

Abstract
Chemotherapy drugs themselves may act as stressors to induce adaptive responses to promote the chemoresistance of cancer cells. Our previous research showed that sirtuin 1 (SIRT1) was overexpressed in pancreatic cancer patients and deregulation of SIRT1 with RNAi could enhance chemosensitivity. Thus, we hypothesized that SIRT1 might facilitate chemoresistance in pancreatic cancer cells through regulating the adaptive response to chemotherapy-induced stress. In the present study, SIRT1 in PANC-1, BXPC-3, and ASPC-1 cells was upregulated after treatment with gemcitabine. Moreover, the decrease in SIRT1 activity with special inhibitor EX527 had a synergic effect on chemotherapy with gemcitabine in PANC-1 and ASPC-1 cell lines, which significantly promoted apoptosis, senescence, and G0/G1 cycle arrest. Western blot results also showed that SIRT1, acetylated-p53, FOXO3a, and p21 were upregulated after combined treatment, whereas no obvious change was evident in total p53 protein. To further confirm the role of SIRT1 in clinical chemotherapy, SIRT1 was detected in eight pancreatic cancer tissues acquired by endoscopy ultrasonography guided fine needle aspiration biopsy before and after chemotherapy. Compared to before chemotherapy, SIRT1 was significantly increased after treatment with gemcitabine in six cases. Thus, our results indicated a special role for SIRT1 in the regulation of adaptive response to chemotherapy-induced stress, which is involved in chemoresistance. Moreover, it indicates that blocking SIRT1 activity with targeting drugs might be a novel strategy to reverse the chemoresistance of pancreatic cancer.
Keywords: Adaptive response, EX527, gemcitabine, pancreatic cancer, SIRT1
Pancreatic cancer is one of the most aggressive of all human malignancies. It has an extremely low 5-year survival rate of <1% and is the fourth leading cause of cancer-related death.(1–3) The poor prognosis is in large part due to the fact that early symptoms of pancreatic cancer are occult and early diagnosis is difficult. Thereafter, at the time of diagnosis, only 10–20% of tumors are resectable due to its early dissemination and aggressive growth. Therefore, chemotherapy is one of the most important treatments to prolong the lives of pancreatic cancer patients. Unfortunately, compared to other cancers, pancreatic cancer shows extraordinary resistance to chemotherapy including gemcitabine (GEM) and 5-fluorouracil.(4) It is urgent to discover novel mechanisms and targets to reverse the resistance of pancreatic cancer to chemotherapy.
Recently, research has indicated that adaptive responses to DNA damage induced by chemotherapy drugs may help cancer cells to resist such genetic stress and be involved in chemoresistance through heat shock protein, nuclear factor-κB (NF-κB), or other pathways.(5–8) Research has revealed that sirtuin 1 (SIRT1), a mammalian stress-responsive deacetylase of class III histone deacetylases, regulates aging and resistance to oxidative and DNA damage stress by inhibiting cellular apoptosis or senescence.(9–12) An increasing number of proteins have been identified as substrates of SIRT1, including p53, FOXO transcription factors, and NF-κB. SIRT1 can deacetylate and inhibit the activities of such critical factors in stress response and apoptosis regulation.(13–16) The p53 protein becomes acetylated in response to DNA damage, and the acetylated form has been reported to increase its transcriptional activity, enhance site-specific DNA binding, and prevent ubiquitination of key lysine residues.(17,18) Reports has also revealed that SIRT1-deficient mice showed p53 hyperacetylation and increased ionizing radiation-induced apoptosis,(19) and siRNA-mediated knockdown of SIRT1 reduced drug resistance and induced growth inhibition in epithelial cancer cells in vitro.(20) Our previous study showed that SIRT1 was overexpressed in pancreatic cancer samples and the PANC-1 cell line, and SIRT1 deregulation with shRNA inhibited the proliferation of PANC-1 cells and enhanced their chemosensitivity.(21) Hence, we presume that SIRT1 may be involved in the adaptive reaction of pancreatic cancer cells to chemotherapy-induced DNA damage stress.
First, we detected changes in SIRT1 expression in PANC-1, BXPC-3, and ASPC-1 cell lines after GEM treatment. Second, we applied EX527, a specific small molecule inhibitor,(22) to observe the effects of SIRT1 downregulation on pancreatic cancer cells treated with GEM. Third, the apoptosis, senescence, cell cycle, and relevant signal pathways were investigated to reveal the preliminary mechanism of deregulation of SIRT1 on chemosensitivity. Finally, to further confirm the effect of SIRT1 on clinical chemotherapy, the SIRT1 expression of pancreatic cancer fine needle aspiration samples guided with endoscopy ultrasonography (EUS-FNA) were assessed before and after chemotherapy treatment.
Materials and Methods
Cell culture
The PANC-1, BXPC-3, ASPC-1, and 293T cell lines were purchased from ATCC (Manassas, VA, USA). Cells were grown in RPMI-1640 medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin in a humidified atmosphere containing 5% CO2. Cells in the logarithmic phase of growth were used for all studies described.
