

Abstract
Bone metastasis is a multistep process that includes cancer cell dissemination, colonization, and metastatic growth. Furthermore, this process involves complex, reciprocal interactions between cancer cells and the bone microenvironment. Bone resorption is known to be involved in both osteolytic and osteoblastic bone metastasis. However, the precise roles of the bone resorption in the multistep process of osteoblastic bone metastasis remain unidentified. In this study, we show that bone resorption plays important roles in cancer cell colonization during the initial stage of osteoblastic bone metastasis. We applied bioluminescence/X-ray computed tomography multimodal imaging that allows us to spatiotemporally analyze metastasized cancer cells and bone status in osteoblastic bone metastasis models. We found that treatment with receptor activator of factor-κB ligand (RANKL) increased osteoblastic bone metastasis when given at the same time as intracardiac injection of cancer cells, but failed to increase metastasis when given 4 days after cancer cell injection, suggesting that RANKL-induced bone resorption facilitates growth of cancer cells colonized in the bone. We show that insulin-like growth factor-1 released from the bone during bone resorption and hypoxia-inducible factor activity in cancer cells cooperatively promoted survival and proliferation of cancer cells in bone marrow. These results suggest a mechanism that bone resorption and hypoxic stress in the bone microenvironment cooperatively play an important role in establishing osteoblastic metastasis.
Keywords: Bone resorption, hypoxia, IGF/IGFR signal, multimodality in vivo imaging, osteoblastic bone metastasis
Bone is one of the most common sites for cancer metastasis.1,2 Presently, increased incidence of bone metastasis from primary cancers occurring in various organs and tissues is attributed to the prolonged survival rate of the patient due to improved cancer control at the disease sites.3,4 These facts emphasize the need for developing novel therapeutic strategies based on understanding the basic biology of bone metastasis. Aberrant bone remodeling caused by reciprocal interaction among disseminated cancer cells, osteoblasts, and osteoclasts is an important event in establishment of bone metastasis.1,5 Receptor activator of nuclear factor-κB ligand (RANKL) is a tumor necrosis factor ligand superfamily member that is essential for the formation, activation, and function of osteoclasts.6 In osteolytic bone metastasis, cancer cells directly or indirectly activate osteoclasts through the RANKL signaling pathway, resulting in progression of bone resorption. The bone resorption triggers the release of growth factors stored in the bone matrix, such as transforming growth factor-β (TGF-β) and insulin-like growth factor (IGF), which interact with the cancer cells and enhance their growth during osteolytic bone metastasis.7,8
Osteoblastic bone metastasis is a process that induces aberrant bone formation due to stimulation of osteoblasts by cancer cells.9 In addition to finding osteolytic lesions in osteoblastic bone metastasis,10,11 it was recently found that denosumab, a human mAb against RANKL, significantly increased bone metastasis-free survival of prostate cancer patients; osteoblastic metastasis affects the vast majority of these patients.12–14 This clinical evidence strongly suggests that bone resorption plays an important role in osteoblastic bone metastasis. However, the precise mechanism of bone resorption and its involvement in osteoblastic bone metastasis are still poorly understood.
Hypoxia is a key regulatory factor in accelerating malignant processes of cancers. Hypoxia inducible factors (HIFs) are activated in hypoxia and orchestrate a vast array of gene products related to the processes for cellular adaptation to hypoxia.15 Earlier studies have indicated that HIFs play a critical role in cancer progression during bone metastasis.16,17 The activity of HIFs is regulated by the cellular oxygen sensors, prolyl hydroxylases.18 As each tissue cell has its own optimal partial pressure of oxygen (pO2), the oxygen concentration that activates HIFs is different for every cell. The pO2 of the bone marrow is relatively low compared to other organs,19 indicating that cancer cells migrating to the bone marrow would be exposed to hypoxic stress, leading to an increase of HIF activity. Therefore, understanding the role of HIFs in metastatic cancer cells in the bone is important for exploring the mechanism of bone metastasis.
In this study, we explore the role of bone resorption in establishment of osteoblastic bone metastasis of osteosarcoma cells in the hypoxic bone marrow environment by using multimodal (bioluminescence and X-ray computed tomography [CT]) imaging. The results show that the RANKL-induced bone resorption facilitates the colonization of cancer cells in bone marrow through the cooperative effects of IGF-1 released during bone resorption and HIF activation in the hypoxic bone marrow microenvironment.
Materials and Methods
Cell culture and isolation of LM8/luc and LM8/HRE-luc
The murine osteosarcoma cell line LM8 was gifted from Dr. Hideki Yoshikawa (Osaka University, Osaka, Japan). The LM8/luc and LM8/HRE-luc cells were isolated after transfection with plasmid pEF/luc and pEF/HRE-luc, respectively,20 by the calcium phosphate method.21 The cells were maintained at 37°C in 5% FCS-DMEM (Nacalai Tesque, Kyoto, Japan) supplemented with penicillin (100 U/mL) and streptomycin (100 μg/mL). Hypoxic cell cultures were done under 5% CO2 and 1%, 3%, or 5% O2.
