

Abstract
We have reported that interferon (IFN)-α can attack cancer cells by multiple antitumor mechanisms including the induction of direct cancer cell death and the enhancement of an immune response in several pancreatic cancer models. However, an immunotolerant microenvironment in the tumors is often responsible for the failure of the cancer immunotherapy. Here we examined whether the suppression of regulatory T cells (Tregs) within tumors can enhance an antitumor immunity induced by an intratumoral IFN-α gene transfer. First we showed that an intraperitoneal administration of an agonistic anti-glucocorticoid induced TNF receptor (GITR) monoclonal antibody (mAb), which is reported to suppress the function of Tregs, significantly inhibited subcutaneous tumor growth in a murine pancreatic cancer model. The anti-GITR mAb was then combined with the intratumoral injection of the IFN-α-adenovirus vector. The treatment with the antibody synergistically augmented the antitumor effect of IFN-α gene therapy not only in the vector-injected tumors but also in the vector-uninjected tumors. Immunostaining showed that the anti-GITR mAb decreased Foxp3+ cells infiltrating in the tumors, while the intratumoral IFN-α gene transfer increased CD4+ and CD8+ T cells in the tumors. Therefore, the combination therapy strongly inclined the immune balance of the tumor microenvironment in an antitumor direction, leading to a marked systemic antitumor effect. The CCR5 expression on Tregs was downregulated in the antibody-treated mice, which may explain the decrease of tumor-infiltrating Tregs. The combination of Treg-suppression by GITR mAb and the tumor immunity induction by IFN-α gene therapy could be a promising therapeutic strategy for pancreatic cancer.
Keywords: Anti-glucocorticoid induced TNF receptor antibody, interferon-α gene therapy, pancreatic cancer
Overcoming pancreatic cancer remains one of the most formidable challenges in oncology today despite recent advances in therapeutic and diagnostic modalities.(1–3) It is expected to continue to be one of the five leading causes of cancer-related death in Japan, with <5% of patients alive 5 years after diagnosis.(4) Its high mortality is due to the high incidence of metastatic disease at the time of diagnosis, a fulminant clinical course, and the lack of adequate systemic therapies.(1–3) Pancreatic cancer must be considered as a systemic disease, and new therapeutic approaches that can effectively target this cancer spread in vivo are urgently needed.(2,3,5)
The interferon (IFN)-α protein is a pleiotropic cytokine regulating anti-proliferation, induction of cell death, anti-angiogenesis and immunomodulation, and has been used for treatment in a variety of cancers such as chronic myeloid leukemia, melanoma and renal cancer.(6–8) Although IFN-α was long thought to act mainly by suppressing tumor cell proliferation in vivo, more recently it has been established that type I IFNs have important roles in regulating the innate and adaptive arms of the immune system: upregulation of major histocompatibility complex class I gene, promotion of the priming and survival of T cells, enhancement of humoral immunity, and increase of the cytotoxic activity of natural killer (NK) cells and CD8+ T cells.(9,10) We also reported that an intratumoral IFN-α gene transfer elicits a systemic tumor-specific immunity in several animal models,(11–15) and showed that dendritic cells (DCs) in tumors have a critical role: (i) intratumoral expression of IFN-α effectively induces cell death of cancer cells and exposes tumor associated antigens in large quantity to DCs; (ii) IFN-α promotes maturation of CD11c+ cells, which facilitates the presentation of TAAs on CD11c+ cells; (iii) CD11c+ cells in tumors transduced with the IFN-α gene produce a large quantity of immune-stimulatory cytokines such as IL-12.(14,16,17) Furthermore, intratumoral gene transfer allows an increased and sustained local concentration of IFN-α in tumors, with minimal leakage of the cytokine into the systemic blood circulation.(11,12,14) Our data showed that, due to the effective induction of antitumor immunity and the lower toxicity, an intratumoral route of the IFN vector is superior to an intravenous administration.(14)
The development of an effective cancer immunotherapy is, however, often difficult because cancer generates an immunotolerant microenvironment against the host immune system.(18) Regarding this microenvironment, extensive studies have shown that CD4+Foxp3+ regulatory T cells (Tregs) are critical in controlling antitumor immune responses.(19) Therefore, Treg modulation is a promising strategy for enhancing the efficacy of cancer immunotherapy.(20) Recently, it has been reported that anti-glucocorticoid induced tumor necrosis factor receptor (GITR), a type I transmembrane protein with homology to tumor necrosis factor receptor family members, was a molecule that inhibits T-cell receptor-induced apoptosis.(21) GITR is a co-stimulatory molecule expressed at different levels in resting CD4+ and CD8+ T cells and is upregulated after T-cell activation.(21,22) Anti-glucocorticoid induced tumor necrosis factor receptor is also constitutively expressed on CD4+CD25+ Tregs at high levels, and it has been shown that activation of GITR signaling by GITR ligand or agonistic antibody can inhibit the suppressive activity of Tregs attributable to both the co-stimulatory activity of GITR on responder CD4+CD25− T cells and a direct effect on CD4+CD25+ Tregs.(21–23)
In this study, we examined antitumor effects of agonistic anti-GITR monoclonal antibody (mAb) and intratumoral IFN-α gene transfer in a murine pancreatic cancer model, and showed that the treatment with anti-GITR mAb synergistically enhances the antitumor effect of IFN-α gene therapy for pancreatic cancer. The combination of an induction of tumor immunity and a blockade of the immunotolerant microenvironment could be a promising therapeutic strategy for patients with pancreatic cancer.
