

Abstract
The aim of the present study was to establish cancer stem-like cell/cancer-initiating cell (CSC/CIC)-targeting immunotherapy. The CSC/CIC are thought to be essential for tumor maintenance, recurrence and distant metastasis. Therefore they are reasonable targets for cancer therapy. In the present study, we found that a heat shock protein (HSP) 40 family member, DnaJ (Hsp40) homolog, subfamily B, member 8 (DNAJB8), is preferentially expressed in CSC/CIC derived from colorectal cancer (CRC) cells rather than in non-CSC/CIC. Overexpression of DNAJB8 enhanced the expression of stem cell markers and tumorigenicity, indicating that DNAJB8 has a role in CRC CSC/CIC. A DNAJB8-specific cytotoxic T lymphocyte (CTL) response could be induced by a DNAJB8-derived antigenic peptide. A CTL clone specific for DNAJB8 peptide showed higher killing activity to CRC CSC/CIC compared with non-CSC/CIC, and CTL adoptive transfer into CRC CSC/CIC showed an antitumor effect in vivo. Taken together, the results indicate that DNAJB8 is expressed and has role in CRC CSC/CIC and that DNAJB8 is a novel target of CRC CSC/CIC-targeting immunotherapy.
Keywords: Cancer immunotherapy, cancer stem-like cell, colorectal cancer, DNAJB8, tumor antigen
Colorectal cancer (CRC) is one of the major malignant diseases worldwide and has the second-highest mortality rate in the United States.(1) Although recent approaches have achieved improvements in CRC treatment including novel combination chemotherapies and molecular-targeting therapies, the prognosis of patients with metastasis and recurrence is still poor. Thus, development of therapy for advanced CRC is an urgent task. Cancer stem-like cells/cancer-initiating cells (CSC/CIC) are defined by their ability of tumor initiation, self-renewal and differentiation.(2) It has been reported that CSC/CIC are resistant to chemotherapy and radiotherapy.(3) The CSC/CIC with these features are considered to be related to metastasis and recurrence and to be important therapeutic targets.
Immunotherapy for cancer has attracted much attention as a new strategy compared with traditional therapies such as chemotherapy and radiotherapy. Identification of the first human tumor-associated antigens (TAA) in the early 1990s enabled cancer immunotherapies using antigenic peptides derived from TAA.(4) Some peptide vaccination trials showed fascinating results; however, there are also pessimistic opinions.(5,6) Our previous trials using a Survivn 2B-derived peptide vaccine showed partial clinical benefits for some cancer patients, but the results were not sufficiently satisfactory.(7–12) There are many reasons why peptide vaccine therapies are not so effective in clinical settings, one of the main reasons being antigen loss on cancer cells. Therefore, the ideal targets for cancer immunotherapy are thought to be TAA that have critical functions for CSC/CIC maintenance, since CSC/CIC have a high tumor-initiating ability.(13)
DnaJ (Hsp40) homolog, subfamily B, member 8 (DNAJB8) belongs to the heat shock protein (HSP) 40 family. The HSP40 family proteins are co-chaperones of HSP70 and DNAJB8 has a role in suppression of misfolded toxic protein aggregation.(14) Recently, it has been reported that some members of the HSP40 family are related to the development and metastasis of cancers and that their expression was detected in breast cancer stem cells.(15) We found that DNAJB8 was expressed preferentially in CSC/CIC derived from renal cell carcinoma. DNAJB8 was expressed only in the testis among normal tissues and thus it is a novel cancer/testis antigen. Moreover, we confirmed its immunogenicity by using a mice DNA vaccination model, indicating that DNAJB8 is a promising target of CSC/CIC-targeting immunotherapy.(16) However, it is not clear whether DNAJB8 is expressed in human CSC/CIC of other cancers, including CRC. In the present study, we analyzed the expression and functions of DNAJB8 in CRC and the potency of DNAJB8 as a target for CRC CSC/CIC-targeting immunotherapy.
Materials and Methods
Cell lines
Colon adenocarcinoma cell lines SW480 (HLA-A*0201/2402), HT29 (HLA-A*0101/2403) and HCT15 (HLA-A*0201/2402) were kind gifts from Dr K. Imai (Sapporo, Japan). All cells were cultured in DMEM (Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Carlsbad, CA, USA). The erythroleukemia cell line K562 was purchased from ATCC (Manassas, VA, USA) and was cultured in RPMI-1640 (Sigma-Aldrich) supplemented with 10% FBS. The HLA-A*2402 stably transfected transporter associated with antigen processing (TAP)-deficient cell line T2A-A*2402 (T2-A24) was a kind gift from Dr K. Kuzushima (Nagoya, Japan) and was cultured in RPMI-1640 supplemented with 10% FBS and 0.8 mg/mL G418 (Life Technologies).