Tumor xenografts
All the experimental procedures using mice were approved by the Animal Experiment Committees of the Tokyo Institute of Technology (Tokyo, Japan) (authorization number 2010006-3) and carried out in accordance with relevant national and international guidelines. LM8 cells (1 × 105 cells/100 μL) were injected into the left cardiac ventricle of 7-week-old male BALB/c-nu/nu nude mice (Oriental Yeast, Tokyo, Japan) as described previously.22 Recombinant human soluble RANKL23 (1 mg/kg; Oriental Yeast) or PBS was injected i.p. at 24-h intervals for 5 days. Acriflavine (4 mg/kg; Sigma-Aldrich, St Louis, MO, USA) or saline was injected daily for 10 days from 1 h after cancer cell injection.24
Micro X-ray CT imaging
Micro X-ray CT imaging was carried out using the R_mCT2 system (Rigaku, Tokyo, Japan). Hind limb bones were imaged at voxel size 10 × 10 μm using the following parameters: 90 kV X-ray tube voltage, 160 μA X-ray tube current, and 500 ms per frame. X-ray CT images were analyzed by ImageJ 1.44 (U.S. National Institutes of Health, Bethesda, MD, USA).
In vivo and ex vivo bioluminescence imaging
Bioluminescence (BL) images of tumor-bearing mice were acquired with the IVIS Spectrum imaging system (Caliper Life Sciences, Alameda, CA, USA) 15 min after i.p. injection of D-luciferin (100 mg/kg; Promega, Madison, WI, USA) using the conditions described in Data S1. Ex vivo imaging was acquired immediately after the last in vivo imaging. The BL intensity of bone metastasis was measured by region of interest analysis of hind limbs. Bone metastasis frequency was assessed 14 days after cancer cell injection by counting the bone metastatic sites with BL signals.
Multimodal imaging
Three-dimensional BL imaging and micro X-ray CT images were obtained and analyzed as described in Data S1.
Histological analysis
Hind limb bones of mice were excised and fixed in 70% ethanol for 48 h, decalcified in 10% EDTA for 2 weeks, processed, and embedded in paraffin. Sectioned bones (5-μm thickness) were stained with H&E. For von Kossa staining, hind limbs were excised from mice immediately following micro X-ray CT imaging and treated as described in Data S1.
Proliferation assay
LM8/luc cells (2 × 103/well) were seeded in 96-well plates with 1% FCS-DMEM and after 20 h of incubation the cells were further cultured for 0, 24, 48, and 72 h with IGF-1 (PeproTech, Rocky Hill, NJ, USA), TGF-β1 (PeproTech), and soluble RANKL under normoxic (21% O2) or hypoxic (1% O2) conditions. After the culture, cell proliferation rates were assessed with WST-1 reagent (Roche Diagnostics, Basel, Switzerland) according to the manufacturer's protocol.
Quantitative RT-PCR
LM8/luc cells were cultured for 24 h in DMEM with 5% FCS under normoxic (21% O2) or hypoxic (1% O2) conditions. Total RNA extraction from culture cells and bone tissues, and quantitative RT-PCR (qRT-PCR) were carried out as described in Data S1. The primers for mouse genes are shown in Table S1.
Western blotting
Cell lysate was collected and subjected to Western blot analysis with anti-HIF-1α rabbit polyclonal Ab (Novus Biologicals, Littleton, CO, USA), anti-IGF1 receptor β rabbit polyclonal Ab (Cell Signaling Technology, Beverly, MA, USA), anti-phospho-IGF1 receptor β (Tyr1135) rabbit monoclonal Ab (Cell Signaling Technology), anti-RANK rabbit polyclonal Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-β-actin mouse mAb (Sigma-Aldrich).
Luciferase reporter assays
LM8/luc or LM8/HRE-luc cells (5.0 × 104) were seeded onto 24-well plates and cultured overnight. The cells were further cultured under normoxic or hypoxic conditions for the indicated time. The cells were lysed and their luciferase activity was measured using a Luciferase Assay Kit (Promega).
Colony formation assay
Anchorage-independent growth was assessed by colony formation in soft agar as described.25 Five hundreds cells were cultured in 60-mm plates for 14 days in 21% or 1% O2. At the end of culture, colonies >50 μm in diameter were counted.
Statistical analysis
Statistical analyses were carried out using Student's t-test. Values of P < 0.05 were considered statistically significant.