Materials and Methods
Tumor cell lines and recombinant adenovirus vectors
Pan02 (H-2b: National Cancer Institute, Frederick, MD, USA) and CT26 (H-2d: American Type Culture Collection, Rockville, MD, USA) are a C57BL/6-derived pancreatic cancer cell line and a BALB/c-derived colon cancer cell line, respectively. They were maintained in an RPMI1640 medium (Nissui Pharmaceutical, Tokyo, Japan) containing 10% fetal bovine serum (FBS), 2 μM L-glutamine and 0.15% sodium bicarbonate (complete RPMI). The recombinant adenovirus vectors expressing mouse interferon-α (Ad-mIFN) and alkaline phosphatase cDNA (Ad-AP) were prepared as described.(12) The recombinant adenoviruses are based on serotype 5 with deletions of the entire E1 and a part of the E3 regions, and have the CAG promoter, which is a hybrid of the cytomegalovirus immediate early enhancer sequence and the chicken β-actin/rabbit β-globin promoter.(24) A cesium chloride-purified virus was desalted using a sterile Bio-Gel P-6 DG chromatography column (Econopac DG 10; BioRad, Hercules, CA, USA) and diluted for storage in a 13% glycerol/PBS solution. All viral preparations were confirmed by PCR assay to be free of the E1+ adenovirus.
Animals and tumor inoculation
Five-to-seven week-old female C57BL/6 (H-2b) and BALB/c (H-2d) mice were purchased from Charles River Japan (Kanagawa, Japan). Female (C57BL/6 × BALB/c) F1 mice (H-2b/d) were purchased from Japan SLC, Inc. (Hamamatsu, Japan), and were housed under sterilized conditions. Animal studies were carried out according to the Guideline for Animal Experiments of the National Cancer Center Research Institute and approved by the Institutional Committee for Ethics in Animal Experimentation. Pan02 (5 × 106) and CT26 cells (1 × 106) were injected subcutaneously into the legs of C57BL/6 and BALB/c mice, respectively. When a subcutaneous tumor was established (˜1.0 cm in a diameter), it was directly injected once with Ad-mIFN or control vector (Ad-AP). The tumor volume was calculated using the formula: tumor volume = 1/2 × ([the shortest diameter]2 × [the longest diameter]). Data are presented as mean ± standard deviation (SD).
ELISpot assays
Interferon-γ ELISpot kit (BD Biosciences, San Jose, CA, USA) was used according to the manufacturer's instructions. Briefly, splenocytes (1 × 105) and mitomycin C (MMC)-treated tumor cells (1 × 104) or syngeneic lymphocytes were co-cultured in 96-well plates pre-coated with mouse IFN-γ (BD Biosciences) for 20 h at 37°C in complete RPMI medium in triplicate. The syngeneic lymphocytes was used to examine whether the therapy induces an autoimmune reaction. After the wells were washed, biotinylated anti-mouse IFN-γ antibody (2 μg/mL) was added and incubated for 2 h at room temperature. A streptavidin-horseradish peroxidase solution was then added and incubated for 1 h at room temperature. After the addition of an aminoethyl carbozole substrate solution, spots were counted under a stereomicroscope.
Flow cytometry of cell surface marker and intracellular cytokine staining
Allo-phycocyanin (APC)-conjugated monoclonal antibody (mAb) to identify mouse IFN-γ and fluorescein isothiocyanate (FITC)-conjugated mAb to detect CD4, CD8 and CCR5 were purchased from BD Biosciences. Splenocytes (1 × 106) were incubated with medium alone (control) or Pan02 (1 × 105) cells for 2 days; brefeldin-A (10 μg/mL) was then added for 2 h of incubation. After being washed, the cells were incubated with the CD4, CD8 or CCR5 mAbs in a total volume of 100 μL PBS with 5% FBS for 30 min at 4°C. CD4 or CD8 stained cells were then fixed and permeabilized with a permeabilization buffer (BD Biosciences), and were finally stained with antibody to IFN-γ for 15 min at room temperature, washed again and analyzed by FACSCalibur (BD Biosciences). Irrelevant IgG mAbs were used as a negative control. Ten thousand live events were acquired for analysis.
Immunohistochemistry
Immunostaining was performed using streptavidin-biotin-peroxidase complex techniques (Nichirei, Tokyo, Japan). Consecutive cryostat tissue sections (6 μm) were mounted on glass slides and fixed in ethanol for 20 min. After blocking with normal rabbit serum, the sections were stained with rat anti-mouse CD4, CD8 and Foxp3 antibodies (BD Biosciences). Parallel negative controls with antibodies of the same isotype were examined in all cases. The sections were counter-stained with methylgreen.
RT-PCR of CCL3, CCL4 and CCL5 genes
To examine the expression of CCR5 ligands such as CCL3, CCL4 and CCL5 in Pan02 subcutaneous tumors, reverse transcriptase (RT)-PCR amplification was carried out using total RNA from the tumors in a 50 μL PCR mixture with the following primer sets: CCL3 upstream (5′-TTCTGCTGACAAGCTCACCCT-3′), CCL3 downstream (5′-ATGGCGCTGAGAAGACTTGGT-3′), CCL4 upstream (5′-CCCACTTCCTGCTGTTTCTC-3′), CCL4 downstream (5′-GAGGAGGCCTCTCCTGA -3′), CCL5 upstream (5′-TTCCCTGTCATTGCTTGCTCT-3′), CCL5 downstream (5′-GCAGCTGAGATGCCCA-3′) primers. In total, 30 cycles (β-actin: 25 cycles) of the PCR were carried out at 95°C for 30 s, 60°C for 30 s and 72°C for 1 min. The PCR products were fractionated on 2.0% agarose gel.