Western blot analysis
Western blot was carried out as previously described,(23) and 30 μg tissue or cellular protein lysates were subjected to electrophoresis on 10% or 12% SDS–polyacrylamide gels and transferred to PVDF membranes (Millipore, Billerica, MA, USA). After blocking in 5% skim milk, the membranes were incubated overnight at 4°C with antibodies specific to p53 and acetylated-p53 (Cell Signaling Technology, Danvers, MA, USA), FOXO3a (Abcam Biotechnology, Cambridge, UK), SIRT1, p21, and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing three times in Tris-buffered saline (10 mM Tris–HCl [pH 7.5] and 150 mM NaCl) containing 1% Tween 20, HRP-conjugated goat anti-mouse or goat anti-rabbit antibodies were applied. After repeated washes, bound antibodies were detected using the ECL Western blot detection system (Amersham GE Healthcare, Buckinghamshire, UK) and exposed to X-OMAT film (Kodak, Rochester, NY, USA). Intensities of the bands were quantified by Alpha DigiDoc 1201 (Alpha Innotech, San Leandro, CA, USA).
Quantitative RT-PCR
SIRT1 was examined in pancreatic cancer cells and tissues before and after GEM treatment using quantitative RT-PCR (qRT-PCR). Briefly, Total RNA (1 μg) extracted (TRIzol reagent; Invitrogen, Carlsbad, CA, USA) was reverse transcribed to cDNA according to the manufacturer's instructions. Real-time PCR was carried out using the SYBR Green quantitative PCR kit (Invitrogen) according to the manufacturer's instructions using an Applied Biosystems 7300 real-time PCR system (Carlsbad, CA, USA). SIRT1 forward primer, 5′-CGGAAACAATACCTCCACCTGA-3′; reverse primer, 5′-GAAGTCTACAGCAAGGCGAGC A-3′ (amplicon size, 242 bp). All data were normalized to the GAPDH (forward primer, 5′-GACAACTTTGGCATCGT-3′; reverse primer, 5′-ATGCAGGGATGATGTTCTGG-3′; amplicon size, 133 bp). Relative SIRT1 mRNA expression was calculated using the 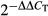 method.
method.
Deacetylation assays
SIRT1 activity was measured using the Fluor de Lys kit according to the manufacturer's instructions (Biomol International, Plymouth Meeting, PA, USA). The fluorescence was assayed by excitation at 360 nm and emission at 460 nm. The SIRT1 activity was expressed in relative fluorescence units.
Cell viability assay
Cells (5000/well) were plated in 96-well plates and treated with various concentrations of drugs. After incubating for the indicated concentrations and intervals, cell viability was determined by 0.5 mg/mL MTT assay. The resulting formazan crystals were solubilized with DMSO and the absorbance of each well was measured at 570 nm using a multiscanner autoreader (Dynatech MR5000, Edgewood, NY, USA). Each concentration and group was repeated three times in five replicates.
Short hairpin RNA transfection of cells
The SIRT1 (NM-012238) shRNA was designed by GeneChem RNA Technologies (GeneChem, Shanghai, China). Target sequences that effectively mediated silencing and negative control were as follows: 5′-CCTTCTGTTCGGTGATGAA-3′ (19 bp, SIRT1-RNAi) and 5′-CGTACGCGGAATACTTCA-3′. The shRNA was transiently transfected into pancreatic cancer cells using Lipofectamine 2000 diluted with OptiMEM (Invitrogen). The transfection procedure was carried out according to the manufacturer's recommendations. Transfected cells were harvested 48 h after transfection and analyzed by RT-PCR and Western blot.
Apoptosis and cell cycle analysis by flow cytometry
To determine cell apoptosis, DMSO (control group), EX527, and/or GEM treated cells were collected, washed twice in cold PBS, and resuspended in 100 μL annexin binding buffer. Then 5 μL annexin V–FITC conjugate and 2 μL propidium iodide (PI; 1 mg/mL) were added and this suspension was incubated for 15 min at room temperature. The samples were then further diluted with 400 μL annexin binding buffer. Cells were identified by a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). Cells for cycle analysis were washed in cold PBS, fixed in cold ethanol (70%), and stored at 4°C overnight. The fixed cells were then washed twice with cold PBS and stained with 50 μg/mL PI in the presence of 25 μg/mL RNase A. The samples were analyzed by FACS. All experiments were carried out in duplicate and repeated twice.
Senescence assay
Senescence induced by EX527 and/or GEM in tumor cells was evaluated by senescence-associated-β-gal (SA-β-gal) staining assay. The SA-β-gal staining was assayed at pH 6.0 as previously described.(24) Briefly, the freshly adhering cells were fixed in 3% paraformaldehyde for 3 min, washed three times with PBS, and incubated at 37°C with fresh SA-β-gal staining solution. According to the requirements of manufacturers, staining was evident at 37°C overnight. The percentage of positively blue stained cells was determined by scoring six random high power fields per 25-cm3 flask.
Patients and biopsies
Ten samples of unresectable pancreatic cancer were acquired by EUS-FNA from patients attending the Pancreatic Disease Institute, Union Hospital (Wuhan, China) between February 2010 and November 2010. The patients include six males and four females with a median age of 56 years (range, 35–76 years). None of the patients had received chemotherapy or radiotherapy before EUS-FNA. These patients received chemotherapy with GEM, including three cycles of GEM (1000 mg/m2) at days 1, 8, and 15 every month. Specimens were obtained with EUS-FNA once again at the 6-month follow-up visit. Because of disease progression, one male and one female died at 67 and 120 days, respectively. The samples were finally available from eight cases. Immediately after EUS-FNA and resection, some fractions of tissue samples were either snap-frozen in liquid nitrogen (for protein extraction) or fixed in 10% buffered formalin solution and then embedded in paraffin (for histological analysis). Informed consent was obtained from all patients before biopsy. The study protocol was approved by the ethics committee of Union Hospital, Huazhong University of Science and Technology.