Results
LM8 osteoblastic bone metastasis model
A cell line derived from Dunn murine osteosarcoma, LM8 was established as a highly lung metastatic subline by in vivo selection through pulmonary metastasis.26 Furthermore, LM8 formed osteoblastic lesions in the bone marrow after intratibial injection.27 These facts motivated us to assess bone metastasis formation following intracardiac (i.c.) injection of LM8. We isolated an LM8 subclone (LM8/luc), which stably expresses a firefly luciferase and preferentially metastasizing to the bone. Bioluminescence signals were typically obtained around the hind limbs of nude mice within 1 week of i.c. injection of LM8/luc (Fig.1a). We confirmed LM8/luc metastasis in the bone marrow by ex vivo imaging and histological analysis of hind limb bones (Fig.1b,c). The multimodal imaging of LM8/luc bone metastasis revealed the precise location of bone metastasis in the femur and ilium (Figs1d,S1) and aberrant bone formation at the metastatic sites 21 days after LM8/luc transplantation (Figs1e,S2).
Figure 1.
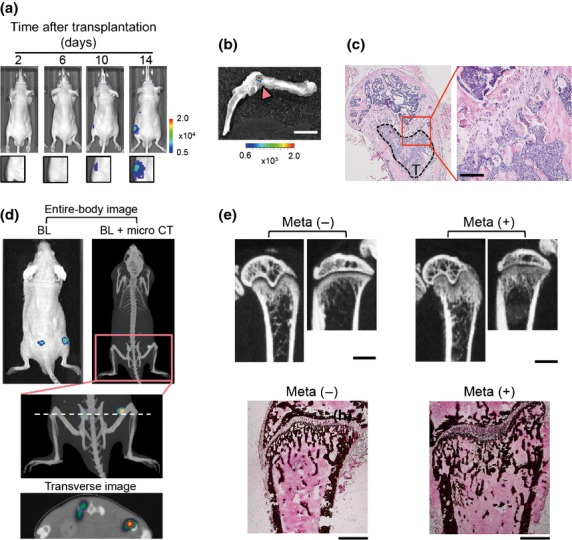
Murine osteosarcoma LM8 cells develop osteoblastic bone metastasis. (a) Representative time course bioluminescence (BL) images after intracardiac (i.c.) transplantation of LM8/luc. (b) Ex vivo imaging of LM8/luc tumor-bearing hind limb shown in (a) (14 days after LM8/luc injection). LM8/luc metastasis signal is indicated by an arrowhead. Scale bar = 5 mm. (c) Hematoxylin–eosin staining of hind limb bone with LM8 metastasis (T) of (b). Scale bar = 100 μm. (d) Multimodal imaging. Images were obtained 14 days after i.c. transplantation of LM8/luc. The dashed line indicates imaging section of the transverse image. Micro CT, micro X-ray computed tomography. (e) Aberrant bone formation due to osteoblastic bone metastasis in the femur and tibia. Micro X-ray CT images were obtained 21 days after i.c. injection of LM8 (upper panels). Scale bar = 1 mm. The lower panels indicate that von Kossa staining of the same metastasis-free (Meta−) and bone metastatic (Meta+) femurs as the upper panels. Scale bar = 500 μm.
Bone resorption induced by RANKL promotes LM8/luc metastatic colonization in bone marrow
Although osteoblastic metastasis lesions often contain osteolytic lesions in clinical patients,1,11 the role of bone resorption in osteoblastic bone metastasis has not been extensively studied. A clinical study showed that inhibition of RANKL increased bone metastasis-free survival of prostate cancer patients,14 implying that bone resorption might play important roles in establishment of osteoblastic bone metastasis. Our previous study revealed that RANKL-induced bone resorption started within 24 h of inoculation, and obvious bone loss was observed after 50 h.23 Therefore, in order to elucidate the role of bone resorption in establishment of osteoblastic bone metastasis, we used two different schedules of i.p. administration of RANKL: 5 days of consecutive administration initiated 1 h (RANKL1) or 4 days (RANKL2) after i.c. injection of LM8/luc (Fig.2a). An analysis using multimodal imaging revealed that treatment with RANKL clearly reduced bone mass 3 days after inoculation (Fig.2b), and that significantly higher BL signals (Fig.2c) and a significantly increased number of metastatic sites (Fig.2d) were observed in the bone of early RANKL-treated (RANKL1) mice than in those of untreated and late RANKL-treated (RANKL2) mice. Circulating cancer cells were dramatically reduced 24 h after cancer cell injection and undetectable on day 4, the day RANKL was given in the RANKL2 schedule (data not shown), suggesting that the chance of cancer cells entering the bone after day 4 was very low. These results strongly suggest that RANKL influences the fate of cancer cells that homed in the bone during the first 3 days after i.c. injection. The enhanced metastatic progression of LM8/luc cells by RANKL was not a direct effect of RANKL on LM8/luc cells, as LM8 cells do not express RANK, a receptor of RANKL (Fig. S3). These results indicate that RANKL-induced bone resorption promotes the colonization process of LM8 metastasis by influencing the microenvironment in the bone marrow.
Figure 2.