Retroviral gene transduction and generation of stable transformants
Transduction of genes into cells was carried out using a retrovirus-mediated method as described previously.(16) PLAT-A cells (a kind gift from Dr T. Kitamura, Tokyo, Japan), which are amphotropic packaging cells, were transiently transduced with a pMXs-puro retroviral vector expressing DNAJB8 and a control plasmid using Lipofectamine 2000 reagent (Life Technologies) following the manufacturer's protocol. Retroviral supernatants were harvested 48 h after transfection. The supernatant was used for infection of HT29 cells in the presence of 8 mg/mL of polybrene (Sigma-Aldrich) overnight. For the generation of stable transformants, the infected cells were selected with 1 μg/mL puromycin. DNAJB8 expression was confirmed using western blot analysis.
Side population (SP) analysis
The SP analysis was performed as described previously.(16–18) The cells were labeled with Hoechst 33342 dye (Lonza, Walkersville, MD, USA) for 90 min at concentrations of 10 μg/mL for HCT15, 7.5 μg/mL for HT29 and 5 μg/mL for SW480 with or without Verapamil (Sigma-Aldrich), which is an inhibitor of ATP-binding cassette (ABC) transporters, at concentrations of 100 μM for HT29 and 50 μM for SW480 and HCT15. Cells were counterstained with 1 μg/mL propidium iodide (Sigma-Aldrich) for labeling dead cells. Viable cells were sorted using a BD FACS Aria II Cell-Sorting System (BD, Franklin Lakes, NJ, USA).
Xenograft model
All mouse procedures were carried out in accordance with institutional protocol guidelines at Sapporo Medical University School of Medicine. The SP cells and presorted cells from colon cancer cell lines were mixed with Matrigel (BD) at a 1:1 volume and injected subcutaneously into the back of 4–8-week-old female non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice. Tumor size was assessed weekly using a caliper and calculated using the following formula: tumor size (mm3) = (longest diameter × shortest diameter2)/2.
RT-PCR analysis and quantitative RT-PCR analysis
RT-PCR analysis was performed as described previously.(16) Total RNA (tRNA) were isolated from SP, main population (MP) and unsorted cells using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocol. Complementary DNA (cDNA) was synthesized from 2 μg of total RNA by reverse transcription using Superscript III reverse transferase (Invitrogen, Palo Alto, CA, USA). A cDNA panel for a set of normal human adult tissues and fetal tissues was purchased from Clontech (Mountain View, CA, USA). RT-PCR was performed in 20 μL of PCR mixture containing 1 μL of cDNA mixture, 0.5 μL of Taq DNA polymerase (Qiagen, Valencia, CA, USA) and 4 pmol of primers. The PCR mixture was initially incubated at 94°C for 2 min, followed by 35 cycles of denaturation at 94°C for 15 s, annealing at 58°C for 30 s and extension at 72°C for 30 s. The primer pairs used for RT-PCR analysis were 5′-CATGATGGAGACGGAGCTGA-3′ and 5′-ACCCCGCTCGCCATGCTATT-3′ for SOX2 with an expected PCR product size of 410 base pairs (bp), 5′-AGCTCTGTGGACTGCTGGTT-3′ and 5′-GGACGCCAGTTGCAAAGTAT-3′ for DNAJB8 with an expected PCR product size of 409 bp, 5′-CCCGACAAGAACCCTGACAAT-3′ and 5′-AGGTGGATGAGAAGGTGGTG-3′ for POU5F1 with an expected PCR product size of 163 bp, 5′-CTCTTCCTCAAACCGTCTGC-3′ and 5′-GATCGGAGGCTAAGCAACTG-3′ for LGR5 with an expected PCR product size of 181 bp and 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) with an expected product size of 452 bp. Quantitative real-time PCR was performed using an ABI PRISM 7000 sequence detection system (Life Technologies) according to the manufacturer's protocol. Primers and probes were designed by the manufacturer (TaqMan gene expression assays; Life Technologies). Thermal cycling was performed using 40 cycles of denaturation at 95°C for 15 s followed by annealing at 60°C for 1 min. Each experiment was performed in triplicate, with normalization to the GAPDH gene as an internal control.