Immunohistochemistry
Sections (4-μm thick) of paraffin-embedded pancreatic tissues were mounted onto treated slides for immunohistochemical staining. The streptavidin–peroxidase method was used for immunostaining with rabbit polyclonal antibody to SIRT1 (1:100; Santa Cruz Biotechnology). Biotinylated secondary antibody and 3, 3-diaminobenzidine staining procedures were carried out at room temperature. A negative control was obtained by replacing the primary antibody with a normal rabbit IgG. Two experienced pathologists independently analyzed SIRT1 staining while blinded to the clinicopathological data and clinical outcomes of the patients. The level of SIRT1 expression was calculated by combining estimates of quantity scores and staining intensity scores as follows: score 0, no staining; score 1, 1–10% of positive staining cells; 2, 10–50%; 3, 50–70%; 4, 70–100%. Staining intensity was evaluated as follows: 0, negative (no color); 1, weak (weak yellow); 2, moderate (yellow); 3, strong (brown). The immunohistochemical score was recorded by multiplying the quantity and staining intensity scores. The total score >3 in a section was considered as positive expression.(25)
Statistical analysis
Results are expressed as means ± standard deviation. All statistical analyses were made with a two-sided Student's t-test or one-way anova test, and P < 0.05 was considered significant.
Results
Increased SIRT1 in pancreatic cancer cell lines treated with GEM
PANC-1, BXPC-3, and ASPC-1 cell lines were treated with GEM (0, 5, 25 μg/mL). The Western blot results showed that SIRT1 expression in those three cell lines were all significantly increased, and the same results were also found by qRT-PCR in RNA levels (Fig. 1).
Fig. 1.
Induction of sirtuin 1 (SIRT1) in pancreatic cancer cell lines treated with gemcitabine (GEM). PANC-1, BXPC-3, and ASPC-1 cells were incubated with GEM (0, 5, 25 μg/mL) for 48 h. SIRT1 expression was monitored by Western blot analysis (a) and quantitative RT-PCR (b). The experiments were repeated twice and a representative result is shown. *P < 0.05, **P < 0.01 versus control group.
Chemosensitivity of pancreatic cancer cell lines to GEM enhanced by EX527 through specifically deregulating activity of SIRT1
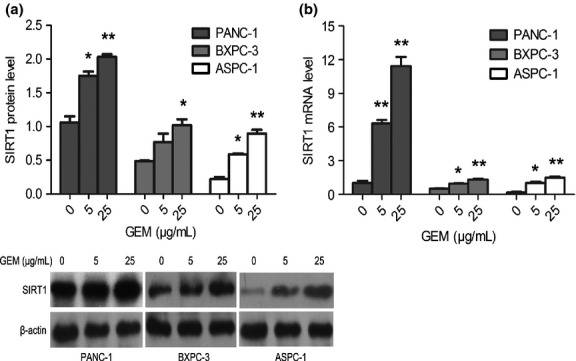
We explored the effect of EX527 on SIRT1 activity using the in vitro Fluor de Lys deacetylation assay. The SIRT1 activity of three pancreatic cancer cell lines was significantly deregulated by EX527 (2 μM), whereas no obvious deregulation of SIRT1 was shown in 293T cells (Fig. 2a). The proliferation of the cell lines was also evaluated by MTT test. Compared to 293T cells, the proliferation of PANC-1, BXPC-3, and ASPC-1 cells was significantly inhibited in a dose-dependent manner (IC50 = 8.78 ± 0.06, 7.97 ± 0.03, and 5.34 ± 0.04 μM, respectively; Fig. 2b).
Fig. 2.
EX527-mediated inhibition of sirtuin 1 (SIRT1) activity suppresses pancreatic cancer cell proliferation. (a) EX527 inhibits deacetylase activity of SIRT1 in pancreatic cancer cells. PANC-1, BXPC-3, ASPC-1, and 293T (control) cells were exposed to EX527 (2 μM) for 48 h. Deacetylase activity of SIRT1 was measured by the Fluor de Lys assay. (b) Growth inhibitory effect of EX527 on pancreatic cancer cell lines but not 293T cells. PANC-1, BXPC-3, ASPC-1, and 293T cells were exposed to different concentrations of EX527 for 48 h, and MTT assay was used to determine cell viability. *P < 0.05 versus control group.
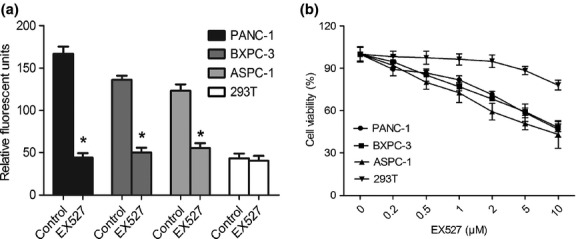
To explore whether EX527 had a synergic effect with GEM, pancreatic cancer cells were treated with EX527 (1 μM) at a concentration of IC20 and various concentrations of GEM. Compared with GEM alone, a significant decrease in cell viability was observed in the cells treated with EX527 plus GEM, as shown in the MTT assay (Fig. 3). After treatment with EX527, the GEM concentration causing 50% growth inhibition (IC50) was significantly decreased in PANC-1 (56.70 ± 2.73 vs 19.87 ± 5.38 μg/mL, P < 0.01), BXPC-3 (17.86 ± 2.51 vs 8.99 ± 1.54 μg/mL, P < 0.01), and ASPC-1 cells (21.67 ± 4.48 vs 8.07 ± 2.11 μg/mL, P < 0.01). We also observed that EX527 enhanced the chemosensitivity of pancreatic cancer cell lines to cisplatin (Fig. S1).