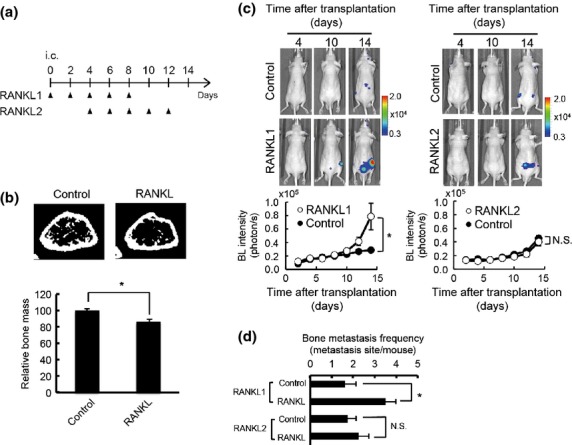
Receptor activator of factor-κB ligand (RANKL) promotes metastatic colonization of murine osteosarcoma LM8 cells in the bone. (a) Experimental protocol. PBS (Control) or RANKL (1 mg/kg) was injected i.p. at the indicated days after intracardiac (i.c.) transplantation of LM8/luc. Two treatment schedules, RANKL1 and RANKL2, are indicated. Mice were dissected 14 days after i.c. injection of LM8. (b) Representative micro X-ray computed tomography cross-section images of mouse femur 3 days after daily injection of PBS (control) or RANKL (1 mg/kg) (n = 5). *P < 0.05. (c) Representative bioluminescence (BL) in vivo images and quantitative analysis of bone metastasis in hind limbs. RANKL1 (left panel): n = 7 for Control; n = 19 for RANKL1); RANKL2 (right panel): n = 6 for both Control and RANKL2. *P < 0.05. (d) Bone metastasis frequency. RANKL1: n = 7 for Control; n = 11 for RANKL1; RANKL2: n = 4 for Control and RANKL2. *P < 0.05. N.S., not significant.
Hypoxia-inducible factor activity of LM8 increases in bone microenvironment
The pO2 in bone marrow has been estimated at 23–40 mmHg (3–5% O2),19 which is relatively lower than other organs and tissues. To understand the status of LM8 cells that have migrated to the bone marrow, we examined their hypoxic response in vitro and in vivo. The most abundant HIF, HIF-1, is a central regulator of the hypoxic response in many cell types15 and reported as a key factor in promoting homing, colonization, and progression of bone metastasis.28,29 In LM8 cells, the protein level of HIF-1α, the α subunit of HIF-1, increased as hypoxic treatment was prolonged (Fig.3a) and decreased as the O2 concentration increased (Fig.3b). The HIF transcriptional activity was monitored by using LM8/HRE-luc cells, which express firefly luciferase in a HIF-dependent manner. The activity of HIF in LM8/HRE-luc cells varied in response to hypoxic treatments (Fig.3c,d) in good correlation with HIF-1α expression (Fig.3a,b). In vivo BL signaling from LM8/luc, corresponding to tumor burden, and LM8/HRE-luc, corresponding to HIF activity, was monitored after i.c. injection of LM8 cells (Fig.3e). In s.c. tumors of LM8, HIF activity increased much more slowly than tumor burden (Fig. S4), suggesting that HIF activity in s.c. LM8 tumors reflects a gradual increase in hypoxic regions as tumors grow. However, in the bone metastatic sites, the HIF activity and tumor burden showed a parallel increase during the first week after LM8 injection (Fig.3e), indicating that HIF in LM8 was activated in the hypoxic microenvironment immediately after migration of LM8 to the bone marrow. Furthermore, HIF activity drastically increased in bone metastatic sites during the second week after LM8 injection (Fig.3e) and inhibition of HIF transcriptional activity by acriflavine24 significantly suppressed growth of LM8 bone metastasis (Fig.3f,g). These results support the idea that HIF is activated in LM8 homing to the hypoxic bone marrow and plays an important role in LM8 colonization and the progression of bone metastasis.
Figure 3.
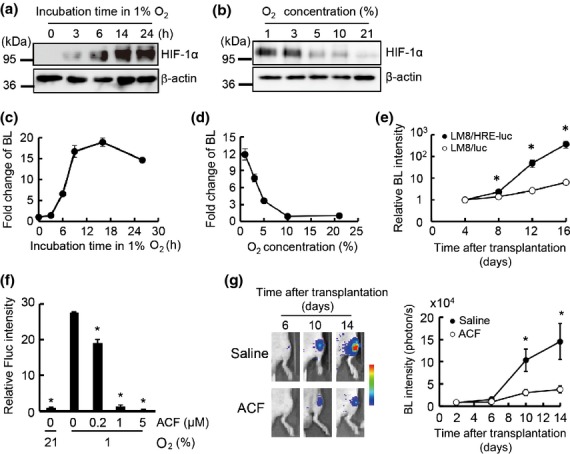
Hypoxia-inducible factor (HIF) activity of murine osteosarcoma LM8 cells is increased in the bone microenvironment. (a) HIF-1α protein levels in LM8 in 1% O2. (b) HIF-1α protein levels in LM8 under various O2 concentrations for 16 h. (c) Luciferase activity of LM8/HRE-luc in 1% O2. Relative luciferase activities to the one at 0 h are shown. (d) Luciferase activity of LM8/HRE-luc under various O2 concentrations for 16 h. Relative luciferase activity to the one in 21% O2 are indicated. (e) HIF activity in bone metastasis sites. Bioluminescence (BL) intensity in LM8/HRE-luc or LM8/luc metastases was measured at the indicated times after transplantation. To calculate relative BL intensities, BL intensities at indicated times were divided by those at day 4 (3.17 × 103 photons/s/ROI for LM8/HRE-luc and 3.43 × 103 photons/s/ROI for LM8/luc); n = 4 for LM8/HRE-luc, n = 6 for LM8/luc. *P < 0.01. (f) Inhibition of HIF activity in LM8 cells using acriflavine. HRE-Fluc activity was assessed with indicated concentrations of acriflavine (ACF). *P < 0.05. (g) Antitumor effect of HIF inhibitor. Luciferase activity of LM8/luc bone metastasis was measured at indicated times after transplantation. Relative luciferase activities to the ones on day 2 are indicated; n = 8 for each group. *P < 0.01.