Sphere formation assay
To assay sphere formation efficiency, 103 cells were plated in Ultra Low Attachment six-well plates (Corning Incorporated Life Sciences, Acton, MA, USA) and cultured in Dulbecco's modified Eagle's medium/F12 (Life Technologies) supplemented with 20 ng/mL epidermal growth factor and 20 ng/mL basic fibroblast growth factor (R&D Systems, Minneapolis, MN, USA). The cells were incubated in a 5% CO2 incubator for 2 weeks and the number of spheres was counted under a microscope in 15 low-power fields and then the average was calculated.
Synthetic peptides and peptide binding assay
Putative antigenic peptides can be designed using several website programs, such as BIMAS (http://www-bimas.cit.nih.gov/molbio/hla_bind/). Four peptides, DNAJB8_22(8) (AYRKLALRW), DNAJB8_90(10) (GYTFRNPEDI), DNAJB8_99(9) (IFREFFGGL) and DNAJB8_143(9) (AFMEAFSSF), were designed from the amino acid sequence of DNAJB8 according to the HLA-A24-binding motifs.
Peptide binding affinity to HLA-A24 was assessed using HLA-A24 a stabilization assay as described previously.(19) Survivin-2B_80(9) (AYACNTSTL) peptide was used as a positive control and SL8C (SIINFEKL), which is a H2-Kb-binding peptide derived from Ovalbumin protein, was used as a negative control.
Cytotoxic T lymphocyte (CTL) induction and establishment of CTL clone
The PBMC were isolated from two healthy volunteer donors, from whom we obtained informed consent according to the guidelines of the Declaration of Helsinki, using standard density gradient centrifugation on Lymphoprep (Nycomed, Oslo, Norway). Isolation of CD8+ cells and establishment of phytohaemagglutinin (PHA) blast were performed as described previously.(20,21)
The CTL induction was performed as described previously.(20,21) CD8+ T cells were stimulated with a peptide-pulsed PHA blast for 2–3 times weekly. On day 21, CD8+ T cell reactivity was assessed using interferon (IFN)-γ enzyme-linked immunospot (ELISpot) assay. On day 28, the cytotoxic activity of T cells was assessed using a conventional 6-h 51Cr release assay. To obtain CTL clones, standard limiting dilution was performed as described previously.(21)
Interferon-γ ELISpot assay and cytotoxicity assay
An IFN-γ ELISpot assay was performed as described previously.(21) CD8+ T cells, 2 × 105, were incubated with 5 × 104/well T2-A24 cells pulsed with each peptide (50 μg/mL) or K562 cells. After incubation for 40 h at 37°C, IFN-γ spots were developed and counted as per the manufacturer's instructions.
The lytic activity of CTL was tested using a 51Cr release assay as described previously.(20,21) 51Cr-labeled target cells (2000 cells/well) were incubated with various numbers of effector cells for 6 h at 37°C in 96-well microtiter plates and the cytotoxicity was calculated with radioactivity of the culture supernatants.
Cytotoxic T lymphocyte adoptive transfer
Fifteen NOD/SCID mice were inoculated subcutaneously in the back with 1 × 105 HT29 cells. Three weeks later, when the tumors were palpable, 1 × 104 or 1 × 103 DNAJB8_143(9)-specific CTL clone cells or PBS was injected intravenously into five mice for each group. The same adoptive transfer procedure was performed 2 weeks after inoculation with SP cells. Tumor size was assessed weekly.
Statistical analysis
In the xenograft model, cytotoxicity assay and CTL adoptive transfer model, samples were analyzed using Student's t-test, with P < 0.05 conferring statistical significance.
Results
Expression of DNAJB8 in CRC CSC/CIC
Human CRC CSC/CIC have previously been isolated from CRC cell lines and primary CRC samples using several methods, including the use of cell surface markers (e.g. CD133), SP cells using Hoechst 33342 analysis and the Aldefluor assay.(22–26) In the present study, we isolated CRC CSC/CIC using SP cell analysis. Although previous studies showed that some colon cancer SP cells were not enriched with a CSC/CIC population,(27) our previous study confirmed that SW480, HT29 and HCT15 SP cells had high tumor-initiating ability, high expression levels of stem cell markers such as SOX2 and POU5F1 and strong resistance to chemotherapeutic agents compared with MP cells.(18) The ratio of SW480 SP cells was 4.0%, that of HCT15 cells was 5.5% and that of HT29 cells was 0.8%. All of these SP cells were specifically inhibited by verapamil, an ABC transporter inhibitor (Fig. 1a). Expression of DNAJB8 mRNA in SP cells and MP cells was examined using RT-PCR. DNAJB8 mRNA was detected in SP cells derived from SW480, HCT15 and HT29 cells, whereas DNAJB8 mRNA was not detected in MP cells (Fig. 1b). Thus, DNAJB8 is one candidate of CSC/CIC-specific antigens in CRC as well as in renal cell carcinomas. DNAJB8 mRNA was detected in SP cells derived from HT29 cells at the highest level (Fig. 1b) and therefore HT29 cells were used for further analysis. The quality of SP cells derived from HT29 cells as a source of CRC CSC/CIC was confirmed by higher expression levels of stem cell markers, including SOX2, POU5F1 and LGR5, using RT-PCR and by a higher tumor-initiating ability in a xenograft model than those of MP cells (Fig. 1c, d).