Fig. 3.
EX527 enhanced chemosensitivity of pancreatic cancer cell lines to gemcitabine (GEM). PANC-1, ASPC-1, and BXPC-3 cells were treated with increasing concentrations of GEM in the presence or absence of 1 μM EX527, and cell viability was measured at 48 h. All data are presented as means ± SD of three independent experiments. *P < 0.05 versus control group.
To further verify that the effect of EX527 on the chemosensitivity of pancreatic cancer cell lines is mainly due to inhibition of the SIRT1 pathway, EX527 and chemotherapy drugs were used to treated cell lines in which SIRT1 expression was deregulated by SIRT1 siRNA. Compared to control cells, the IC50 value of GEM was remarkably decreased in SIRT1-RNAi-PANC-1 cells (52.66 ± 2.65 vs 8.99 ± 3.02 μg/mL, P < 0.01) and SIRT1-RNAi-ASPC-1 cells (20.20 ± 1.98 vs 4.55 ± 2.29 μg/mL, P < 0.01). There was no further inhibition apparent in EX527-treated SIRT1-RNAi-PANC-1 and SIRT1-RNAi-ASPC-1 cells (IC50, 7.16 ± 2.92 and 3.57 ± 1.42 μg/mL, respectively, Fig. 4a). Furthermore, the Western blot results showed that EX527 had not further deregulated the SIRT1 expression in SIRT-RNAi transfected cells (Fig. 4b). These results revealed that the enhanced chemosensitivity of EX527 was critically through inhibiting SIRT1 activity.
Fig. 4.
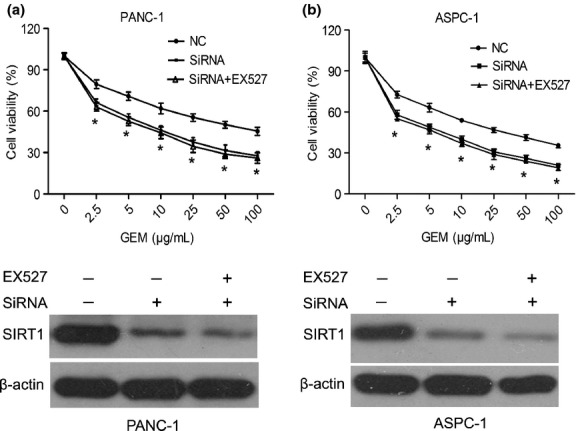
Chemosensitivity was induced in pancreatic cancer cells by EX527, through specifically deregulating the activity of sirtuin 1 (SIRT1). Downregulation of endogenous SIRT1 using RNA interference following EX527 (2 μM) treatment in PANC-1 (a) and ASPC-1 cells (b). The SIRT1 expression of siRNA plus EX527 treatment was also measured by Western blot analysis. All data are presented as means ± SD of three independent experiments. *P < 0.05 versus control group. NC, negative control.
Effects of EX527 and GEM on apoptosis and cell cycle of pancreatic cancer cells
To assess the mechanism by which EX527 sensitized PANC-1 and ASPC-1 cells to GEM, apoptosis and the cell cycle were analyzed by FACS. As shown in Figure 5(a), compared with the PANC-1 control group (3.46 ± 0.71%), apoptosis was increased in PANC-1 cells treated with GEM (50 μg/mL, 4.81 ± 0.68%) or EX527 (2 μM, 6.06 ± 0.63%); the combination of GEM and EX527 treatment caused a significant increase in apoptosis (11.07 ± 0.90%, P < 0.01). Similar results were observed in ASPC-1 cells (Fig. 5b), which suggested that EX527 had synergic effects on induction of apoptosis with GEM in pancreatic cancer cells.
Fig. 5.

Effect of EX527 and (or) gemcitabine (GEM) on apoptosis of pancreatic cancer cells. PANC-1 (a) and ASPC-1 (b) cells were incubated with EX527 (2 μM) and (or) GEM (50 μg/mL) for 48 h. Cell apoptosis was monitored by annexin V staining and flow cytometry. The right lower quadrant of each plot represents early apoptotic cells and the right upper quadrant represents late apoptotic cells. The experiments were repeated twice with similar results. *P < 0.05, **P < 0.01 versus control group; #P < 0.01 versus single drug treatment.
When treated with GEM or EX527, the distribution of PANC-1 cells in G0/G1 phase notably increased (57.85 ± 2.29% vs 66.89 ± 1.99%, 76.79 ± 2.01%, P < 0.01; Fig. 6a). Moreover, compared to EX527 or GEM alone, treatment with a combination of both resulted in further significant increase of cells in G0/G1 phase (84.39 ± 1.30%, P < 0.01). These results were also confirmed in ASPC-1 cells (Fig. 6b).
Fig. 6.

Effect of EX527 and (or) gemcitabine (GEM) on cell cycle distribution in pancreatic cancer cells. Cycle phase distributions of PANC-1 (a) and ASPC-1 (b) cells were analyzed by flow cytometry after staining with propidium iodide. Percentage of cells in each phase of the cell cycle (G0/G1, G2/M and S-phase) is indicated. The experiments were repeated twice with similar results.