Insulin-like growth factor-1 stimulates LM8 progression under hypoxic conditions
Treatment with RANKL induced significant bone resorption (Fig.2b) that triggers the release of growth factors stored in the bone. Therefore, the contribution of growth factors in cooperation with HIF activity to LM8 metastatic progression was examined. As TGF-β and IGF-1 are the major growth factors stored in the bone matrix, we first examined their effects on LM8 proliferation. Transforming growth factor-β as well as RANKL did not show any proliferative effects on LM8/luc under normoxia or hypoxia (Fig.4a), which was explained by the results of the qRT-PCR analysis indicating that LM8 did not express the TGF-β2 receptor (Fig. S5a). We further confirmed that the TGF-β/Smad pathway was not activated in LM8 after TGF-β treatment (Fig. S5b, Data S1). Thus, it was concluded that TGF-β did not have a direct effect on LM8 for RANKL-induced promotion of LM8 metastasis. In contrast, IGF/IGFR signaling seemed important because IGF-1 was able to promote proliferation of LM8 cells under both normoxia and hypoxia (Fig.4a). Furthermore, IGF-1 promoted anchorage-independent growth of LM8 cells in hypoxia (Fig.4b). Because the effect of IGF-1 on LM8 proliferation was more significant under hypoxia than normoxia, the activity of IGF/IGFR signals in LM8 cells was further examined in hypoxia. Remarkably, the Igf1r expression and IGF1R phosphorylation after IGF stimulation were significantly increased under hypoxic conditions (Fig.4c,d). We then assessed the involvement of IGF-1 in HIF activation in LM8 cells and found that IGF-1 treatment significantly enhanced the protein stability of HIF-1α and transcriptional activity of HIF in LM8 under hypoxic conditions (Fig.4e,f). Furthermore, we examined the expression level of downstream genes of the IGF/IGFR signal and HIF (Fig.4g). Under hypoxic conditions, Igf1 expression significantly increased (Fig.4g), suggesting that autocrine IGF-1 made the proliferation effect of exogenous IGF-1 on LM8 less significant in hypoxia (Fig.4a).
Figure 4.
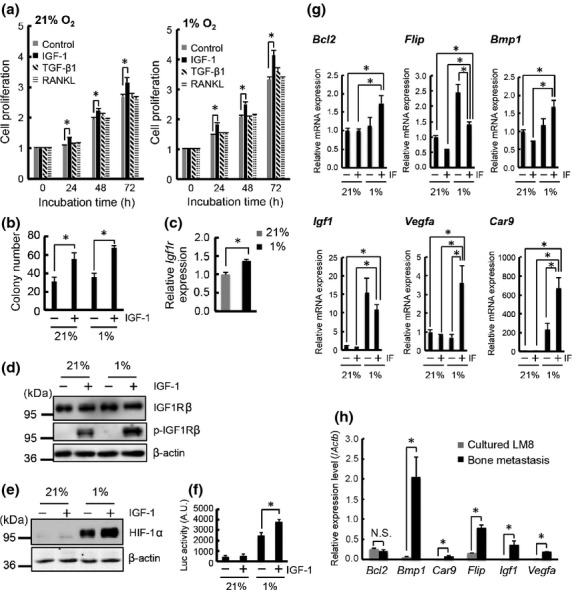
Insulin-like growth factor (IGF)/IGF receptor (IGFR) signaling and hypoxia cooperatively stimulate progression of murine osteosarcoma LM8 cells. (a) Proliferation assay of LM8/luc with IGF-1 (100 ng/mL), transforming growth factor-β1 (TGF-β1; 10 ng/mL), and receptor activator of factor-κB ligand (RANKL; 100 ng/mL) in 21% or 1% O2. (b) Colony formation assay of LM8/luc with IGF-1 (100 ng/mL) for 14 days in 21% or 1% O2. (c) Quantitative RT-PCR (qRT-PCR) analysis of Igf1r expression in LM8/luc cultured for 16 h in 21% and 1% O2. (d) Protein expression and phosphorylation levels of IGF1R. The cells were precultured for 16 h in 21% or 1% O2 then treated with IGF-1 (100 ng/mL) for 1 h. (e) Effect of IGF-1 on hypoxia-inducible factor-1α (HIF-1α) protein level. LM8 cells were treated with IGF-1 (100 ng/mL) for 12 h in 21% and 1% O2. (f) Effect of IGF-1 on HIF transcriptional activity. LM8/HRE-luc cells were treated with IGF-1 (100 ng/mL) for 12 h in 21% and 1% O2. *P < 0.05. (g) qRT-PCR analysis of downstream genes of IGF/IGFR signaling and HIF. LM8/luc cells were treated with IGF-1 (IF) (100 ng/mL) for 6 h in 21% and 1% O2. *P < 0.05. (h) qRT-PCR analysis of downstream genes of IGF/IGFR signaling and HIF in bone metastasis at 14 days after LM8 injection. *P < 0.05.