Fig. 1.
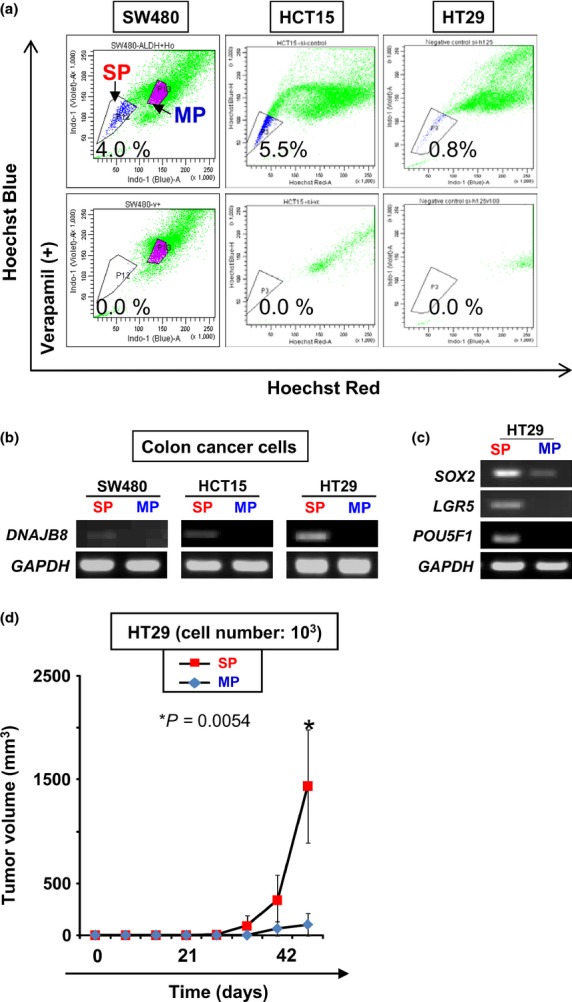
Isolation of colon cancer stem-like cells/cancer-initiating cells (CSC/CIC) as side population (SP) cells. (a) Isolation of SP cells from colon cancer cell lines. SW480, HCT15 and HT29 were stained with Hoechst 33342 dye with or without verapamil and analyzed using a FACSAria II cells sorter. (b) RT-PCR of DNAJB8. MP, main population. (c) RT-PCR of CSC/CIC markers in HT29 cells. (d) Tumor growth of HT29 SP cells and MP cells. HT29 SP cells, 103, and MP cells were inoculated subcutaneously into the backs of five non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice and tumor growth was measured weekly. Data represent means ± SD. The difference between SW480 SP and MP cells was examined for statistical significance using the Student's t-test.
Role of DNAJB8 in CRC CSC/CIC
To evaluate the functions of DNAJB8 in HT29 cells, we established DNAJB8-overexpressed cells. We confirmed the overexpression of DNAJB8 mRNA using quantitative RT-PCR (Fig. 2a). Expression levels of the stem cell markers SOX2, LGR5 and POU5F1 were increased by 2.2-fold, 2.3-fold and 2.1-fold, respectively, in DNAJB8-overexpressed HT29 cells compared with the level in the control HT29 cells (Fig. 2a). The percentage of SP cells in DNAJB8-overexpressed HT29 cells was higher than that in control HT29 cells (Fig. 2b). To confirm the tumorigenicity in vivo of DNAJB8-overexpressed HT29 cells, we used a xenograft transplantation model. The DNAJB8-overexpressed HT29 cells showed higher tumor-initiating ability compared with HT29 control cells (Fig. 2c). A sphere formation assay was used to evaluate CSC/CIC-like features. DNAJB8-overexpressed HT29 cells showed higher sphere-forming ability than that of HT29 control cells (Fig. 2d), indicating that overexpression of DNAJB8 induced CSC/CIC properties.
Fig. 2.