Statistical analysis
Comparative analyses of the data were performed by the Student's t-test, using spss statistical software (SPSS Japan, Tokyo, Japan). P < 0.05 was considered as a significant difference.
Results
Antitumor effect of intratumoral injection of Ad-mIFN
To examine the in vivo antitumor effect of the IFN-α gene transduction, various amounts (1 × 107, 5 × 107 and 5 × 108 PFU) of Ad-mIFN were injected into the right tumors in the mice with Pan02 tumors on both legs. The injection showed remarkable tumor suppressive effects not only in the vector-injected right tumors but also in the vector-uninjected left tumors in a dose-dependent manner (Fig. 1). The tumor suppressive effect was stronger in the right tumors than in the left tumors, possibly due to the direct anti-proliferative effect of IFN-α in Pan02 cells (data not shown) in addition to an induction of antitumor immunity. Tumor volumes were not changed in the mice treated by intratumoral injection of Ad-AP at 5 × 108 PFU as compared with the no treatment group (Fig. 1).
Fig. 1.
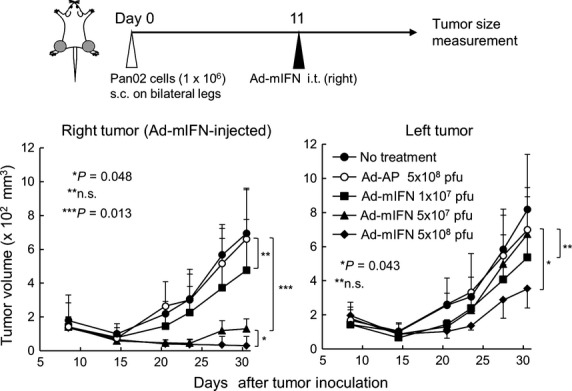
Adenovirus-mediated intratumoral inter-feron (IFN)-α gene transfer induces a systemic antitumor effect. Pan02 cells were inoculated on both legs in C57BL/6 mice, and 11 days later various amounts (1 × 107, 5 × 107, 5 × 108 PFU) of Ad-mIFN or Ad-AP were injected into the subcutaneous tumor on the right leg (n = 4). Data are representative of two separate experiments with similar results.
Intraperitoneal administration of anti-glucocorticoid induced TNF receptor monoclonal antibody suppressed the tumor growth
To examine whether the blockade of GITR-GITR ligand interaction was able to inhibit the tumor growth of Pan02 subcutaneous tumors, an agonistic anti-GITR mAb (DTA-1: 100 μg) was intraperitoneally injected into the mice with right-leg Pan02 tumors. This significantly suppressed tumor growth as compared with the control IgG injection (Fig. 2a). Then, to examine the expansion of tumor-responsive lymphocytes after the injection of GITR mAb, the splenocytes were harvested 14 days after the antibody administration, and stimulated with MMC-treated Pan02 cells or syngeneic lymphocytes. An ELISpot assay showed that the anti-GITR mAb significantly increased the number of IFN-γ-secreting cells in response to Pan02 cells but not to syngeneic lymphocytes compared with the control IgG treatment (Fig. 2b), suggesting that the blockade of GITR effectively expanded tumor-responsive immune cells.
Fig. 2.
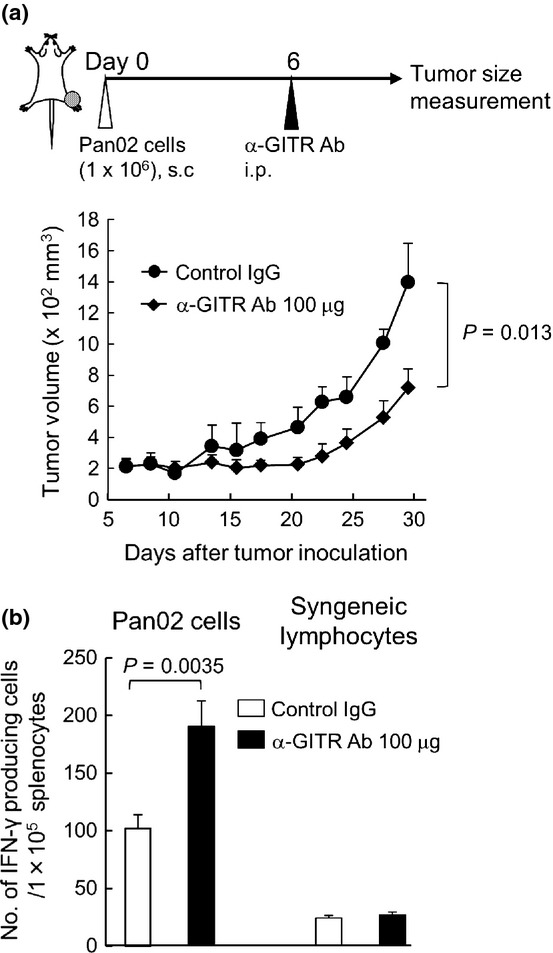
Intraperitoneal injection of anti-glucocorticoid induced TNF receptor (GITR) monoclonal antibody (mAb) suppresses the growth of pancreatic cancer tumors. (a) Pan02 cells were inoculated on the right legs in C57BL/6 mice, and 6 days later 100 μg of anti-GITR mAb was intraperitoneally injected into the mice (n = 3). Data are representative of two separate experiments with similar results. (b) The increase of interferon (IFN)-γ-positive cells in response to stimulation of Pan02 cells by an ELISpot assay. Two weeks after the injection of anti-GITR mAb, splenocytes were isolated from treated mice, and co-cultured with Pan02 cells or control lymphocytes (n = 3).