In addition to positive feedback regulation of Igf1, anti-apoptotic genes Bcl2 and Flip, and genes for adaptation in hypoxic bone marrow, Bmp1, Vegfa, and Car9, were upregulated with IGF-1 and hypoxia costimulation (Fig.4g). We further confirmed that most of these genes were significantly upregulated in the bone metastasis tissues (Fig.4h). These results indicate that IGF-1 and hypoxia cooperatively promote LM8 survival and proliferation and also suggest an in vivo mechanism for osteoblastic metastasis, in which bone resorption and hypoxia cooperatively enhance cancer cell colonization and growth.
Discussion
Understanding the mechanism of bone metastasis is becoming increasingly important for establishing an effective cancer therapy. In this study, we show that RANKL-induced bone resorption during the early stage of osteoblastic bone metastasis facilitated metastatic colonization of cancer cells.
Injection of RANKL in the early stage of metastasis facilitated the growth of metastatic cancers and increased the number of metastatic sites (Fig.2). Because LM8 does not express RANK (Fig. S3), RANKL does not directly influence the recruiting and homing processes of LM8 to the bone. Furthermore, although the same number of cancer cells was expected to have homed in the bone shortly after cancer cell injection in both RANKL1 and RANKL2 schedules, RANKL1 showed a higher incidence of metastasis and faster growth of metastatic cancer cells (Fig.2c,d). These results strongly suggest that RANKL-induced bone resorption plays an important role in colonization of cancer cells shortly after they enter the bone. We showed that IGF-1 increased the colony-forming ability of LM8 (Fig.4b) and that genes related to cell survival and proliferation were upregulated under hypoxic conditions similar to those in the bone marrow (Fig.4g) as well as in bone metastasis tissues (Fig.4h). The overall results are consistent with a previous study showing that IGF/IGFR signal activation reduces apoptosis of cancer cells in the bone marrow.30 Although further studies are necessary to determine the role of IGF-1 in the osteoblastic metastasis process, data from previous reports and ours strongly support the idea that bone resorption and subsequent release of IGF-1 play an important role in colonization and establishment of osteoblastic bone metastasis. Therefore, bone undergoing resorption would have a higher chance for bone metastasis and blocking of IGF/IGFR signaling would be an effective treatment for osteoblastic bone metastasis.
In osteoblastic bone metastasis, activated osteoblasts are thought to release growth factors leading to the proliferation of cancer cells, however, these growth factors have not yet been identified.9 In this work, we suggest that IGF-1 is the most reasonable candidate because IGF-1 promoted LM8 colonization at the metastatic sites, and because the IGF/IGFR signaling pathway, such as Igf1r expression and IGFR phosphorylation, was strongly activated in LM8 cells under hypoxic conditions (Fig.4c,d). We also showed that HIF was strongly activated in metastasized LM8 in the physiologically hypoxic bone microenvironment (Fig.3e), and that IGF-1 enhanced HIF expression and activity under hypoxic conditions (Fig.4e,f). These findings are consistent with previous studies showing that activation of HIF enhances cancer cell homing and metastatic progression in the bone marrow.16,17,28,29 Together with previous reports that IGF-1 activates HIF in various cancer cells,31,32 the cross-talk between IGF/IGFR signaling and hypoxia would be a key event that promotes osteoblastic bone metastasis (Fig.5), and is a potential target for effective therapy for bone metastasis.
Figure 5.