Effect of SIRT1 inhibitor and GEM on senescence of pancreatic cancer cells
According to the results of the SA-β-gal assay, compared to untreated PANC-1 and ASPC-1 cells, SA-β-gal-positive cells were mildly increased in GEM-treated PANC-1 and ASPC-1 cells, and moderately increased in EX527-treated cells. Moreover, compared to GEM or EX527 alone, the combination of GEM and EX527 significantly increased the number of SA-β-gal-positive cells in PANC-1 and ASPC-1 cells even further (Fig. 7).
Fig. 7.
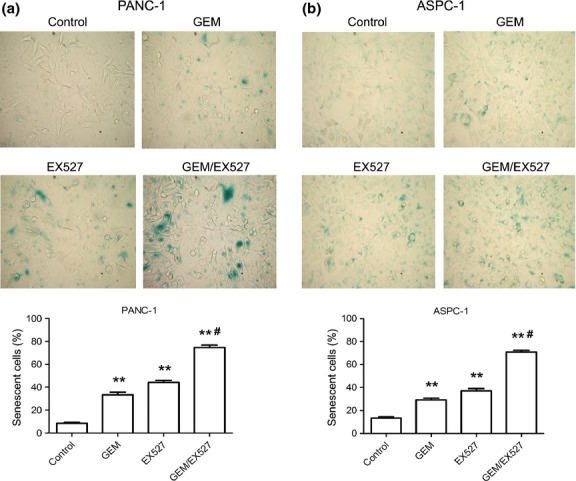
Induction of cellular senescence in pancreatic cancer cells treated with EX527 and (or) gemcitabine (GEM). PANC-1 (a) and ASPC-1 (b) cells were treated with EX527 (2 μM) and (or) GEM (50 μg/mL) for 48 h, and senescence-associated-β-gal activity (blue) was measured (×400). The graphs show the relationship between cellular senescence and each drug. Error bars represent mean ± SD. **P < 0.01 versus control group; #P < 0.01 versus single drug treatment.
Expression of SIRT1, p53, AC-p53, FOXO3a, and p21 protein in PANC-1 cells treated with GEM and EX527
To further explore the mechanism of apoptosis and senescence, the proteins involved in SIRT1 signaling were analyzed. There was no significant change in SIRT1 expression with EX527 treatment, and EX527 did not decrease SIRT1 expression in GEM + EX527 treatment. Total p53 protein had no notable change with regard to EX527 or GEM treatment. AC-p53 protein of PANC-1 cells was significantly upregulated with EX527 treatment, but not in GEM-treated PANC-1 cells. Expression levels p21 were significantly enhanced when the cells were treated with EX527 or GEM. Moreover, FOXO3a expression was upregulated in EX527-treated cells, but no significant change of FOXO3a expression was shown after treatment with GEM. Compared to EX527 alone, although AC-p53 had no further increase, both FOXO3a and p21 expression were significantly increased (Fig. 8a). These results were also confirmed in ASPC-1 cells (Fig. 8b).
Fig. 8.
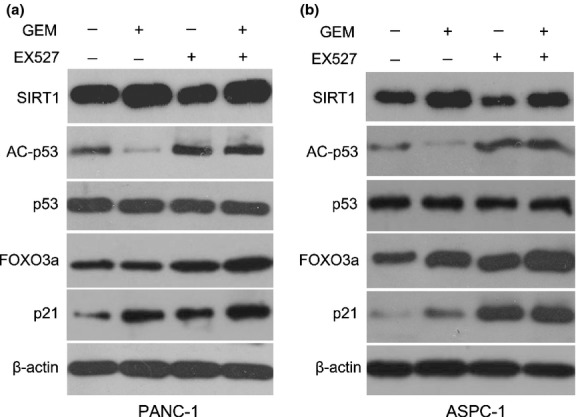
Effect of EX527 and (or) gemcitabine (GEM) on protein levels of sirtuin 1 (SIRT1), acetylated-p53 (AC-p53), p53, FOXO3a, and p21. PANC-1 (a) and ASPC-1 (b) pancreatic cancer cells were treated with EX527 (2 μM) and (or) GEM (50 μg/mL) for 48 h. The expression levels of apoptosis and senescence-associated proteins were analyzed by Western blot. A representative result is shown.
Expression of SIRT1 unregulated in pancreatic cancer tissues after chemotherapy with GEM
The SIRT1 expression in clinical pancreatic cancer samples were detected by immunohistochemistry (IHC). Compared to samples before chemotherapy, the results showed that the number of SIRT1-positive cells was significantly higher in pancreatic cancer samples after chemotherapy than those before chemotherapy (83.17 ± 5.42% vs 66.83 ± 6.01%, P < 0.05). Simultaneously, the IHC results also showed that SIRT1 did not only locate primarily to the nucleus but also had some minor cytoplasmic localization in pancreatic cancer cells (Fig. 9a). Furthermore, the SIRT1 expression was accurately evaluated by qRT-PCR and Western blot analysis. Compared to pancreatic cancer tissues before chemotherapy, the SIRT1 expression was clearly upregulated in pancreatic cancer after chemotherapy in mRNA (12.14 ± 6.68 vs 2.96 ± 2.06, P < 0.05) and protein levels (0.88 ± 0.41 vs 0.22 ± 0.10, P < 0.05) (Fig. 9b,c). The results roughly coincide with the IHC analysis.
Fig. 9.