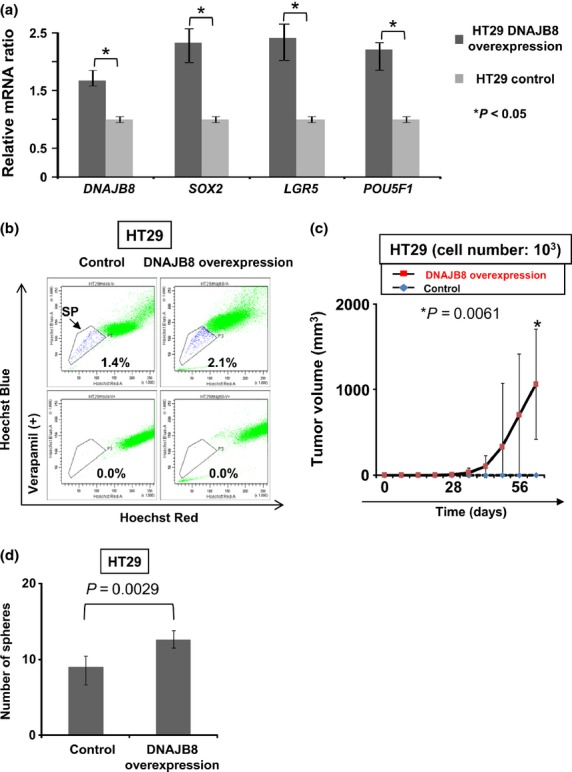
DNAJB8 gene overexpression experiments in HT29 cells. (a) Quantitative RT-PCR of DNAJB8 and colon cancer stem-like cells/cancer-initiating cells (CSC/CIC) markers. Data are shown as a comparison with the expression level in HT29 control cells. Data represent means ± SD. (b) Isolation of side population (SP) cells. HT29 DNAJB8-overexpressed cells and control cells were stained with Hoechst 33342 dye with or without verapamil and analyzed using a FACSAria II cell sorter. (c) Tumor growth of HT29 DNAJB8-overexpressed cells and control cells. HT29 DNAJB8-overexpressed cells, 103, and control cells were inoculated subcutaneously into the backs of five non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice and tumor growth was measured weekly. Data represent means ± SD. The difference between HT29 DNAJB8-overexpression cells and control cells was examined for statistical significance using the Student's t-test. (d) Sphere formation assay. To assay sphere formation efficiency, 103 cells were cultured in Ultra Low Attachment six-well plates for 2 weeks and the number of spheres was counted under a microscope in 15 low-power fields and then the average was calculated.
Establishment of DNAJB8-targeting immunotherapy
To verify the immunogenicity for peptides from DNAJB8 protein, DNAJB8-specific CTL were induced using HLA-A*2402-positive healthy volunteer donors. Candidate antigenic peptides carrying the HLA-A*2402-binding anchor motif were screened according to the amino acid sequence of DNAJB8 protein and there were four candidate peptides (DNAJB8_22[8], DNAJB8_90[10], DNAJB8_99[9] and DNAJB8_143[9]) (Fig. 3). The HLA-A24 binding ability was then assessed using a HLA-A24 binding assay with SL8C peptide as a negative control and Survivin-2B_80(9) as a positive control. DNAJB8_22(9), DNAJB8_99(9) and DNAJB8_143(9) showed ability to bind to HLA-A24, whereas DNAJB8_90(10) did not (Fig. 3). Therefore, DNAJB8_22(9), DNAJB8_99(9) and DNAJB8_143(9) peptides were used for further CTL induction experiments.
Fig. 3.

DNAJB8 peptides carrying a HLA-A24 binding motif. (a) Candidate of DNAJB8 peptides carrying a HLA-A24 binding motif. (b) Peptide-binding assay. Binding affinity was evaluated by comparing mean fluorescence intensity of HLA-A24 expression in the presence of peptide pulsation to mean fluorescence intensity in the absence of the peptide. Survivin-2B_80(9) (AYACNTSTL) peptide was used as a positive control and SL8C (SIINFEKL) peptide was used as a negative control.
