JK. p53 mutations in squamous cell carcinoma of the head and neck predominate in a subgroup of former smokers with a low frequency of genetic instability. Cancer Res. 1997;57:4070–4. [PubMed] [Google Scholar]
- 37.Brennan JA, Boyle JO, Koch WM, et al. Association between cigarette smoking and mutation of the p53 gene in squamous cell carcinoma of the head and neck. N Engl J Med. 1995;332:712–7. doi: 10.1056/NEJM199503163321104. [DOI] [PubMed] [Google Scholar]
- 38.Le Calvez F, Mukeria A, Hunt JD, et al. TP53 and KRAS mutation load and types in lung cancers in relation to tobacco smoke: distinct pattern in never, former, and current smokers. Cancer Res. 2005;65:5076–83. doi: 10.1158/0008-5472.CAN-05-0551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Details of p53 mutations in virus-unrelated head and neck squamous cell carcinomas.