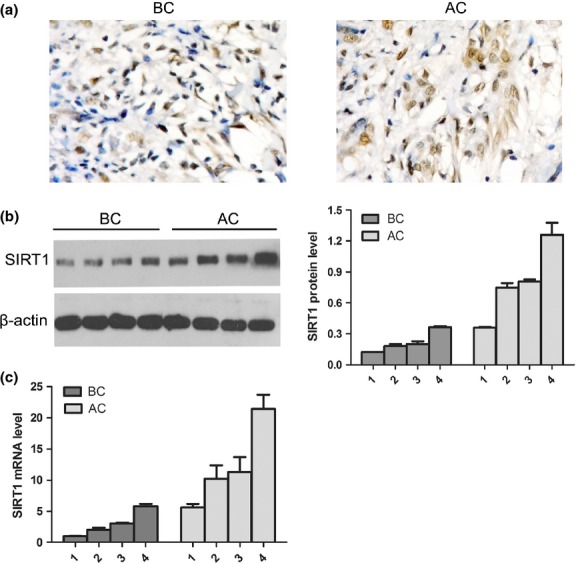
Sirtuin 1 (SIRT1) expression was evaluated in pancreatic cancer samples before and after chemotherapy. (a) SIRT1 expression in pancreatic cancer samples before chemotherapy (BC) and after chemotherapy (AC) (n = 8) was analyzed by immunohistochemical staining with an anti-SIRT1 antibody in paraffin-embedded tissue sections. (b) Representative expression of SIRT1 was analyzed in pancreatic cancer samples before (n = 4) and after chemotherapy (n = 4) by Western blot analysis. The β-actin level was assessed as a loading control. (c) Expression of SIRT1 was also detected in RNA levels by quantitative RT-PCR. Representative results are shown.
Discussion
The major findings of our study are that SIRT1 may regulate the adaptive reactions of cancer cells exposed to chemotherapy, and be involved in the chemoresistance of pancreatic cancer. Several aspects of our study are intriguing. Previous studies have reported that levels of SIRT1 were elevated in prostate cancer, acute myeloid leukemia, and primary colon cancer,(26–28) but downregulated in ovarian cancers and bladder cancer.(29) It remains controversial whether SIRT1 acts as a tumor promoter or suppressor. For example, SIRT1-mediated deacetylation suppresses the functions of several tumor suppressors including HIC1, p53, and p73, suggesting that SIRT1 has a promoting activity in tumor development and progression.(30–32) In contrast, SIRT1 may have a suppressive function on tumor cells by suppressing NF-κB,(33–35) the dysregulation of which leads to the onset of tumorigenesis.(36) These contradictory results suggest that the dual function of SIRT1 in different tissues may depend on the type of tumor and the mechanism of tumorigenesis, as well as the temporal distribution and abundance of different SIRT1 downstream targets and factors that regulate SIRT1. Therefore, future research is obliged to reveal a conclusive answer. Nevertheless, although there are contradictory results about inhibiting or promoting the proliferation of cancer cells, most studies including our previous data showed that SIRT1 inhibition did have a synergic effect with chemotherapy.(21,37,38) However, the exact mechanism of SIRT1 affecting chemotherapy remains unstated. Our data showed that SIRT1 was significantly elevated in pancreatic cancer cells treated by GEM. It implied that SIRT1 was involved in the adaptive response to DNA damage stress in pancreatic cancer chemotherapy. In addition, our studies suggest that specific SIRT1 inhibitor EX527 can modulate the chemoresistance of pancreatic cancer cells to GEM. Our data reinforce the work of others who have shown that inhibition of SIRT1 can alter the chemoresistance of other carcinomas.(37,38)
Our research showed that EX527 significantly decreased the SIRT1 catalytic activity in PANC-1, BXPC-3, and ASPC-1 cell lines, but had little effect on 293T cells. After treatment with EX527, the proliferation of pancreatic cancer cells was also significantly retarded in a dose-dependent manner. The 293T cells with lower SIRT1 activity showed inhibitory effects only with a very high concentration (10 μM) of EX527. It indicated that overexpressed SIRT1 activity could favor pancreatic cancer cell growth, and the inhibitory effect of EX527 on the proliferation of pancreatic cancer cells was SIRT1-dependent.
The chemotherapy results showed that PANC-1 cells had the highest IC50 of GEM and ASPC-1 cells had the lowest IC50. Interestingly, the SIRT1 expression was strongest in PANC-1 cells but weakest in ASPC-1 cells. The positive relation between SIRT1 and IC50 implied that SIRT1 might act as a promoter of chemoresistance in pancreatic cancer. Furthermore, after treatment with EX527, the IC50 values of the three pancreatic cancer cell lines were all significantly decreased. After transfection with SIRT-RNAi, the IC50 also notably decreased in SIRT1-RNAi-PANC-1 and SIRT1-RNAi-ASPC-1 cells. These results verified that SIRT1 inhibition had a synergic effect with GEM. Nevertheless, no further inhibitory effects were shown in SIRT-RNAi-PANC-1 or SIRT1-RNAi-ASPC-1 cells after treatment with EX527, which confirmed that the effect of EX527 was caused by inhibition of SIRT1 activity but no other proteins. To clarify that SIRT1 downregulation is not selectively affected by GEM treatment, we also observed that EX527 enhanced the chemosensitivity of pancreatic cancer cell lines to the additional therapeutic agent cisplatin.
The differences in SIRT1 expression and sensitivity to GEM between the three cell lines may be due to different cellular contexts or its targets in specific signaling pathways, depending on different ATP synthesis, nucleotide metabolism, and RNA production.