The PBMC of two healthy volunteer donors (A and B) were stimulated using a mixture of DNAJB8_22(9), DNAJB8_99(9) and DNAJB8_143(9) and then the reactivity for each peptide was evaluated using a IFN-γ ELISpot assay and 51Cr release assay. Interferon-γ secretion was observed for DNAJB8_22(8)-pulsed and DNAJB8_143(9)-pulsed target cells from both donors using a IFN-γ ELISpot assay (Fig. 4a), whereas cytotoxic activity was detectable for only DNAJB8_143(9)-pulsed target cells using a 51Cr release assay (Fig. 4b). Therefore, DNAJB8_143(9) peptide is a candidate for DNAJB8-targeting immunotherapy. We generated four CTL clones specific for DNAJB8_143(9) from donor A (CTL clone #21, 67 and 84) and one clone from donor B (CTL clone #70) and performed further analysis using CTL clone #84. The DNAJB8_143(9)-specific CTL clone showed cytotoxic activity for T2-A24 cells pulsed with DNAJB8_143(9) peptide but not for T2-A24 cells without the peptide or for K562 cells (Fig. 4c). To verify whether this CTL clone can recognize endogenously presented DNAJB8_143(9) peptide of DNAJB8-positive CRC CSC/CIC, we performed a 51Cr release assay using SP cells derived from HT29 cells. The DNAJB8_143(9)-specific CTL clone showed greater cytotoxic activity for HLA-A*2402+ HT29-SP cells than for HLA-A*2402+ HT29-MP cells or HLA-A*2402-DNAJB8- K562 cells (Fig. 4d). These results indicate that DNAJB8_143(9) peptide is an immunogenic epitope and that the endogenously processed peptide is presented on the surface of SP cells.
Fig. 4.
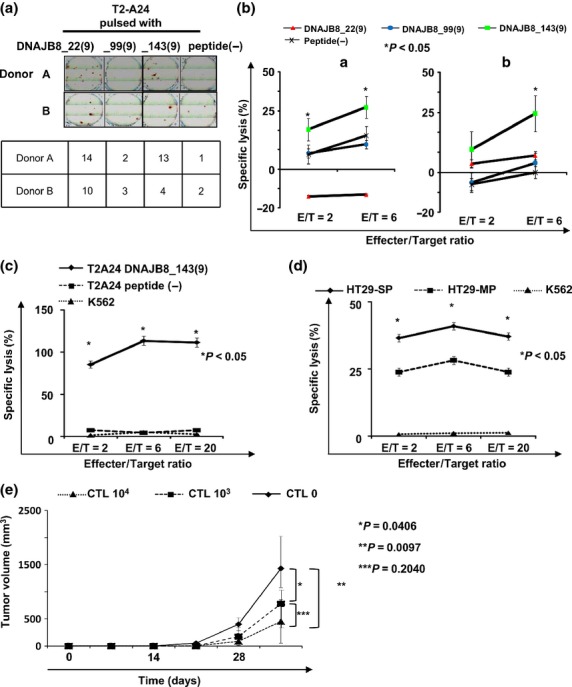
Antitumor effect of DNAJB8-specific cytotoxic T lymphocyte (CTL) clone. (a) Interferon-γ enzyme-linked immunospot assay. (b) 51Cr release assay. We evaluated specific cytotoxic activity against peptide-pulsed T2-A24 cells in healthy donors. (c, d) 51Cr release assay using the DNAJB8_143(9)-specific CTL clone. We established CTL clones recognizing DNAJB8_143(9) and evaluated cytotoxic activity against side population (SP) cells and main population (MP) cells derived from HT29 cells. (e) Tumor growth of HT29 cells in a therapeutic adoptive transfer model. HT29 cells were inoculated subcutaneously into the back of five NOD/SCID mice and CTL clone cells or PBS was injected intravenously 3 weeks later. Tumor growth was measured weekly. Data represent means ± SD. Differences between groups were examined for statistical significance using the Student's t-test.
Finally, we evaluated the antitumor effect in vivo of the DNAJB8-specific CTL clone using a therapeutic CTL adoptive transfer model. The DNAJB8-CTL clone-transferred group showed a significant antitumor effect compared with that in the control group (Fig. 4e). The group with 104 CTL injection showed a tendency for a greater antitumor effect than that in the group with 103 CTL injection. These results indicate that DNAJB8_143(9) peptide is an immunogenic epitope and is a candidate for CRC CSC/CIC-targeting immunotherapy.