Combination therapy of anti-glucocorticoid induced TNF receptor monoclonal antibody and intratumoral IFN-α gene transfer showed an augmented antitumor activity
To ascertain whether the combination of anti-GITR mAb enhances an antitumor immunity induced by the intratumoral IFN-α gene transfer, the antibody was intrapertitoneally administered at day 6 after the subcutaneous inoculation of Pan02 cells followed by intratumoral injection of Ad-mIFN at day 11. The injection of a lower dose (1 × 107 PFU) of Ad-mIFN showed the tumor suppressive effects not only in vector-injected right tumors but also in the vector-uninjected left tumors in anti-GITR mAb-treated mice compared with the injection of Ad-AP (Fig. 3a). Furthermore, the higher dose (5 × 107 PFU) of Ad-mIFN with anti-GITR mAb showed the stronger suppressive effect of Pan02 tumor growth at both sites compared with the lower dose adenovirus injection (Fig. 3b).
Fig. 3.

Anti-glucocorticoid induced TNF receptor (GITR) monoclonal antibody (mAb) enhances the antitumor immunity of intratumoral interferon (IFN)-α gene transfer. Data are representative of at least two separate experiments with similar results. (a) Growth suppression of subcutaneous tumors by anti-GITR mAb and a lower dose of Ad-mIFN. Pan02 cells were inoculated on both legs in C57BL/6 mice. The 100 μg of GITR mAb was intrapertioneally injected once at day 6, and then 1 × 107 PFU of Ad-mIFN or Ad-AP was injected once into the tumors at day 11 (n = 8). (b) Antitumor effect by anti-GITR mAb and a higher dose of Ad-mIFN. Pan02 cells were inoculated on both legs in C57BL/6 mice. The 100 μg of GITR mAb was intrapertioneally injected once at day 6, and then 5 × 107 PFU of Ad-mIFN or Ad-AP was injected once into the tumors at day 11 (n = 10). (c) Growth suppression of CT26 subcutaneous tumors by anti-GITR mAb and Ad-mIFN. CT26 cells were inoculated on right legs in BALB/c mice. The 30 μg of GITR mAb was intrepertioneally injected once at day 8, and then 3 × 107 PFU of Ad-mIFN or Ad-AP were injected once into the tumors at day 11 (n = 9). (d) Growth of different tumors by a combination therapy. Pan02 cells were inoculated on right legs and CT26 cells were inoculated on left legs in (C57BL/6 × BALB/c) F1 mice. The 30 μg of anti-GITR mAb antibody was intraperitoneally injected into the mice at day 6, and 5 × 107 PFU of Ad-mIFN was injected into the right Pan02 tumors at day 11.
To confirm the antitumor activity of the combination therapy in a different tumor cell line, 3 × 107 PFU of Ad-mIFN was injected into CT26 tumor-bearing mice after the administration of anti-GITR mAb. The antitumor activity of combination therapy was also evident against the colon cancer cells (Fig. 3c). Then, we subcutaneously inoculated Pan02 cells on the right legs and CT26 cells on the left legs of (C57BL/6 × BALB/c) F1 mice. The 30 μg of DTA-1 antibody was intraperitoneally injected into the mice at day 6, and then 5 × 107 PFU of Ad-mIFN was injected into the right Pan02 tumors at day 11. The combination therapy significantly suppressed the Pan02 tumor growth compared with the no treatment, GITR antibody alone and the Ad-mIFN alone groups, whereas the effect was not recognized in the CT26 tumors (Fig. 3d), suggesting that the combination therapy induced the tumor-specific antitumor immunity.
Combination therapy of anti-glucocorticoid induced TNF receptor monoclonal antibody and intratumoral IFN-α gene transfer increased the tumor-responsive T cells
To examine the in vivo immune reaction by the combination therapy, splenocytes were collected from the treated mice and cultured with Pan02 cells. An ELISpot assay showed that the average number of IFN-γ-producing splenocytes in response to Pan02 cells was increased in the Ad-mIFN alone group, whereas the combination therapy further increased the IFN-γ-positive spots (Fig. 4a). The numbers of spots in splenocytes co-cultured with syngeneic lymphocytes were not changed in the treated groups (Fig. 4a). In fact, all treated mice looked healthy during the course of experiments and blood chemistry showed no abnormal values in the treated mice at 4 weeks (data not shown), indicating that the combination therapy did not induce autoimmunity. To analyze the subset of activated lymphocytes, the frequency of tumor-reactive immune cells was determined by intracellular cytokine staining and flow cytometry. The percentage of CD4+ and CD8+ T cells stimulated to produce IFN-γ in response to Pan02 cells increased significantly in the mice treated by the combination therapy (Fig. 4b), indicating that this therapy enhances systemic antitumor immunity.