Cellular senescence, a process of cell aging in which cells irreversibly exit the cell cycle and cease to divide, is accompanied by changes in SIRT1 activity. It is established that SIRT1 overexpression antagonizes stress-induced senescence as a result of direct p53 deacetylation.(39) Ota et al.(40) found that SIRT1 inhibition by other inhibitors, sirtinol and splitomicin, and siRNA caused senescence in human cancer MCF-7 and H1299 cells. Our previous results also showed that SIRT1-RNAi promoted senescence and apoptosis in pancreatic cancer cells.(21) Thus, senescence and apoptosis were evaluated for identifying the effect of SIRT1 on pancreatic cancer cells in chemotherapy. Our results showed that either EX527 or GEM clearly induced a senescence-like phenotype in PANC-1 and ASPC-1 cells, which implied that senescence was also a very important effect of chemotherapy on cancer cells. In addition, senescence was further increased with the combination of EX527 and GEM, which further confirmed that the reason for chemoresistance could be partly attributable to the involvement of SIRT1 in antisenescence. The FACS results showed that EX527 or GEM treatment separately produced a block in the G0/G1 phase of the cell cycle versus control cells, but it was significantly enhanced after the combination of EX527 and GEM. Peck et al.(41) also reported that EX527 induced significant cell cycle arrest in breast cancer cells. The results also indicated that either EX527 or GEM caused a mild increase in apoptosis, and it was significantly enhanced after treatment with both EX527 and GEM. These results suggested that inhibition of SIRT1 combined with GEM had synergic effects on senescence, cell cycle arrest, and apoptosis of pancreatic cancer cells.
It was also shown that EX527 only decreased SIRT1 activity but not its expression, and total p53 expression had no obviously change in PANC-1 cells with GEM, EX527 alone, or combination treatment. Levels of acetylated-p53, the deacetylated target of SIRT1, were significantly decreased in GEM-treated PANC-1 cells, coinciding with increased SIRT1 expression. These results indicated that the GEM-induced SIRT1 increase could downregulate acetylated-p53 and resistance to apoptosis. Nevertheless, only the cell cycle inhibitor p21,(42) but not the apoptosis inducer FOXO3a,(43) significantly increased after GEM treatment. Lin et al.(44) also showed that knockdown of p21 in mouse embryonic fibroblasts partially reversed cellular senescence and cell arrest. These data indicated that apoptosis and senescence of pancreatic cancer cells induced by GEM, at least in part, were mediated through inhibition of p21. In contrast, acetylated-53, p21, and FOXO3a were all significantly increased in EX527-treated PANC-1 cells. Moreover, compared to GEM, the combination treatment significantly increased the expression of acetylated-p53, p21, and FOXO3a, which inhibited GEM-induced adaptive responses and then enhanced the chemosensitivity of pancreatic cancer cells. Our findings seem to be at odds with the results reported by Solomon et al.,(45) who failed to see any effect of in vitro acetylation of p53 on altering cell survival following DNA damage. These differences might be due to the heterogeneity of cancer cells or the status of p53.
To further verify our hypothesis regarding SIRT1 involvement in chemoresistance, we collected pancreatic cancer tissues through EUS from patients who received GEM treatment. The SIRT1 expression was evaluated pre- and post-chemotherapy. The IHC results showed that SIRT1 expression was significantly increased after chemotherapy, also confirmed by qPCR and Western blot. These results strongly indicated that SIRT1 is involved in the chemoresistance of pancreatic cancer. Interestingly, EX527, for the treatment of Huntington's disease to inhibit neuronal death, is currently in a Phase 1a combined single and multiple ascending dose study in the European Union to assess safety, tolerability, and pharmacokinetics in healthy volunteers.(46) Therefore, EX527 may also be applied as a novel treatment for pancreatic cancer after wide preclinical in vivo experiments demonstrating highly favorable safety profiles and pharmaceutical properties. Further studies should address the specificity of EX527 for pancreatic cancer therapy and chemotherapy in vivo.
Novel targets and more effective, less toxic therapeutic strategies should be explored to improve the poor prognosis of pancreatic cancer. Although the role of SIRT1 in cancer is controversial, our results preliminarily demonstrated that SIRT1 might regulate adaptive responses to chemotherapy-induced stress and then facilitate chemoresistance. It indicated that the combination of SIRT1 inhibitor and GEM or other chemotherapeutic drugs would be a novel treatment to reverse chemoresistance in pancreatic cancer.
Acknowledgments
This study was supported by grants from the National Science Foundation Committee (NSFC) of China (Grant nos. 30972900 and 81372666), and the Research Special Fund for Public Welfare Industry of Health of China (Grant no. 201202007).
Disclosure Statement
The authors have no conflicts of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1 EX527 enhanced chemosensitivity of pancreatic cancer cell lines to cisplatin.