Discussion
The CSC/CIC are resistant to standard cancer therapies including chemotherapy and radiotherapy,(3) and one of the major topics in research on CSC/CIC is how to target CSC/CIC. Cancer immunotherapy is a novel approach for targeting CSC/CIC. Some reports, including our previous reports, have indicated a relationship between CSC/CIC and immunotherapies. Todaro and colleagues reported that human colon CSC/CIC were killed by γδ T lymphocytes in vitro.(28) Ning and colleagues reported that the vaccinations of dendritic cell (DC) pulsed with mice melanoma CSC/CIC and mice squamous cell CSC/CIC conferred effective antitumor effects in vivo.(29) However, there have been no reports showing direct killing of human CSC/CIC by CTL. Thus, we analyzed the susceptibility of CRC CSC/CIC specific for CEP55, a novel TAA of CRC.(18) We showed that a CEP55 peptide-specific CTL clone could efficiently recognize SP cells as well as MP cells of human CRC and could kill xenograft tumors derived from SP cells in vivo.(18) In the present study, we identified DNAJB8 as a novel CRC CSC/CIC antigen and showed that CTL specific for DNAJB8-derived peptide efficiently recognized SP cells derived from HT29 cells. The CTL clone specific for CEP55 recognized both SP and MP cells at similar levels,(18) while the CTL clone specific for DNAJB8 recognized SP cells at a higher level than that of MP cells. CEP55 is expressed in both SP and MP cells at similar levels, while DNAJB8 is preferentially expressed in SP cells. Since DNAJB8 is preferentially expressed in CSC/CIC, the combination of DNAJB8-targeting immunotherapy with standard therapies including chemotherapy and radiotherapy might be an effective approach to eradicate cancer. The CTL clone specific for DNAJB8 showed lower, but significant cytotoxicity for MP cells (Fig. 4d), whereas MP cells did not express DNAJB8 mRNA (Fig. 1b). The SP cells might have the ability to differentiate MP cells and DNAJB8 mRNA is suppressed according to SP cell differentiation into MP cells. The protein or antigenic peptides of DNAJB8 may remain for a while after differentiation. Therefore, the cytotoxicity for MP cells may be specific for DNAJB8 peptide.
Both the CEP55-specific CTL clone and the DNAJB8-specific CTL clone efficiently recognize CRC CSC/CIC. In our previous review article, we reported that TAA can be distinguished according to the expression patterns in CSC/CIC and non-CSC/CIC, which are: (i) CSC/CIC antigens, which are expressed preferentially in CSC/CIC; (ii) shared antigens, which are expressed in both CSC/CIC and non-CSC/CIC at similar levels; and (iii) non-CSC/CIC antigens, which are preferentially expressed in non-CSC/CIC.(30) In the mouse DNA vaccination model, Dnajb8 showed a greater antitumor effect than that of Survivin.(16) Dnajb8 is a CSC/CIC antigen and Survivin is a shared antigen. CSC/CIC antigens might be better than TAA at achieving an antitumor effect and DNAJB8 might be a better target than CEP55 or Survivin. In our previous clinical trials, we used Survivin-2B-derived antigenic peptides.(7–12) Therefore, use of the CSC/CIC antigen DNAJB8 may therefore improve the clinical outcome of a peptide vaccination trial.
DNAJB8 is a cancer/testis antigen and many other cancer/testis antigens are preferentially expressed in CSC/CIC.(31) However, the functions of most known cancer/testis antigens in CSC/CIC are not known, indicating that there is a risk of antigen loss when targeted by CTL. DNAJB8 has a role in the maintenance of CSC/CIC and it might therefore be a better candidate for CSC/CIC-targeting immunotherapy. In the present study, we used DNAJB8 stably overexpressed cells to analyze the functions of DNAJB8 and most of the cells are supposed to express DNJAB8. However, the increase in rates of SP cells was partial (Fig. 2b). DNAJB8 might be essential for the maintenance of CSC/CIC; however DNAJB8 might not be sufficient and other factor(s) may be necessary to induce CSC/CIC.
In summary, we identified DNAJB8 as a novel functional CSC/CIC antigen in CRC. A DNAJB8-specific CTL clone derived from a HLA-A*2402 donor showed antitumor ability both in vitro and in vivo. DNAJB8 is a candidate for CSC/CIC-targeting immunotherapy.
Acknowledgments
The present study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to N. S.), program for developing the supporting system for upgrading education and research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to N. S.) and Takeda Science Foundation (to Y. H.) and Research Fellowship of Japan Society for the Promotion of Science for Young Scientists (to R.M.).