Fig. 4.
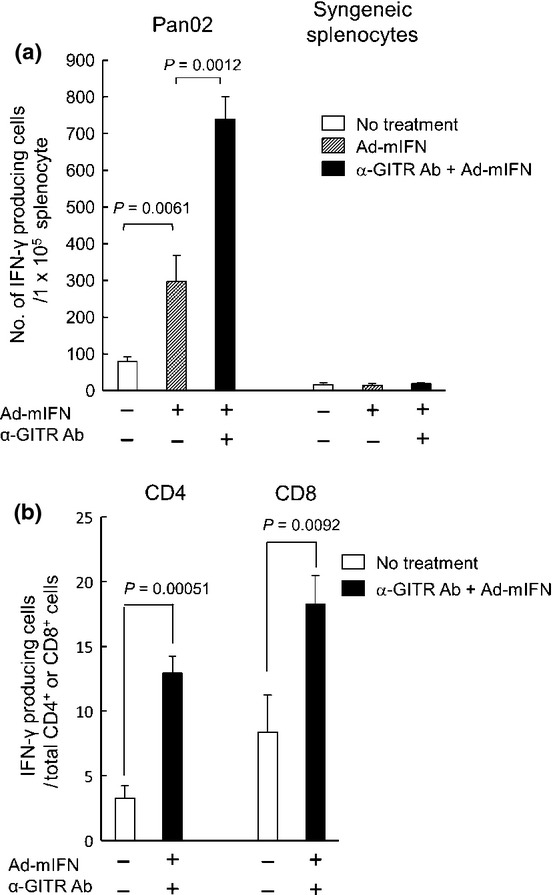
The increase of interferon (IFN)-γ-positive cells in response to stimulation of Pan02 cells by the combination therapy. (a) ELISpot assay of IFN-γ-producing cells in response to stimulation of Pan02 cells. Fourteen days after virus injection, splenocytes were isolated from mice, and co-cultured with Pan02 cells or control lymphocytes (n = 3). Data are representative of three separate experiments with similar results. (b) Intracellular cytokine staining of IFN-γ-producing cells in response to stimulation of Pan02 cells. The splenocytes from treated mice were incubated with Pan02 cells and stained by APC-anti-mouse IFN-γ. The activated cell fractions were analyzed by staining with fluorescein isothiocyanate (FITC)-anti-mouse CD4 or CD8 antibody (n = 3).
Anti-glucocorticoid induced TNF receptor monoclonal antibody reduced Treg-accumulation in tumors
It is known that a large number of tumor-infiltrating lymphocytes (TILs) result in a better antitumor effect for cancer patients.(25) To examine whether an increase of TILs in treated tumors is related to tumor growth suppression, immunohistochemical staining of CD4- and CD8-positive cells was performed in the Pan02 tumors 13 days after the injection of Ad-mIFN. The intratumoral IFN-α expression significantly increased the infiltration of CD4+ and CD8+ T cells in both vector-injected and vector-uninjected tumors, and the anti-GITR mAb also increased the number of CD4+ cells in the both tumors (Fig. 5a,b). Furthermore, the combination therapy of anti-GITR mAb and IFN-α gene transfer further increased the infiltration of CD4+ and CD8+ cells compared with the Ad-mIFN or anti-GITR mAb alone groups (Fig. 5a,b). Next, the infiltration of Foxp3+ cells was assessed in the treated Pan02 tumors. Interferon-α gene transfer and anti-GITR mAb administration decreased the number of Foxp3+ cells in the tumors, and the combination therapy markedly decreased Foxp3+ cells (Fig. 5c). Since a high effector T cells/Tregs ratio is associated with a favorable prognosis for cancer patients,(26) we calculated the ratio of CD4+ or CD8+ cells per Foxp3+ cells in the tumors. The ratios were markedly high in the combination therapy-treated tumors (Fig. 5d).
Fig. 5.

The combination therapy suppresses the accumulation of Tregs in tumors. Thirteen days after the injection of adenoviruses, fresh frozen sections of subcutaneous tumors were processed for immunohistochemistry. Data are representative of three separate experiments with similar results. (a) Immunostaining of CD4+ T cells. Rt.: right leg, Lt.: left leg. (b) Immunostaining of CD8+ T cells. (c) Immunostaining of Foxp3+ cells. (d) Relative ratio of CD4+ or CD8+ T cells per Foxp3+ cells in tumors. The number of CD4+ (left panel) and CD8+ T cells (right panel) in tumors was compared with that of Foxp3+ cells. (e) Proliferative status of CD4+ and CD8+ T cells infiltrated into Pan02 tumors. Tumors were harvested at 2 weeks after the combination therapy, processed into single-cell suspension, and the frequencies of Ki-67+ cells per CD4+ or CD8+ T cells were analyzed by flow-cytometry with PE-anti mouse Ki-67 antibody (eBioscience, San Diego, CA, USA; n = 3). We gated the lymphocyte regions in fluorescence-activated cell sorting (FACS) plots, and developed spots in two dimensions. A hundred thousand live cells were analyzed.