References
- 1.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–30. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 4.Nakano Y, Tanno S, Koizumi K, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96:457–63. doi: 10.1038/sj.bjc.6603559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kohno K, Uchiumi T, Niina I, et al. Transcription factors and drug resistance. Eur J Cancer. 2005;41:2577–86. doi: 10.1016/j.ejca.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Liontos M, Velimezi G, Pateras IS, et al. The roles of p27(Kip1) and DNA damage signaling in the chemotherapy-induced delayed cell cycle checkpoint. J Cell Mol Med. 2010;14:2264–7. doi: 10.1111/j.1582-4934.2010.01145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiligada E. Chemotherapy: induction of stress responses. Endocr Relat Cancer. 2006;13(Suppl 1):S115–24. doi: 10.1677/erc.1.01272. [DOI] [PubMed] [Google Scholar]
- 8.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death–a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hori YS, Kuno A, Hosoda R, Horio Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS ONE. 2013;8:e73875. doi: 10.1371/journal.pone.0073875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang C, Wang MW, Tashiro S, Onodera S, Ikejima T. Roles of SIRT1 and phosphoinositide 3-OH kinase/protein kinase C pathways in evodiamine-induced human melanoma A375-S2 cell death. J Pharmacol Sci. 2005;97:494–500. doi: 10.1254/jphs.fpj04055x. [DOI] [PubMed] [Google Scholar]
- 11.Back JH, Rezvani HR, Zhu Y, et al. Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1. J Biol Chem. 2011;286:19100–8. doi: 10.1074/jbc.M111.240598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahyo T, Mostoslavsky R, Goto M, Setou M. Sirtuin-mediated deacetylation pathway stabilizes Werner syndrome protein. FEBS Lett. 2008;582:2479–83. doi: 10.1016/j.febslet.2008.06.031. [DOI] [PubMed] [Google Scholar]
- 13.Langley E, Pearson M, Faretta M, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–96. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–41. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 16.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939–48. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 17.Barlev NA, Liu L, Chehab NH, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–54. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- 18.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–26. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng HL, Mostoslavsky R, Saito S, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–9. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457–63. doi: 10.1158/0008-5472.CAN-05-1923. [DOI] [PubMed] [Google Scholar]
- 21.Zhao G, Cui J, Zhang JG, et al. SIRT1 RNAi knockdown induces apoptosis and senescence, inhibits invasion and enhances chemosensitivity in pancreatic cancer cells. Gene Ther. 2011;18:920–8. doi: 10.1038/gt.2011.81. [DOI] [PubMed] [Google Scholar]
- 22.Milne JC, Lambert PD, Schenk S, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–6. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao G, Zhang J-G, Liu Y, et al. MiR-148b functions as a tumor suppressor in pancreatic cancer by targeting AMPKα1. Mol Cancer Ther. 2013;12:83–93. doi: 10.1158/1535-7163.MCT-12-0534-T. [DOI] [PubMed] [Google Scholar]
- 24.Zhao G, Qin Q, Zhang J, et al. Hypermethylation of HIC1 promoter and aberrant expression of HIC1/SIRT1 might contribute to the carcinogenesis of pancreatic cancer. Ann Surg Oncol. 2013;20:301–11. doi: 10.1245/s10434-012-2364-9. [DOI] [PubMed] [Google Scholar]
- 25.Friedrichs K, Gluba S, Eidtmann H, Jonat W. Overexpression of p53 and prognosis in breast cancer. Cancer. 1993;72:3641–7. doi: 10.1002/1097-0142(19931215)72:12<3641::aid-cncr2820721215>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 26.Huffman DM, Grizzle WE, Bamman MM, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–8. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 27.Bradbury CA, Khanim FL, Hayden R, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–9. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 28.Stunkel W, Peh BK, Tan YC, et al. Function of the SIRT1 protein deacetylase in cancer. Biotechnol J. 2007;2:1360–8. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- 29.Wang RH, Sengupta K, Li C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–23. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen WY, Wang DH, Yen RC, et al. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–48. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Vaziri H, Dessain SK, Ng Eaton E, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–59. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 32.Dai JM, Wang ZY, Sun DC, Lin RX, Wang SQ. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J Cell Physiol. 2007;210:161–6. doi: 10.1002/jcp.20831. [DOI] [PubMed] [Google Scholar]
- 33.Kong S, Kim SJ, Sandal B, et al. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J Biol Chem. 2011;286:16967–75. doi: 10.1074/jbc.M111.218206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schug TT, Xu Q, Gao H, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. 2010;30:4712–21. doi: 10.1128/MCB.00657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–80. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kiernan R, Bres V, Ng RW, et al. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem. 2003;278:2758–66. doi: 10.1074/jbc.M209572200. [DOI] [PubMed] [Google Scholar]
- 37.Kabra N, Li Z, Chen L, et al. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J Biol Chem. 2009;284:18210–7. doi: 10.1074/jbc.M109.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh WK, Cho KB, Hien TT, et al. Amurensin G, a potent natural SIRT1 inhibitor, rescues doxorubicin responsiveness via down-regulation of multidrug resistance 1. Mol Pharmacol. 2010;78:855–64. doi: 10.1124/mol.110.065961. [DOI] [PubMed] [Google Scholar]
- 39.Pearson M, Carbone R, Sebastiani C, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–10. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 40.Ota H, Tokunaga E, Chang K, et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006;25:176–85. doi: 10.1038/sj.onc.1209049. [DOI] [PubMed] [Google Scholar]
- 41.Peck B, Chen CY, Ho KK, et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 2010;9:844–55. doi: 10.1158/1535-7163.MCT-09-0971. [DOI] [PubMed] [Google Scholar]
- 42.Xia M, Knezevic D, Vassilev LT. p21 does not protect cancer cells from apoptosis induced by nongenotoxic p53 activation. Oncogene. 2011;30:346–55. doi: 10.1038/onc.2010.413. [DOI] [PubMed] [Google Scholar]
- 43.Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–9. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin HK, Chen Z, Wang G, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–9. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solomon JM, Pasupuleti R, Xu L, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26:28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carafa V, Nebbioso A, Altucci L. Sirtuins and disease: the road ahead. Front Pharmacol. 2012;3:4. doi: 10.3389/fphar.2012.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 EX527 enhanced chemosensitivity of pancreatic cancer cell lines to cisplatin.