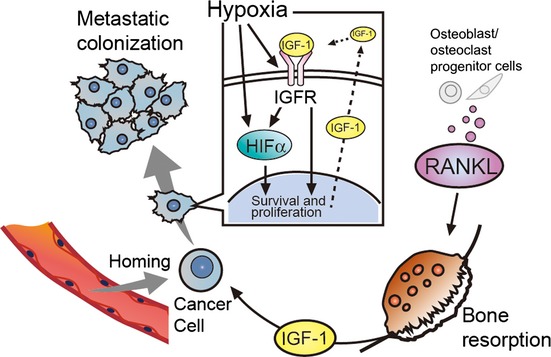
Diagram of receptor activator of factor-κB ligand (RANKL)-induced bone absorption that facilitates colonization of cancer cells through cooperative effects of insulin-like growth factor (IGF)/IGF receptor (IGFR) signaling and hypoxia. HIF, hypoxia-inducible factor.
Androgen deprivation therapy (ADT) is the mainstay of therapy for metastatic prostate cancer. However, use of ADT is associated with a decrease in bone mineral density33–35 and an increased incidence of fractures due to accelerated bone resorption.36 Therapy targeting RANKL has been used to reduce the incidence of osteoporosis and fracture in men with cases of non-metastatic prostate cancer receiving ADT.12 Our results clarify the main function of RANKL in promoting the metastatic colonization of circulating cancer cells in the bone marrow and support the validity of RANKL-targeted therapy for preventing osteoblastic bone metastasis.
In this study, we can rule out the direct involvement of RANKL and TGF-β in cancer cell colonization because LM8 does not express RANK or TGF-β receptor 2 (Figs S3,S5a). Therefore, by using this model, we may be able to clarify the essential function of TGF-β and RANKL on a variety of bone marrow cells, such as mesenchymal stem cells and monocytes, during osteoblastic metastasis. Further understanding of the interaction between these key factors and bone marrow cells elucidate bone metastasis mechanisms and would facilitate prevention therapies for bone metastasis.
Acknowledgments
We are grateful to Shigeaki Watanabe and Machiko Horiuchi (Summit Pharmaceuticals International Corporation, Tokyo, Japan) for technical support with IVIS, Isao Hamanaka and Takafumi Koike (Rigaku Corporation, Tokyo, Japan) for technical support with the R_mCT2 X-ray micro CT system, and Nobuhito Nango (Ratoc System Engineering, Co. Ltd., Tokyo, Japan) for bone analysis. This research was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Integrative Research on Cancer Microenvironment Network” from the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Mitsubishi Foundation (S. Kizaka-Kondoh) and a Grant-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows (T. Kuchimaru).
Disclosure Statement
H. Yasuda is an employee of Oriental Yeast Co. Ltd. None of the other authors have any conflicts of interest.
Funding information
Ministry of Education, Culture, Sports, Science and Technology of Japan. Mitsubishi Foundation. Japan Society for the Promotion of Science (JSPS) Fellows.
Supporting Information
Additional supporting information may be found in the online version of this article:
Coronal images of the entire body image shown in Figure 1(d).
Fig. S2 Histology analysis of LM8 osteoblastic bone metastasis.
Fig. S3 Protein level of receptor activator of factor-κB (RANK) in murine osteosarcoma LM8 cells.
Fig. S4 Quantitative analysis of hypoxia-inducible factor (HIF) activity in s.c. tumors.
Fig. S5 Analysis of transforming growth factor-β (TGF-β)/Smad signaling in murine osteosarcoma LM8 cells.
Primer list for quantitative RT-PCR analysis.
Materials and methods.
References
- 1.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–25. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roodman GD. Mechanism of bone metastasis. N Engl J Med. 2004;350:1655–64. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 3.Fukutomi M, Yokota M, Chuman H, et al. Increased incidence of bone metastasis in hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2001;13:1083–8. doi: 10.1097/00042737-200109000-00015. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Tiwari RC, Murray T, et al. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 5.Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci. 1998;95:3597–602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin JJ, Selander K, Chirgwin JM, et al. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastasis development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoneda T, Hiraga T. Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem Biophys Res Commun. 2005;328:679–87. doi: 10.1016/j.bbrc.2004.11.070. [DOI] [PubMed] [Google Scholar]
- 9.Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5:21–8. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Dai J, Qi Y, et al. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest. 2001;107:1235–44. doi: 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roudier MP, Morrissey C, True LD, Higano CS, Vessella RL, Ott SM. Histopathological assessment of prostate cancer bone osteoblastic metastases. J Urol. 2008;180:1154–60. doi: 10.1016/j.juro.2008.04.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berruti A, Dogliotti L, Gorzegno G, et al. Differential patterns of bone turnover in relation to bone pain and disease extent in bone in cancer patients with skeletal metastases. Clin Chem. 1999;45:1240–7. [PubMed] [Google Scholar]