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–60. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 3.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–13. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–7. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez SA, von Hofe E, Kallinteris NL, et al. A new era in anticancer peptide vaccines. Cancer. 2010;116:2071–80. doi: 10.1002/cncr.24988. [DOI] [PubMed] [Google Scholar]
- 7.Tsuruma T, Hata F, Torigoe T, et al. Phase I clinical study of anti-apoptosis protein, survivin-derived peptide vaccine therapy for patients with advanced or recurrent colorectal cancer. J Transl Med. 2004;2:19. doi: 10.1186/1479-5876-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuruma T, Iwayama Y, Ohmura T, et al. Clinical and immunological evaluation of anti-apoptosis protein, survivin-derived peptide vaccine in phase I clinical study for patients with advanced or recurrent breast cancer. J Transl Med. 2008;6:24. doi: 10.1186/1479-5876-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Honma I, Kitamura H, Torigoe T, et al. Phase I clinical study of anti-apoptosis protein survivin-derived peptide vaccination for patients with advanced or recurrent urothelial cancer. Cancer Immunol Immunother. 2009;58:1801–7. doi: 10.1007/s00262-009-0691-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kameshima H, Tsuruma T, Torigoe T, et al. Immunogenic enhancement and clinical effect by type-I interferon of anti-apoptotic protein, survivin-derived peptide vaccine, in advanced colorectal cancer patients. Cancer Sci. 2011;102:1181–7. doi: 10.1111/j.1349-7006.2011.01918.x. [DOI] [PubMed] [Google Scholar]
- 11.Miyazaki A, Kobayashi J, Torigoe T, et al. Phase I clinical trial of survivin-derived peptide vaccine therapy for patients with advanced or recurrent oral cancer. Cancer Sci. 2011;102:324–9. doi: 10.1111/j.1349-7006.2010.01789.x. [DOI] [PubMed] [Google Scholar]
- 12.Kameshima H, Tsuruma T, Kutomi G, et al. Immunotherapeutic benefit of IFNalpha in survivin2B-derived peptide vaccination for advanced pancreatic cancer patients. Cancer Sci. 2013;104:124–9. doi: 10.1111/cas.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirohashi Y, Torigoe T, Inoda S, et al. The functioning antigens: beyond just as the immunological targets. Cancer Sci. 2009;100:798–806. doi: 10.1111/j.1349-7006.2009.01137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hageman J, Rujano MA, van Waarde MA, et al. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell. 2010;37:355–69. doi: 10.1016/j.molcel.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Sterrenberg JN, Blatch GL, Edkins AL. Human DNAJ in cancer and stem cells. Cancer Lett. 2011;312:129–42. doi: 10.1016/j.canlet.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 16.Nishizawa S, Hirohashi Y, Torigoe T, et al. HSP DNAJB8 controls tumor-initiating ability in renal cancer stem-like cells. Cancer Res. 2012;72:2844–54. doi: 10.1158/0008-5472.CAN-11-3062. [DOI] [PubMed] [Google Scholar]
- 17.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoda S, Hirohashi Y, Torigoe T, et al. Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. Am J Pathol. 2011;178:1805–13. doi: 10.1016/j.ajpath.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakao M, Shichijo S, Imaizumi T, et al. Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J Immunol. 2000;164:2565–74. doi: 10.4049/jimmunol.164.5.2565. [DOI] [PubMed] [Google Scholar]
- 20.Hirohashi Y, Torigoe T, Maeda A, et al. An HLA-A24-restricted cytotoxic T lymphocyte epitope of a tumor-associated protein, survivin. Clin Cancer Res. 2002;8:1731–9. [PubMed] [Google Scholar]
- 21.Inoda S, Hirohashi Y, Torigoe T, et al. Cep55/c10orf3, a tumor antigen derived from a centrosome residing protein in breast carcinoma. J Immunother. 2009;32:474–85. doi: 10.1097/CJI.0b013e3181a1d109. [DOI] [PubMed] [Google Scholar]
- 22.Haraguchi N, Utsunomiya T, Inoue H, et al. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006;24:506–13. doi: 10.1634/stemcells.2005-0282. [DOI] [PubMed] [Google Scholar]
- 23.Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–63. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 25.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 26.Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69:3382–9. doi: 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burkert J, Otto WR, Wright NA. Side populations of gastrointestinal cancers are not enriched in stem cells. J Pathol. 2008;214:564–73. doi: 10.1002/path.2307. [DOI] [PubMed] [Google Scholar]
- 28.Todaro M, D'Asaro M, Caccamo N, et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J Immunol. 2009;182:7287–96. doi: 10.4049/jimmunol.0804288. [DOI] [PubMed] [Google Scholar]
- 29.Ning N, Pan Q, Zheng F, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72:1853–64. doi: 10.1158/0008-5472.CAN-11-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirohashi Y, Torigoe T, Inoda S, Morita R, Kochin V, Sato N. Cytotoxic T lymphocytes: sniping cancer stem cells. Oncoimmunology. 2012;1:123–5. doi: 10.4161/onci.1.1.18075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada R, Takahashi A, Torigoe T, et al. Preferential expression of cancer/testis genes in cancer stem-like cells: proposal of a novel sub-category, cancer/testis/stem gene. Tissue Antigens. 2013;81:428–34. doi: 10.1111/tan.12113. [DOI] [PubMed] [Google Scholar]