Then, to understand the proliferative status of lymphocytes infiltrated into tumors, we examined the Ki-67 expression on CD4+ and CD8+ T cells in regional lymph nodes and Pan02 subcutaneous tumors. Flow-cytometry showed that the frequencies of Ki-67+ cells per CD4+ and CD8+ T cells in the tumors were higher than those in the regional lymph nodes, indicating a significant population of CD4+ and CD8+ T cells in the tumors was in the state of proliferation, which may indicate that tumor-infiltrating T cells responded to tumor-associated antigens (Fig. 5e). Furthermore, the frequencies of Ki-67+CD4+ or CD8+ T cells in the tumors showed a tendency toward an increase in combination therapy-treated mice compared with non-treated mice. Since, in general, the lymphocytes are activated in lymph nodes, the results suggested that Pan02-responsive CD4+ and CD8+ T cells were activated in lymph nodes and infiltrated into the subcutaneous tumors.
GITR mAb suppressed CCR5 expression on Tregs
It was reported that CD4+Foxp3+ Tregs preferentially expressed CCR5 compared with CD4+Foxp3− effector T cells in a murine model of pancreatic cancer.(27) To understand the mechanism of decreased Treg-accumulation in the tumors, the effect of GITR mAb on CCR5 expression of Tregs was analyzed. We first examined the expression of CCR5 ligands such as CCL3, CCL4 and CCL5 in Pan02 subcutaneous tumors. The RT-PCR analysis showed that the Pan02 tumor expressed CCL3, CCL4 and CCL5, whereas the pancreas did not express the ligands except for CCL5 (Fig. 6a). Next, we analyzed the CCR5 expression on Tregs infiltrated into the tumors. Flow-cytometry showed that the percentage of CCR5+ Tregs in the spleens was approximately 7%, whereas that in the tumors was more than 30% (Fig. 6b), suggesting that a particular population of Tregs employed the interaction of CCR5-CCR5 ligands to infiltrate into Pan02 tumors. Then, we examined whether the anti-GITR mAb suppresses the CCR5 expression on Tregs. Flow-cytometry showed that the administration of anti-GITR mAb significantly decreased the CCR5 expression level on Tregs in the spleen, which may explain the reduced number of Tregs infiltrating in the tumors (Fig. 6c). Furthermore, to examine the effect of GITR stimulation on CCR5 expression of Tregs and non-Tregs in vitro, the splenocytes were cultured with DTA-1 antibody at 1 μg/mL for 5 h. Flow-cytometry showed that the frequency of CCR5+ cells per CD4+Foxp3− T cells was decreased approximately 80% of DTA-1-untreated cells, whereas that of CCR5+ cells per Foxp3+ Tregs was decreased <40% (Fig. 6d), suggesting that the GITR mAb suppressed the CCR5 expression on CD4+Foxp3+ Tregs more efficiently than that on CD4+ Foxp3− T cells.
Fig. 6.

Preferential suppression of CCR5 expression on CD4+Foxp3+ cells by anti-glucocorticoid induced TNF receptor monoclonal antibody (anti-GITR mAb). (a) Expression of CCR5 ligands in Pan02 tumors. Total RNA was extracted from Pan02 subcutaneous tumor and subjected to RT-PCR analysis with CCL3, CCL4 and CCL5 specific primers. (b) CCR5+ Tregs in the spleens and tumors. Spleens and tumors were harvested at 3 weeks after tumor inoculation, processed into single-cell suspension, and the percentage of CCR5+CD4+Foxp3+ cells was analyzed by flow-cytometry (n = 4). We gated the CD4+ regions in fluorescence-activated cell sorting (FACS) plots, and developed spots in two dimensions of CCR5 and Foxp3. In the tumors, a hundred thousand live cells were analyzed. (c) Suppression of CCR5 expression on CD4+Foxp3+ cells by anti-GITR mAb. Splenocytes were processed into single-cell suspension, and the cells were analyzed with anti-CD4, anti-Foxp3 and anti-CCR5 antibodies by flow-cytometry (n = 4). (d) Preferential suppression of CCR5 expression on Tregs by anti-GITR mAb. The splenocytes were isolated from the spleens of naïve C57BL/6 mice, and were cultured with anti-GITR mAb at 1 μg/mL for 5 h. The cells were analyzed with anti-CD4, anti-Foxp3 and anti-CCR5 antibodies by flow-cytometry (n = 4).
Discussion
We have reported that an intratumoral type I IFN gene transfer induces a marked regional antitumor effect and systemic tumor immunity by the integrated antitumor effect including direct cytotoxicity, immune stimulation of NK cells and cytotoxic T lymphocytes, maturation and activation of DCs and the antiangiogenesis effect in several pancreatic cancer animal models.(11–13) However, an immunotolerant microenvironment developed by cancer attenuates the antitumor immune response. In pancreatic cancer also, Hiraoka and colleagues(19) reported that Tregs play an important role in controlling the immune response against pancreatic ductal carcinoma from the premalignant stage to established cancer, and that a high prevalence of Tregs is a marker of poor prognosis in pancreatic ductal carcinoma. This study showed that the immune stimulatory reactions by IFN-α gene transfer were significantly reinforced by the inhibition of an immunotolerant environment by anti-GITR mAb.