- 13.Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastasis in men with castration-resistant prostate cancer: a randomized, double-blind study. Lancet. 2011;377:813–22. doi: 10.1016/S0140-6736(10)62344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith MR, Saad F, Coleman R, et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomized, placebo-controlled trial. Lancet. 2012;379:39–46. doi: 10.1016/S0140-6736(11)61226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 16.Hiraga T, Kizaka-Konodoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67:4157–63. doi: 10.1158/0008-5472.CAN-06-2355. [DOI] [PubMed] [Google Scholar]
- 17.Liang Y, Wu H, Lei R, et al. Transcriptional network analysis identifies BACH1 as a master regulator of breast cancer bone metastasis. J Biol Chem. 2012;287:33533–44. doi: 10.1074/jbc.M112.392332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 19.Martin SK, Diamond S, Gronthos S, Peet DJ, Zanettino CW. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia. 2011;25:1533–42. doi: 10.1038/leu.2011.122. [DOI] [PubMed] [Google Scholar]
- 20.Kizaka-Kondoh S, Itasaka S, Zeng L, et al. Selective killing of hypoxia-inducible factor-1-active cells improves survival in a mouse model of invasive and metastatic pancreatic cancer. Clin Cancer Res. 2009;15:3433–41. doi: 10.1158/1078-0432.CCR-08-2267. [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Okayama H. High-efficiency transformation of mammalian cells by DNA-plasmid. Mol Cell Biol. 1987;7:2745–52. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sasaki A, Boyce BF, Story B, et al. Bisphosphonate risedronate reduced metastatic human breast cancer burden in bone in nude mice. Caner Res. 1995;55:3551–7. [PubMed] [Google Scholar]
- 23.Tominori Y, Mori K, Koide M, et al. Evaluation of pharmaceuticals with a novel 50-hour animal model of bone loss. J Bone Miner Res. 2009;24:1194–205. doi: 10.1359/jbmr.090217. [DOI] [PubMed] [Google Scholar]
- 24.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci. 2009;106:17910–5. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Kizaka-Kondoh S, Sato K, Tamura K, Nojima H, Okayama H. Raf-1 protein kinase is an integral component of the oncogenic signal cascade shared by epidermal growth factor and platelet-derived growth factor. Mol Cell Biol. 1992;12:5078–86. doi: 10.1128/mcb.12.11.5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asai T, Ueda T, Itoh K, et al. Establishment and characterization of murine osteosarcoma cell line (LM8) with high metastatic potential to the lung. Int J Cancer. 1998;76:418–22. doi: 10.1002/(sici)1097-0215(19980504)76:3<418::aid-ijc21>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 27.Cao Y, Jia SF, Chakravarty G, Crombrugghe B, Eugenie S, Kleinerman ES. The osterix transcription factor down-regulates interleukin-1α expression in mouse osteosarcoma cell. Mol Cancer Res. 2008;6:119–26. doi: 10.1158/1541-7786.MCR-07-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunn LK, Mohammad KS, Fournier PG, et al. Hypoxia and TGF-β drive breast cancer bone metastasis through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE. 2009;3:e6896. doi: 10.1371/journal.pone.0006896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azab AK, Hu J, Quang P, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119:5782–94. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hiraga T, Myoui A, Hashimoto N, et al. Bone-derived IGF mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res. 2012;72:4238–49. doi: 10.1158/0008-5472.CAN-11-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–11. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 32.Sinha S, Koul N, Dixit D, Sharma V, Sen E. IGF-1 induced HIF-1α-TLR9 cross talk regulates inflammatory responses in glioma. Cell Signal. 2011;23:1869–75. doi: 10.1016/j.cellsig.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 33.Shahinian VB, Kuo YF, Freeman JL, Goodwin JS. Risk of fracture after androgen deprivation for prostate cancer. N Engl J Med. 2005;352:154–64. doi: 10.1056/NEJMoa041943. [DOI] [PubMed] [Google Scholar]
- 34.Smith MR, Lee WC, Brandman J, Wang Q, Botteman M, Pashos CL. Gonadotropin-releasing hormone agonists and fracture risk: a claims-based cohort study of men with nonmetastatic prostate cancer. J Clin Oncol. 2005;23:7897–903. doi: 10.1200/JCO.2004.00.6908. [DOI] [PubMed] [Google Scholar]
- 35.Smith MR, Boyce SP, Moyneur E, Duh MS, Raut MK, Brandman J. Risk of clinical fractures after gonadotropin-releasing hormone agonist therapy for prostate cancer. J Urol. 2006;175:136–9. doi: 10.1016/S0022-5347(05)00033-9. [DOI] [PubMed] [Google Scholar]
- 36.Morgans AK, Smith MR. RANKL-targeted therapies: the next frontier in the treatment of male osteoporosis. J Osteoporos. 2011;2011:941310. doi: 10.4061/2011/941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coronal images of the entire body image shown in Figure 1(d).
Fig. S2 Histology analysis of LM8 osteoblastic bone metastasis.
Fig. S3 Protein level of receptor activator of factor-κB (RANK) in murine osteosarcoma LM8 cells.
Fig. S4 Quantitative analysis of hypoxia-inducible factor (HIF) activity in s.c. tumors.
Fig. S5 Analysis of transforming growth factor-β (TGF-β)/Smad signaling in murine osteosarcoma LM8 cells.
Primer list for quantitative RT-PCR analysis.
Materials and methods.