Antibodies that activate or neutralize immunostimulatory and immunoinhibitory factors show an exciting therapeutic potential in experimental models, and clinical trials with anti-CTLA-4, anti-programmed death-1, anti-programmed death-L1, anti-GITR, indole-2, 3 dioxygenase inhibitors, and other modulators of cancer immunosuppression are now under way. Among these modulators of cancer immunosuppression, we selected an agonistic anti-GITR antibody because, regarding Treg/effector T cell interplay, GITR triggering can have several distinct effects: (i) co-stimulation and activation of effector T cells, which in itself makes them more difficult to be inhibited; (ii) inhibition of Treg activity by decreasing molecules important for suppression; (iii) decrease of the specific sensitivity of effector T cells to Treg-mediated suppression; and (iv) partial depletion of Treg.(21) Another intriguing mechanism is that anti-GITR mAb significantly decreased the number of CD4+CD25+ Tregs infiltrating in tumor sites as shown in Fig. 5(c) and previous reports,(28,29) suggesting that the GITR mAb creates an environment strongly supporting the enhancement of antitumor immunity. Mitsui et al.(29) reported that co-stimulatory signals through GITR may inhibit conversion of CD4+CD25− effector T cells to Tregs, and that another possibility is that CD8+CD25high T cells infiltrating into tumors compete with CD4+CD25+ Treg for availability of IL-2, because of the greater dependence of Tregs on IL-2 compared with effector T cells. In addition, we found that the in vivo CCR5 expression level on Tregs was significantly decreased by the administration of anti-GITR antibody, which may be a novel mechanism of Treg-inhibition in tumors.
A combination treatment of an anti-GITR mAb and intratumoral IFN-α gene transfer exhibited stronger antitumor effects compared with either treatment alone. Immunostaining of treated tumors showed that the anti-GITR mAb increased the number of CD4+ T cells and decreased Foxp3+ cells infiltrating in the tumors, while intratumoral IFN-α gene transfer increased both CD4+ and CD8+ T cells and also decreased the Foxp3+ cell in the tumors (Fig. 5a–c). Therefore, the combination treatment synergistically increased the number of effector T cells and markedly decreased Tregs in the tumors (Fig. 5d), and strongly inclined the immune balance of the tumor microenvironment in an antitumor direction, which should lead to a marked systemic antitumor effect. The precise mechanism for IFN-α-mediated inhibition of Tregs in tumors is not completely understood. Although we previously reported that an intratumoral IFN-α expression stimulates DCs to produce the cytokine IL-6,(17) which is a critical factor for suppressing Tregs, it would also be necessary to examine the direct effect of IFN-α on Tregs in tumors.
In this study, we used a subcutaneous tumor as a model of locally advanced pancreatic cancer, which is the main target of a local IFN-α gene therapy. Although locally advanced pancreatic cancer is defined as surgically unresectable with no evidence of distant metastasis, it is highly prone to metastasize during or after standard chemoradiotherapy. To provide a significant impact on long-term survival, locally advanced cancer also requires the improvement of both regional control and effective systemic treatment against the occurrence of distant metastasis. As shown in Fig. 3(a,b), the combination therapy significantly suppressed the growth of not only vector-injected tumors but also of vector-uninjected tumors, suggesting that the combination is a highly promising strategy for locally advanced pancreatic cancer.
With respect to the safety of the therapy, it is noteworthy that the IFN-α gene transfer showed a strong inhibitory effect and specific cell death induction on cancer cells including pancreatic cancer cells but not on normal cells such as hepatocytes and vascular endothelial cells.(11) In addition, there was a considerable difference in the IFN-α protein concentration between a vector-injected tumor tissue and the serum.(12,14) As one of the reasons for a little leakage of the IFN-α protein from the tumors into blood circulation, we reported that extracellular matrix proteins such as fibronectin in tumors directly interact with IFN-α and retain the cytokine.(30) Therefore, we expect the toxicity of a local IFN-α gene therapy to be tolerable in human clinical trials. On the other hand, although the anti-GITR mAb is effective in suppressing Tregs, a global Treg modulation by anti-GITR mAb may be undesirable, because it may increase susceptibility to autoimmunity.(20) Therefore, for the next research, the development of a strategy to inhibit Tregs in tumors alone will be important for developing an effective and safe immune therapy for cancers.
In summary, our preclinical study suggested that the combination of intratumoral IFN-α gene transfer and anti-GITR mAb is one of the promising new approaches to pancreatic cancer. The strategy may be worthy of an evaluation in a future clinical trial for this highly intractable cancer.
Acknowledgments
We thank Professor Shimon Sakaguchi, Immunology Frontier Research Center, Osaka University, for providing a DTA-1 hybridoma. K Aida and T Udagawa are awardees of a Research Resident Fellowship from the Foundation for Promotion of Cancer Research. This work was supported in part by a grant-in-aid for the 3rd Term Comprehensive 10-year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan, by grants-in-aid for research from the Ministry of Health, Labour and Welfare of Japan, by the program for promotion of Foundation Studies in Health Science of the National Institute of Biomedical Innovation, and by the National Cancer Center Research and Development Fund (23-A-38).
Disclosure Statement
The authors have no conflict of interest.
References
- 1.Hackert T, Buchler MW. Pancreatic cancer: advances in treatment, results and limitations. Dig Dis. 2013;31:51–6. doi: 10.1159/000347178. [DOI] [PubMed] [Google Scholar]
- 2.Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144:1316–26. doi: 10.1053/j.gastro.2013.01.078. [DOI] [PubMed] [Google Scholar]
- 3.Werner J, Combs SE, Springfeld C, Hartwig W, Hackert T, Buchler MW. Advanced-stage pancreatic cancer: therapy options. Nat Rev Clin Oncol. 2013;10:323–33. doi: 10.1038/nrclinonc.2013.66. [DOI] [PubMed] [Google Scholar]
- 4.Okusaka T, Matsumura Y, Aoki K. New approaches for pancreatic cancer in Japan. Cancer Chemother Pharmacol. 2004;54(Suppl. 1):S78–82. doi: 10.1007/s00280-004-0891-1. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida T, Ohnami S, Aoki K. Development of gene therapy to target pancreatic cancer. Cancer Sci. 2004;95:283–9. doi: 10.1111/j.1349-7006.2004.tb03204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talpaz M, Hehlmann R, Quintas-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-alpha in the treatment of chronic myeloid leukemia. Leukemia. 2013;27:803–12. doi: 10.1038/leu.2012.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarhini AA, Gogas H, Kirkwood JM. IFN-alpha in the treatment of melanoma. J Immunol. 2012;189:3789–93. doi: 10.4049/jimmunol.1290060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenblatt J, McDermott DF. Immunotherapy for renal cell carcinoma. Hematol Oncol Clin North Am. 2011;25:793–812. doi: 10.1016/j.hoc.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Santini SM, Lapenta C, Santodonato L, D'Agostino G, Belardelli F, Ferrantini M. IFN-alpha in the generation of dendritic cells for cancer immunotherapy. Handb Exp Pharmacol. 2009;188:295–317. doi: 10.1007/978-3-540-71029-5_14. [DOI] [PubMed] [Google Scholar]
- 10.Ferrantini M, Capone I, Belardelli F. Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie. 2007;89:884–93. doi: 10.1016/j.biochi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Hatanaka K, Suzuki K, Miura Y, et al. Interferon-alpha and antisense K-ras RNA combination gene therapy against pancreatic cancer. J Gene Med. 2004;6:1139–48. doi: 10.1002/jgm.602. [DOI] [PubMed] [Google Scholar]
- 12.Ohashi M, Yoshida K, Kushida M, et al. Adenovirus-mediated interferon alpha gene transfer induces regional direct cytotoxicity and possible systemic immunity against pancreatic cancer. Br J Cancer. 2005;93:441–9. doi: 10.1038/sj.bjc.6602713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hara H, Kobayashi A, Yoshida K, et al. Local interferon-alpha gene therapy elicits systemic immunity in a syngeneic pancreatic cancer model in hamster. Cancer Sci. 2007;98:455–63. doi: 10.1111/j.1349-7006.2007.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narumi K, Kondoh A, Udagawa T, et al. Administration route-dependent induction of antitumor immunity by interferon-alpha gene transfer. Cancer Sci. 2010;101:1686–94. doi: 10.1111/j.1349-7006.2010.01578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Udagawa T, Narumi K, Goto N, et al. Syngeneic hematopoietic stem cell transplantation enhances the antitumor immunity of intratumoral type I interferon gene transfer for sarcoma. Hum Gene Ther. 2012;23:173–86. doi: 10.1089/hum.2011.046. [DOI] [PubMed] [Google Scholar]
- 16.Hara H, Kobayashi A, Narumi K, et al. Intratumoral interferon-alpha gene transfer enhances tumor immunity after allogeneic hematopoietic stem cell transplantation. Cancer Immunol Immunother. 2009;58:1007–21. doi: 10.1007/s00262-008-0616-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narumi K, Udagawa T, Kondoh A, et al. In vivo delivery of interferon-alpha gene enhances tumor immunity and suppresses immunotolerance in reconstituted lymphopenic hosts. Gene Ther. 2012;19:34–48. doi: 10.1038/gt.2011.73. [DOI] [PubMed] [Google Scholar]
- 18.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi N, Hiraoka N, Yamagami W, et al. FOXP3+ regulatory T cells affect the development and progression of hepatocarcinogenesis. Clin Cancer Res. 2007;13:902–11. doi: 10.1158/1078-0432.CCR-06-2363. [DOI] [PubMed] [Google Scholar]
- 20.Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res. 2011;71:6915–20. doi: 10.1158/0008-5472.CAN-11-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nocentini G, Ronchetti S, Cuzzocrea S, Riccardi C. GITR/GITRL: more than an effector T cell co-stimulatory system. Eur J Immunol. 2007;37:1165–9. doi: 10.1002/eji.200636933. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–42. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 23.Nishikawa H, Kato T, Hirayama M, et al. Regulatory T cell-resistant CD8+ T cells induced by glucocorticoid-induced tumor necrosis factor receptor signaling. Cancer Res. 2008;68:5948–54. doi: 10.1158/0008-5472.CAN-07-5839. [DOI] [PubMed] [Google Scholar]
- 24.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 25.Ohtani H. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human colorectal cancer. Cancer Immun. 2007;7:4. [PMC free article] [PubMed] [Google Scholar]
- 26.Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–43. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan MC, Goedegebuure PS, Belt BA, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–55. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen AD, Schaer DA, Liu C, et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS ONE. 2010;5:e10436. doi: 10.1371/journal.pone.0010436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitsui J, Nishikawa H, Muraoka D, et al. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin Cancer Res. 2010;16:2781–91. doi: 10.1158/1078-0432.CCR-09-3243. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida K, Kondoh A, Narumi K, Yoshida T, Aoki K. Extracellular matrix interacts with interferon alpha protein: retention and display of cytotoxicity. Biochem Biophys Res Commun. 2008;376:299–304. doi: 10.1016/j.bbrc.2008.08.132. [DOI] [PubMed] [Google Scholar]